
Abstract
Alpha-1-antitrypsin (AAT) is a protease inhibitor (PI), deficiency of which is associated with emphysema and liver disease. The most common deficiency alleles are the S (p.Glu288Val) and Z (p.Glu366Lys) alleles. The Z allele predisposes the AAT protein to polymerization with accumulation in hepatocytes leading to liver disease in PIZ individuals. Most AAT variants have a characteristic pattern of isoforms by isoelectric focusing (IEF). A novel AAT variant called PIZbristol (p.Thr109Met) with an unusual pattern on IEF was described in 1997. We report a patient with the PIZZbristol phenotype that has not been previously described. A 43-year-old man was referred by his GP to a respiratory clinic for breathlessness. His AAT concentration was 0.50 g/L (reference range 1.0–2.0 g/L). An unusual pattern on IEF was seen and sequencing revealed the presence of the rare variant Zbristol in combination with the Z mutation. This is the second reported case of Zbristol and the first in combination with the Z mutation. The patient maintained plasma AAT concentrations around 0.50–0.70 g/L which suggested that the Zbristol protein contributed to the low plasma concentration of AAT. The clinical symptoms associated with PIZ are usually attributed to the plasma deficiency, but his only respiratory complaint was that of breathlessness. This suggests that the PIZZbristol phenotype may confer an effect on respiratory function but is not involved in liver disease.
Introduction
Alpha-1-antitrypsin (AAT) is a member of the superfamily of serine protease inhibitors, its principle function being the inhibition of neutrophil elastase. The AAT protein is a 52 kDa glycoprotein encoded by a gene SERPINA1, which is highly polymorphic with more than 120 single nucleotide polymorphisms (SNPs) described in the literature. The normal allele is designated M (PIM). The most common deficiency alleles are the S (p.Glu288Val) and Z (p.Glu366Lys) alleles (using the HGVS standard mutation nomenclature) which were previously referred to as p.Glu264Val and p.Glu342Lys, respectively. These alleles are found to occur at a frequency in the UK of 0.005–0.15 for S and <0.04 for Z.1,2
In adults AAT deficiency can often present with non-specific pulmonary symptoms such as coughing and wheezing or the more severe chronic obstructive pulmonary disease (COPD), emphysema or chronic bronchitis. Despite the prevalence of AAT deficiency estimated at 3.4 million worldwide it has been reported that the disease is still often undetected or misdiagnosed. One study reported the average interval between the onset of pulmonary symptoms and diagnosis was 8.3 ± 6.9 years and the average number of physicians seen before AAT deficiency was correctly diagnosed was 2.7 ± 2.4. 3
The Z allele predisposes the AAT protein to polymerization. These polymers accumulate in hepatocytes so that in PIZ individuals only 15% of the AAT molecules produced actually reach the circulation. Carriers of either defective allele, i.e. PIMS or PIMZ were found to be at increased risk for the development of cirrhosis, malignant hepatoma, chronic liver failure as adults and COPD.1,4 PIMZ and PISZ heterozygotes can also be at increased risk of emphysema depending on other environmental factors such as smoking history.
In 1997, Lovegrove et al. described a novel AAT variant called PIZbristol that had an unusual phenotypic pattern on isoelectric focusing. 5 This protein was found to be fully functional but the N-terminal glycosylation site was removed which altered the isoelectric point (pI) of the Zbristol protein causing it to migrate further towards the cathode.
Here we report a patient whom we identified as having the PIZbristol mutation in compound heterozygosity with the PIZ mutation, a genotype which has not previously been described.
Case report
A 43-year-old man was referred by his general practitioner (GP) to a respiratory clinic for breathlessness and a history of chronic fatigue. He had no history of chest infections and had no cough. He was an ex-smoker but his spirometry was supra-normal for his age and height. His AAT concentration had been measured previously and was 0.50 g/L (reference range 1.0–2.0 g/L) and his genotype had been determined as PIZ. His mother had also been tested and was found to have the same genotype. There was no evidence of liver disease.
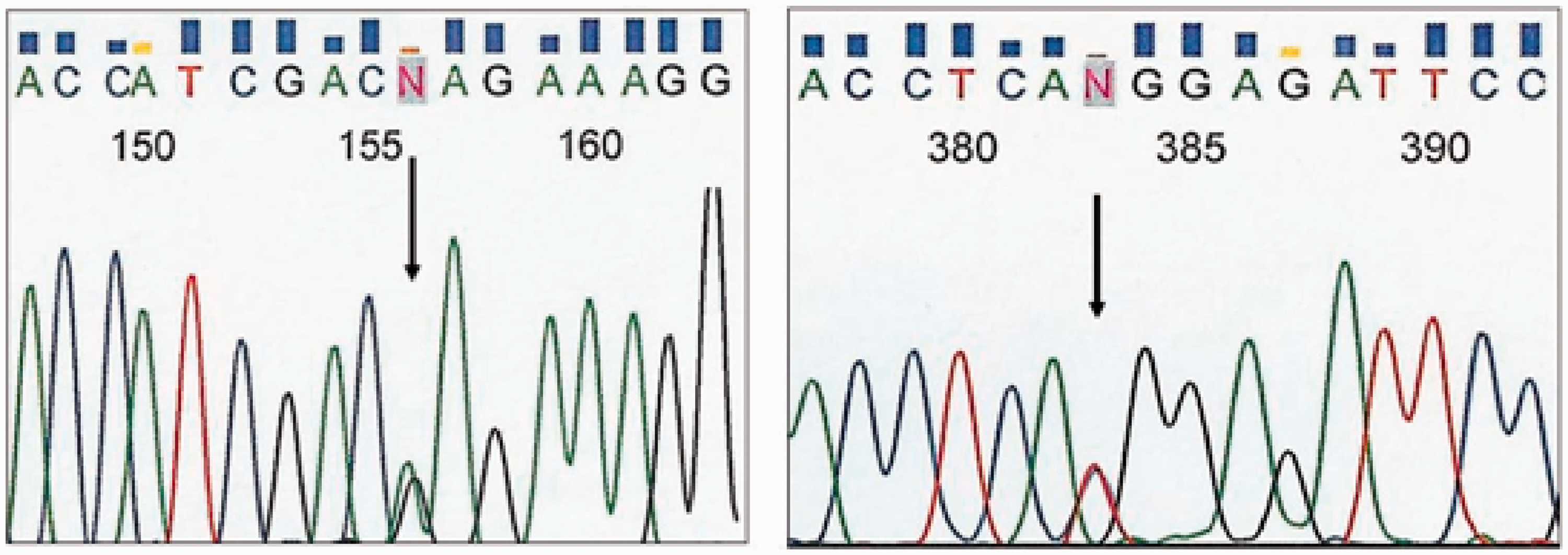
The AAT concentration of 0.50 g/L and no significant respiratory symptoms with well-preserved spirometry was not consistent with a diagnosis of PIZ, thus the AAT genotyping was re-tested at a different location and the patient's phenotype was also tested using isoelectric focusing. Repeat testing indicated a PIZ (Het) genotype (testing was only for the S and Z alleles) and an unusual pattern on the isoelectric focusing (Figure 1), which showed no M bands and indicated the presence of a variant form of AAT together with the Z mutant protein. Sequencing analysis of the SERPINA1 gene was performed which detected the Z allele (nucleotide change c.1096 G > A, p.Glu366Lys) and an additional nucleotide change c.326 C > T (p.Thr109Met using the HGVS nomenclature, previously referred to as p.Thr85Met, Figure 2) which was identified as the rare variant Zbristol.
Isoelectric focusing results showed the presence of the Z protein and an additional band below. No normal (M) protein band was seen in the patient. Sequencing analysis showing the patient was heterozygous for the Z allele (left panel) and for the Zbristol variant (right panel). Coupled with the isoelectric focusing results which showed no M bands to be present, these data demonstrate the patient had the two mutations in trans.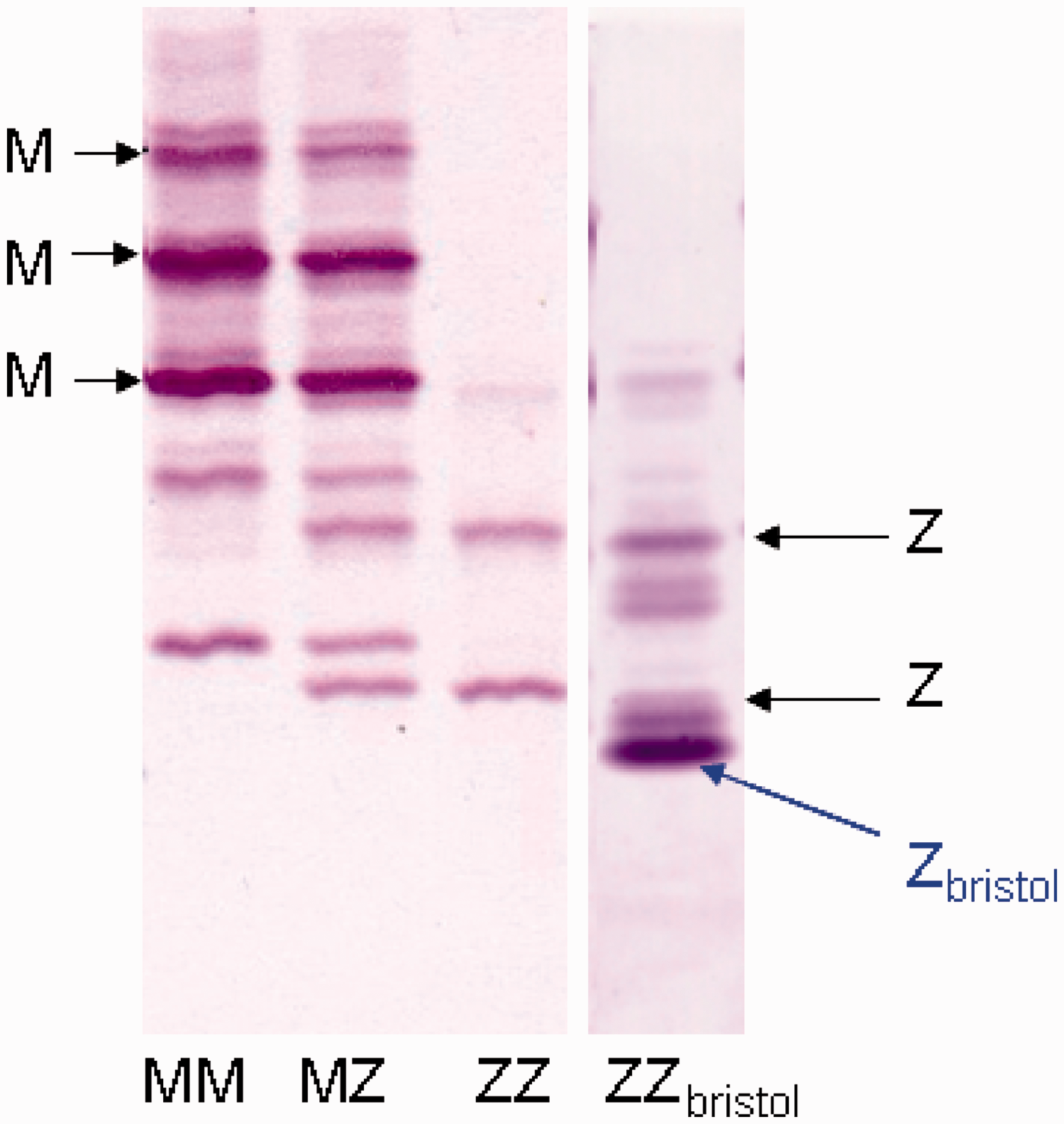
Materials and methods
Taqman PCR was performed on the extracted DNA to test for the AAT S (p.Glu288Val) and Z (p.Glu366Lys) mutations. DNA sequencing was performed on the coding regions and splice junctions of the SERPINA1 gene using Big Dye terminators on an ABI 3130 genetic analyser and aligned with reference sequence NG_008290.1 and NP_000286.3. Serum AAT phenotyping by isoelectric focusing was performed using a Sebia Hydrasys Focusing system. For further method detail see the Supplementary file.
Discussion
Here we describe the second reported instance of the rare AAT variant Zbristol. The first report by Lovegrove et al. 5 detailed one family (three generations) with the PIMZbristol genotype. The Zbristol protein was reported to be functional since it inhibited trypsin activity in vitro and it was stated that the proband in the study had a low plasma concentration of AAT, although the actual AAT concentration was not stated. The proband had three offspring, all of whom died soon after birth due to fulminant liver disease. One of the three offspring was shown to have the MZbristol genotype. It was not certain whether the Zbristol variant had a role in the pathology since only one of the deceased offspring was found to carry the mutation (one offspring had the PIM genotype and one offspring was not tested). Additionally, both the proband and her father (also a PIMZbristol) were healthy in childhood and had not suffered from liver or lung disease as adults.
In this report, we describe the variant Zbristol in compound heterozygosity with the more common Z (p.Glu366Lys) mutation which is well-documented. Typically, PIMZ heterozygotes have AAT concentrations around 60% of normal. 6 The patient described here maintained plasma AAT concentrations around 0.50–0.70 g/L which is lower than expected for a PIMZ genotype, but would be uncharacteristically high for a PIZ genotype and more typical of the PISZ genotype. Samandari and Brown 7 carried out some experiments in vitro using COS-1 cells transfected with AAT proteins containing mutations designed to affect the glycosylation sites. A mutant protein corresponding to the Zbristol mutation described in vivo showed a marked decrease in secretion and intense intracellular ER staining, suggesting the Zbristol variant is largely retained in the ER of hepatocytes. It does, therefore, appear that the Zbristol protein contributed to the low plasma concentration of AAT, possibly caused by retention of Zbristol in the hepatocytes together with the AAT Z protein.
The clinical symptoms associated with PIZ are usually attributed to the plasma deficiency caused by the accumulation of the AAT Z protein in the hepatocytes. This deficiency of AAT can lead to lung injury and eventually emphysema, especially in individuals with a history of smoking. Pulmonary disease caused by AAT deficiency usually has an earlier age of onset (fourth or fifth decade of life) than smoking-related COPD (sixth or seventh decade of life). 8 In this case the patient was in his fifth decade of life with a history of smoking, but the only respiratory complaint was that of breathlessness. This suggests that the PIZZbristol genotype does not confer any significant effect on respiratory function which is likely due to the AAT concentration in this patient being above the protective threshold.
Similar to the AAT Z protein the Zbristol form of AAT is thought to be retained in the endoplasmic reticulum of hepatocytes based on studies performed by Samandari and Brown, 7 although the mechanism for the retention (i.e. whether Zbristol forms polymers that are retained) is unknown. The case study presented here offers no evidence to suggest that the Zbristol protein is involved in liver disease.
This case also highlights the importance of using a combination approach to identify cases of AAT deficiency. Use of the genotyping assay alone in this case would have identified this patient as PIMZ; full gene sequencing was only performed due to the unusual isoelectric focusing result. In a similar case, Zhan et al. 9 identified a novel AAT variant with a pathogenic effect by sequencing the SERPINA1 gene of a patient who was identified as PIMZ by isoelectric focusing but had a disproportionately low plasma AAT concentration of 0.35 g/L. Sequencing revealed the presence of a novel mutation (p.Met409Thr) which was undistinguishable from the normal M allele on isoelectric focusing. These cases demonstrate the importance of using a protocol to identify AAT deficiency using a combination of either AAT plasma concentration or isoelectric focusing in addition to screening for the common AAT mutations by genotyping. Nucleotide sequencing should then be performed if the AAT concentrations or isoelectric focusing results are not consistent with the genotyping results to identify the presence of any rare AAT variant alleles.
Footnotes
Declaration of conflicting interests
Not applicable.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not required.
Guarantor
KJB.
Contributorship
KJB carried out the initial genotyping and wrote the first draft of the paper, MJP carried out the isoelectric focusing, MH and NK carried out and interpreted the sequencing, AB provided the clinical details, BEC advised on the genotyping and RAS edited the manuscript.