
Abstract
A 47-year-old woman, presenting to her family physician with fatigue, was incidentally found to have persistently elevated ferritin. There was clinically no suggestion of iron overload, and laboratory testing showed transferrin saturation at the low end of the reference range. After ruling out acquired causes of hyperferritinaemia, as well as laboratory interference, further questioning revealed a history of bilateral early-onset cataracts, allowing a diagnosis of hyperferritinaemia cataract syndrome to be made. DNA sequencing of the 5′ untranslated region of the L-ferritin gene revealed a novel 4-base deletion in the iron response element, within a region known to be crucial for binding iron regulatory protein.
Introduction
Hereditary hyperferritinaemia cataract syndrome (HHCS) has been increasingly recognized as a cause of hyperferritinaemia without iron overload. 1 It is an autosomal dominant condition caused by mutations in the iron response element (IRE) of the L-ferritin gene (FTL). These mutations in the 5′ untranslated region of FTL interfere with the binding of ferritin mRNA to iron regulatory protein (IRP), leading to translational derepression of ferritin synthesis and constitutively high production of ferritin light chain protein, uncoupled from intracellular iron concentrations. This excess L-ferritin protein may precipitate in the lens of the eye, leading to bilateral early-onset cataracts, the only significant clinical consequence of this condition. The cataracts in HHCS have a distinctive granular appearance which, if recognized at the time of ophthalmologic examination, may lead to initial diagnostic suspicion of the condition. 2 Otherwise, HHCS may present as incidental hyperferritinaemia, as in the present case.
Clinical case
A 47-year-old woman, otherwise well, presented to her family physician with fatigue. She had previously undergone a hysterectomy for menorrhagia; her medical and surgical history was otherwise unremarkable. To rule out iron deficiency, the family physician ordered a serum ferritin. This was found to be 1283 µg/L, approximately 10 times the upper limit of normal. Repeated measurements at different laboratories over several months revealed similarly elevated values. The persistently elevated ferritin was noted during review and validation of results in the laboratory, triggering a detailed investigation.
The differential diagnosis for hyperferritinaemia is broad and includes hereditary haemochromatosis, iatrogenic iron overload, autoimmune and inflammatory conditions, severe infection, haematologic malignancy, liver disease and renal failure.3,4 Additional history did not suggest of any of these conditions. The patient had not received any prior transfusions or parenteral iron therapy. She was not aware of any family history of haemochromatosis or other heritable conditions. Physical examination was essentially normal, including a normal neurologic exam, and no hepatomegaly, splenomegaly or lymphadenopathy. Routine laboratory investigations revealed normal complete blood count and differential, normal liver enzymes and creatinine, normal serum protein electrophoresis pattern and normal C-reactive protein. Serum iron and iron-binding capacity were normal, at 16 µmol/L and 63 µmol/L, respectively, and transferrin saturation was 0.25, at the low end of the reference range (0.20–0.55).
Having ruled out the common acquired causes of hyperferritinaemia, as well as haemochromatosis (given the low-normal transferrin saturation), we were left to consider rare genetic causes such as aceruloplasminaemia, ferroportin disease and HHCS. 4 We also considered spurious hyperferritinaemia caused by heterophile antibody interference. The ferritin measurement was repeated after treating the sample in a heterophilic blocking tube (Scantibodies Laboratory Inc.), and no such interference was detected. Ceruloplasmin concentration was measured and found to be normal. Further questioning revealed that the patient had in fact had bilateral cataracts removed in her early 20 s. The specific appearance of the cataracts was not noted at the time of their removal. The ophthalmologist at that time would not have been able to make the diagnosis, since HHCS was not recognized as a clinical entity until 1995.
Having made the diagnosis of HHCS on clinical grounds, we obtained informed consent from the patient to test for FTL mutations, as follows:
Polymerase chain reaction (PCR) primers (5′-CCGGCGCACCATAAAAGAAGC-3′ and 5′-TTACCCGACCGCACAAAGAAGG-3′) 5 were synthesized, flanking the IRE in the 5′ untranslated region of FTL. PCR was performed on 35 ng of genomic DNA extracted from whole blood (Qiagen M48 DNA kit), using 1.5 U Platinum Taq (Invitrogen), 500 nmol/L each primer, 1.5 mmol/L MgCl2 and 200 µmol/L each dNTP in a 50-µL final reaction volume. Cycling consisted of initial denaturation at 95℃ for 2 min, followed by 30 cycles of 95℃ for 45 s, 56℃ for 30 s, 72℃ for 45 s, and final extension at 72℃ for 5 min. Agarose gel electrophoresis confirmed a single band at approximately 430 base pairs, corresponding to the expected size of the PCR product. Direct sequencing of the PCR product was attempted, using the same primers as above; however, the signal trace in the region of interest was too noisy to be interpretable. Therefore, the product band was extracted from the agarose gel (Qiagen MinElute gel extraction kit) and cloned into the vector pCR2.1-TOPO (Invitrogen). Individual clones were picked, and purified plasmid DNA from these was sequenced with a standard M13-reverse primer. Sequencing was performed at McGill University and Génome Québec Innovation Centre (Montreal, Canada) on an Applied Biosystems 3730xl DNA analyzer.
Approximately half of the clones (2 out of 5) showed wild-type sequence through the IRE, while the remainder showed a 4-base deletion (32_35del4 based on transcription start site), consistent with a heterozygous mutation (Figure 1).
Sequence trace of the 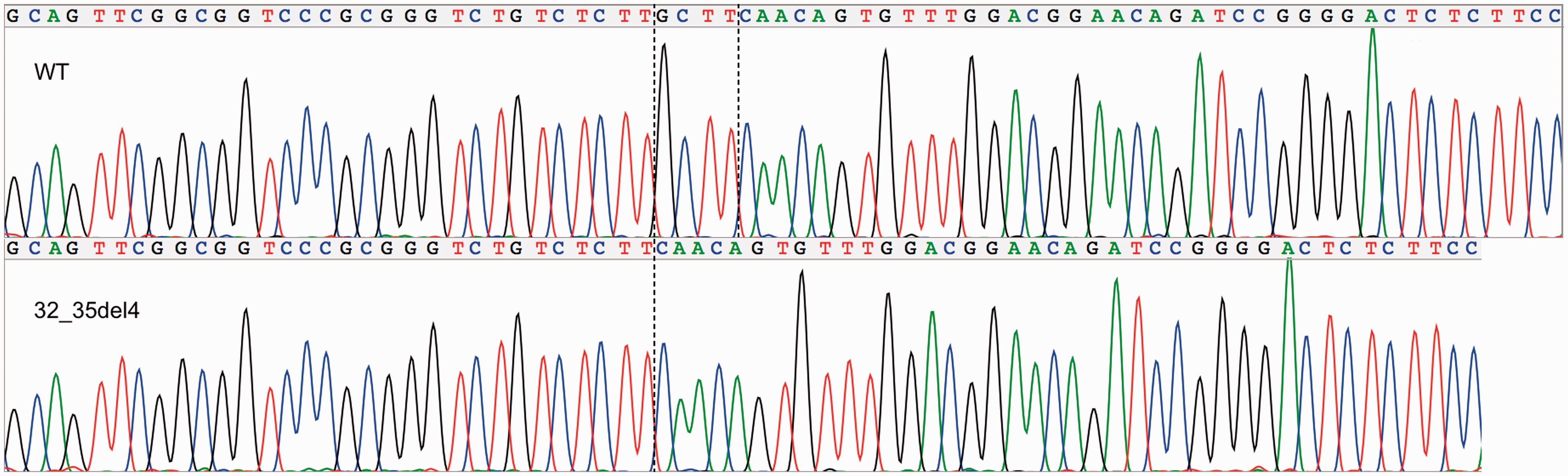
Additional family history was obtained from the patient, as follows. Her mother, age 73, had a ferritin of 159 µg/L and senile cataracts removed approximately five years prior. Her father was deceased at age 69 from myocardial infarction; he had no history of cataracts and no recorded ferritin measurement. She had two siblings: her sister, age 50, had no history of cataracts and a ferritin of 21 µg/L, and her brother had no history of cataracts (declined ferritin testing). Both maternal and paternal grandparents were deceased but had no history of cataracts as far as the patient was aware. Relatives were not tested for the FTL mutation. However, the absence of any family history of early-onset cataracts, and normal ferritin in the two immediate relatives for whom testing was available, raises the possibility that this may be a de novo mutation.
The patient has no children and has had a hysterectomy. She was counselled accordingly with respect to the heritability of the mutation. We reassured her that beyond the cataracts she had already had removed, there are no known clinical consequences of HHCS, and she would not require ongoing monitoring for the condition.
Discussion
HHCS should be considered in the differential diagnosis when there is persistent hyperferritinaemia with normal or low transferrin saturation. Once other congenital and acquired causes of hyperferritinaemia have been ruled out, these laboratory findings, along with a history of early-onset bilateral cataracts, are sufficient to make the clinical diagnosis. Historically, some patients have been misdiagnosed with haemochromatosis, with subsequent therapeutic phlebotomy leading to iatrogenic anemia. 6 In the absence of a history of cataracts, two other rare genetic causes of hyperferritinaemia to consider are aceruloplasminaemia and ferroportin disease. 4 There are also missense mutations in the coding region of FTL (p.Thr30Ile, p. Gln26Ile and p.Ala27Val) known to cause hyperferritinaemia associated with hyperglycosylation of ferritin, without iron overload or cataracts.7,8
HHCS normally follows an autosomal dominant pattern of inheritance. Cases of de novo mutation are also known, which have been referred to as ‘non-hereditary hyperferritinemia cataract syndrome’ by Cao et al. 9 or more simply ‘hyperferritinaemia cataract syndrome’.
There are numerous mutations in FTL known to cause HHCS.
1
They include both point mutations and small deletions affecting the stem-loop structure of the IRE. The 32_35del4 mutation detected in this case has not been previously reported. It encompasses the cytosine bulge, a conserved portion of the IRE stem-loop structure known to be crucial for binding iron-regulatory protein (Figure 2). X-ray crystallography of IRE-IRP complex has shown that this unpaired cytosine at position 33 inserts into a pocket in domain 4 of the IRP-1 protein.
10
In vitro binding experiments indicate that mutations affecting cytosine-33 greatly decrease IRP binding.9,11,12 Known HHCS-causing mutations affecting this position include 33C > A, 33C > U and 10_38del29.
1
Iron response element of L-ferritin, showing RNA secondary structure. Numbering based on transcription start site. The heterozygous deletion 32_35del4 was detected in our patient with hyperferritinaemia cataract syndrome. This encompasses the cytosine bulge at position 33, a highly conserved residue that participates directly in binding to iron regulatory protein.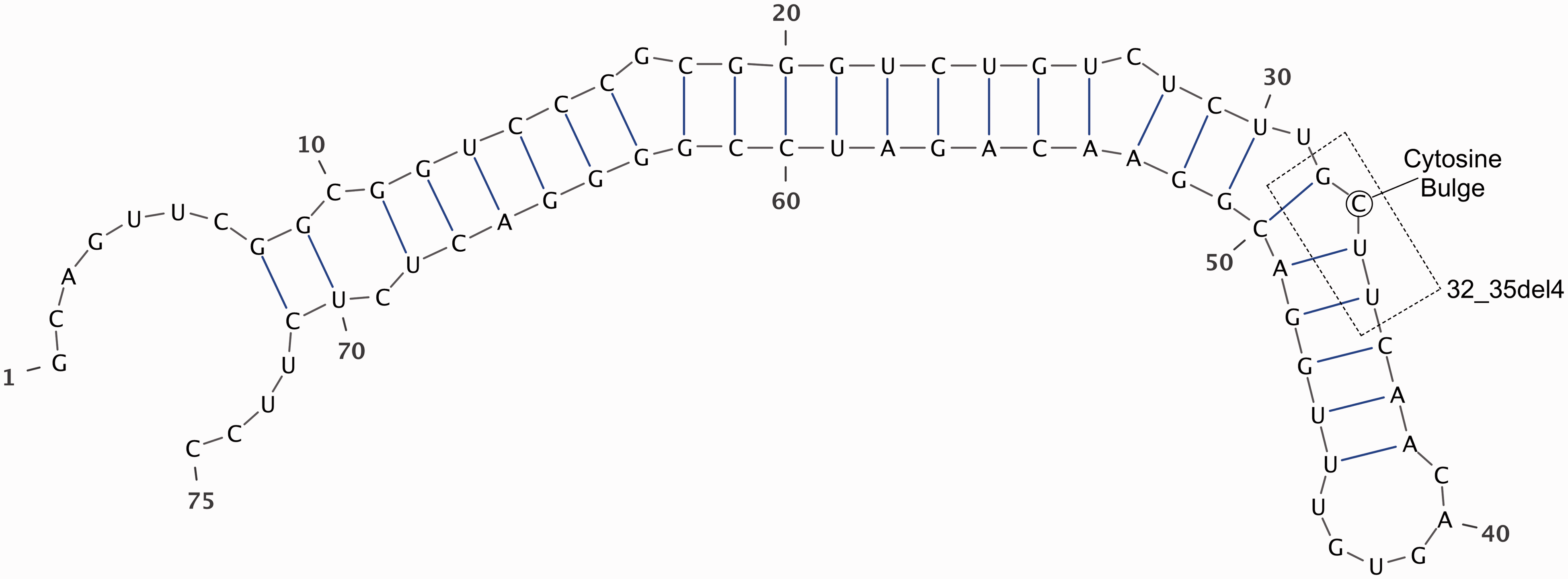
Footnotes
Acknowledgements
We would like to thank Sonja Babovic at the BC Cancer Research Centre (Vancouver, Canada) for invaluable assistance with cloning and sequencing, the patient for her permission to publish this case report and the patient’s family physician for providing additional history and clinical context.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
The patient provided informed consent for all investigations and to have this case report published.
Guarantor
IG.
Contributorship
IG performed literature research, conceived and carried out molecular studies and wrote the manuscript. MP provided lab result consultation, advised on investigation strategy and reviewed the manuscript.