
Abstract
Phaeochromocytoma and extra adrenal paraganglioma are rare neuroendocrine tumours and have the potential to secrete adrenaline, noradrenaline and dopamine causing a myriad of clinical symptoms. Prompt diagnosis is essential for clinicians and requires a multidisciplinary specialist approach for the clinical and laboratory investigation, diagnosis, treatment and follow-up of patients. This paper is an integrated review of the clinical and laboratory evaluation and treatment of patients suspected to have phaeochromocytoma or paraganglioma, highlighting recent developments and best practices from recent published clinical guidelines.
Introduction
Phaeochromocytomas are rare neuroendocrine tumours that arise from sympathetic adrenomedullary chromaffin tissue.1,2 These tumours usually produce one or more catecholamines: noradrenaline (NAd), adrenaline (Ad) and dopamine (DP). In contrast, paragangliomas are derived from extra-adrenal chromaffin tissue of the paravertebral ganglia of abdomen, pelvis and thorax. Differences in clinical features and behaviour suggest that they should be regarded as different diseases. 3
Paragangliomas do not typically secrete Ad due to the reduced expression peripherally of the cortisol-dependant enzyme phenylethanolamine-N-methyltransferase, which converts NAd to Ad. 4
Paragangliomas can also arise from parasympathetic ganglia found along the glossopharyngeal and vagal nerves in the neck, and base of the skull. 2 The latter are usually biochemically silent.
Phaeochromocytomas account for approximately 85%, and sympathetic and parasympathetic paragangliomas 15–20%, of chromaffin cell tumours. 5 For simplicity, throughout this article phaeochromocytomas and paragangliomas will be referred to as PPGL.
The prevalence of PPGL varies according to the population studied. In an outpatient hypertensive population, the prevalence is reported as 0.2–0.6%.6–9 Autopsy studies show that 0.05–0.1% of patients have undiagnosed tumours.10–12 It has been suggested that the prevalence of the latter is falling as more PPGL are being picked up, while people are alive owing to better screening tests and more incidental findings during imaging for other reasons. It is estimated that 1.5–14.0% of incidentally discovered adrenal masses found on computerized tomography (CT) or magnetic resonance imaging (MRI) are phaeochromocytomas.13–15
The most common indications for biochemical screening for PPGL are: (a) the presence of both hypertension and episodic symptoms of catecholamine excess (e.g. palpitations, headache and periods of diaphoresis); (b) treatment of resistant hypertension; (c) assessment of functionality of incidentally identified adrenal masses; (d) predisposition to hereditary PPGL and (e) assessment of recurrence of PPGL.
Clinical presentation
PPGL are notoriously difficult to diagnose, especially for the non-specialist clinician as they are rare in the community. Most clinicians never see a patient with a PPGL, and the symptoms can be mixed and if not clearly analysed are similar to those of many diseases, including anxiety attacks. Moreover, unless the diagnosis is considered, it cannot be reached. Symptoms vary according to which catecholamine is being secreted, and therefore which α- and/or β-receptor(s) are being stimulated. Only 30–40% of patients demonstrate the classic triad of headache, palpitations and diaphoresis associated with functional PPGL.16,17 In many patients, hypertension will be associated with these symptoms. Patients may have been diagnosed with hypertension previously and treated with polypharmacy due to the resistance of the hypertension. This group of patients along with young hypertensive patients (<40 years old) should be screened for causes of secondary hypertension, including PPGL. Catecholamine secretion can be episodic or continuous; this variable symptomatology contributes to the difficulty in making the diagnosis.
Resistant or labile hypertension
Hypertension is present in 70–80% of patients with PPGL.16,17 It may be described as labile or paroxysmal (37%) but is often persistent hypertension (63%). 17 Persistent hypertension is more usual with a predominant NAd-producing tumour, whereas Ad is associated with orthostatic and paroxysmal hypertension. 18 Labile hypertension can cause difficulties in treatment, resulting in the trial of many different antihypertensive drugs at different dosages. This trial of different agents without success should raise the suspicion of the potential presence of PPGL. When hypertension appears resistant to single therapy, one should be suspicious of a possible PPGL. This tends to be more prevalent in the young hypertensive population, and while PPGL only accounts for a small percentage (2–4%) of these patients,19,20 it is curable and so should be screened for.
Normotensive patients
Although frequently associated with hypertension, 5–55% of patients will be normotensive.16,17,21 Due to this, they may be incidentally discovered or go undiagnosed without the paroxysmal nature of the hypertension or the presence of orthostatic hypotension on standing. Lower concentrations of catecholamines may be seen more in normotensive PPGL patients than in those who are hypertensive. While it makes sense that the extent of symptoms is related to the concentration of catecholamines produced, this does not necessarily correspond to size of the tumour on imaging. 17
Palpitations/cardiac events
Palpitations (runs of tachycardia sometimes associated with anxiety but, in the absence of a causative stressful event) may result from PPGL. In such situations, the symptoms may be mistakenly attributed to anxiety 22 or a stress response provoked by a particular event. Palpitations usually result from the stimulus of β1-receptors. PPGL can also present with cardiomyopathies, pulmonary oedema and cardiac arrests. 23
Sweating and pallor
Stimulation of the α1-receptor causes sweating and is seen in a number of patients with PPGL.
A typical loss of colour or pallor is seen in PPGL patients, where they are described as going ‘white’ due to vasoconstriction caused by α1-receptor stimulation. Vasoconstriction can also be peripheral. This can be observed when the α1-receptors are blocked in preparation for surgery, when often there is a visible change in skin colour and temperature.
Headache
Headaches are often precipitated by the increase in blood pressure and vasodilation and are more common in hypertensive PPGL patients than in normotensive patients. 17
Abdominal pain
Abdominal pain can be an associated symptom of PPGL in a stressful event, precipitating an acute crisis. 24
Hyperglycaemia
Diabetic ketoacidosis, type 2 diabetes and hyperglycaemic crisis have been reported as a presenting feature of PPGL, especially when there are atypical features such as labile hypertension or a patient’s failure to respond to insulin therapy.25,26
Catecholamines normally prevent hypoglycaemia by promoting lipolysis, gluconeogenesis and glycogenolysis through stimulation of β2-receptors and are a cause of insulin resistance. The excess uncontrolled secretion of catecholamines from a PPGL can therefore result in hyperglycaemia. Elective surgical treatment can lead to a reduction in dose of diabetic medication or even cure the patient’s ‘diabetes’. 27
Unusual symptoms
As there are numerous effects resulting from α- and β-receptor stimulation, PPGL can less commonly present with symptoms including hypercalcaemia and hypercortisolaemia 28 due to the increased secretion of ACTH and thromboembolic events.29,30
Phaeochromocytoma crisis
PPGL crisis is defined as an acute severe presentation of a catecholamine-induced haemodynamic instability causing end-organ damage or dysfunction. 31 It can be divided into types A and B. Type A represents a limited crisis with haemodynamic instability and end-organ dysfunction or damage of one or more organs. Type A can progress to type B, which is an extensive crisis with the patient going into shock. Type B crisis is characterized by sustained hypotension and multiorgan damage of two or more systems. 32 This diagnosis can be confused with myocardial infarction, septic shock and cardiomyopathy. 30
A crisis can be precipitated by many factors including a general anaesthetic in an unprepared patient, direct trauma to the tumour, drugs including metaclopramide and some contrast agents. Type A crisis carries a 6% mortality, while type B confers a higher mortality of 28%. 32
Investigation
Biochemical screening
Potential advantages and disadvantages of measuring plasma and urine metadrenalines.
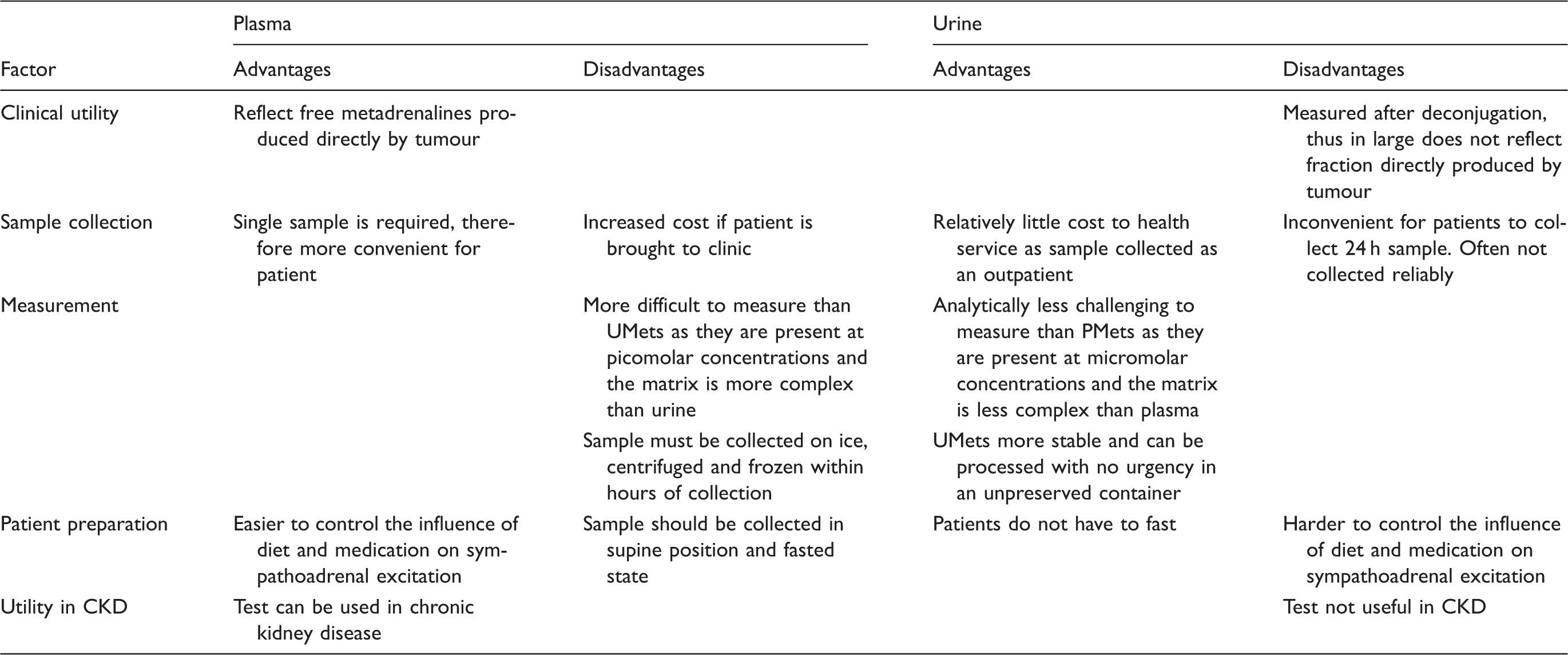
Note: Adapted from Pacak et al. 33
CKD: chronic kidney disease; UMets: total fractionated urine metadrenalines; PMets: plasma-free metadrenalines.
Diagnostic performance characteristics of plasma and urine metadrenalines.
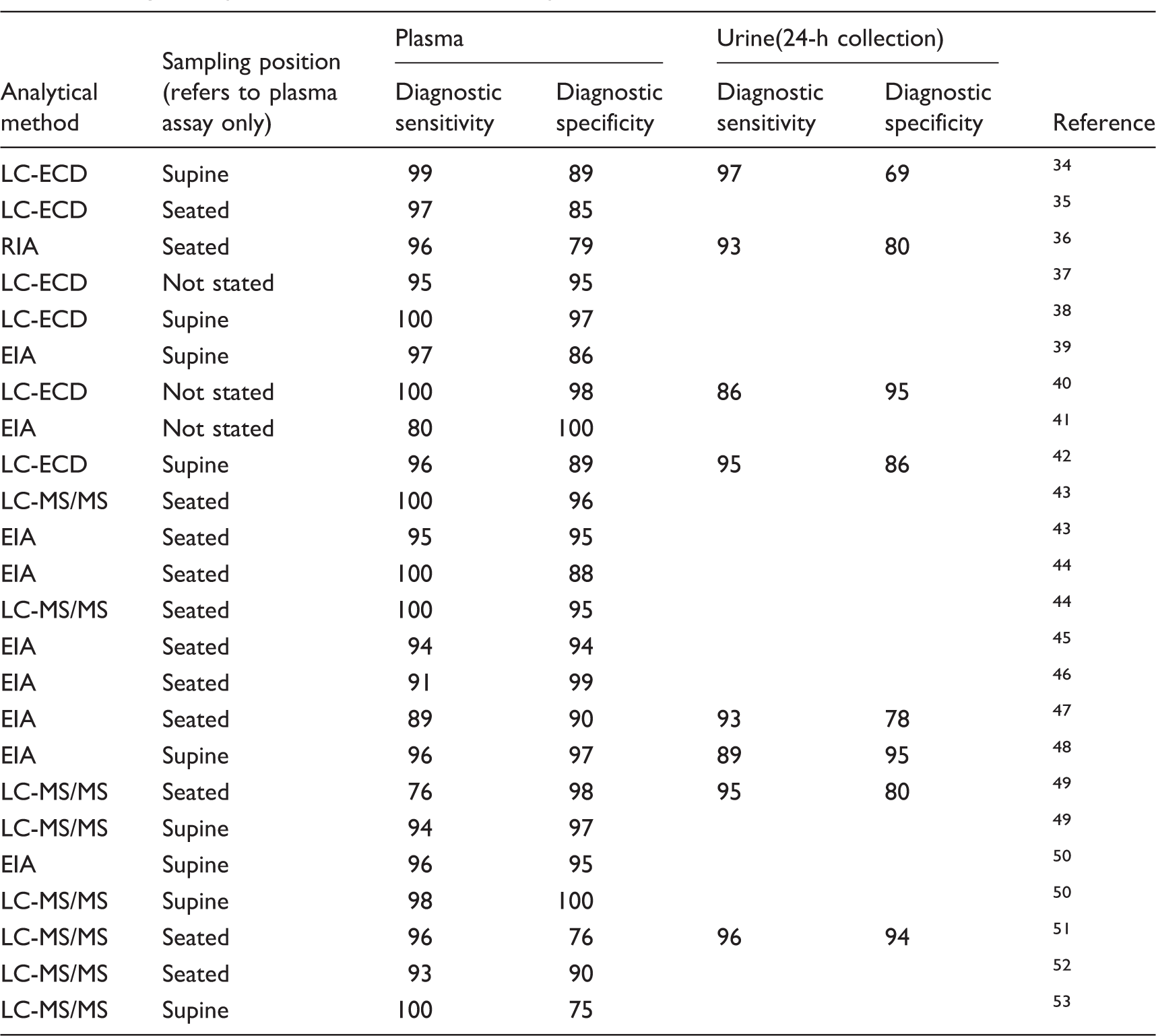
Note: Data from studies published between 2002 and 2017. The performance characteristics vary according to cut-off used for metadrenalines.
LC-ECD: liquid chromatography-electrochemical detection; RIA: radioimmunoassay; EIA: enzyme immunoassay; LC-MS/MS: liquid chromatography tandem mass spectrometry.
While there have been differences observed in the diagnostic sensitivity and specificity of UMets and PMets using different analytical methodologies, to date, there has not been a multicentre comparison using the gold standard method 1 of liquid chromatography tandem mass spectrometry (LC-MS/MS) for analysis.
Measurement of 3-methoxytyramine (3-MT) is not recommended for the investigation of PPGL, but it has the potential to distinguish patients with and without metastases and may be useful in identifying patients with inherited forms of PPGL.54–56
3-MT is a diagnostically more sensitive biomarker of tumoural DP production than plasma or urinary DP in patients with mutations in the succinate dehydrogenase type B (SDHB) gene.54–56
The greatest challenge associated with the interpretation of UMets and PMets results is the ‘borderline-positive reference range’ in patient populations with a low prevalence of PPGL.
Analytical and pharmacological factors responsible for potential interferences in the measurement and interpretation of metadrenalines.
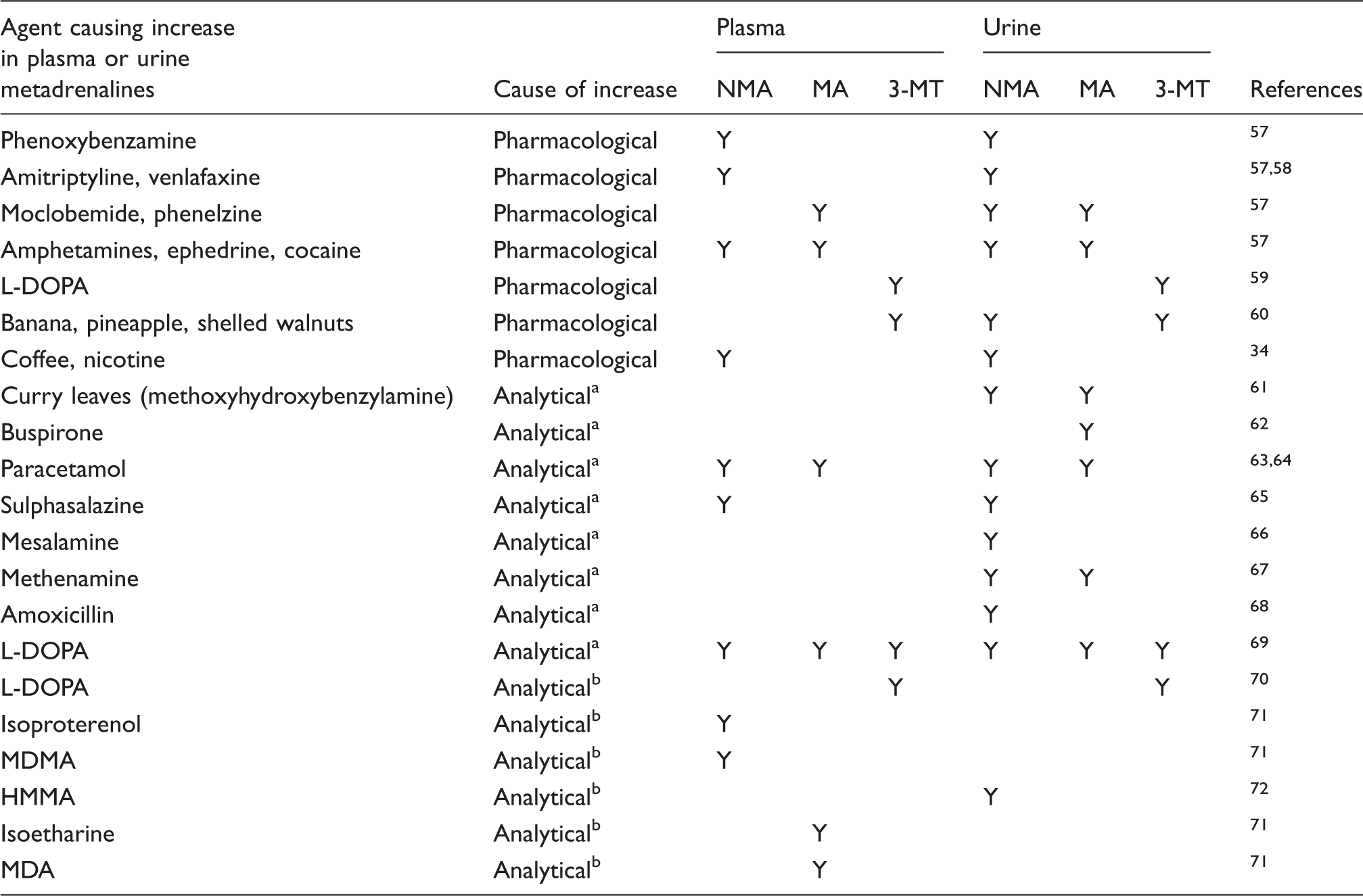
NMA: normetadrenaline; MA: metadrenaline; 3-MT: 3-methoxytyramine; MDMA: 3,4-methylenedioxymethamphetamine; MDA: 3,4-methylenedioxyamphetamine; HMMA: 4-hydroxy-3-methoxymethamphetamine.
Liquid chromatography-electrochemical detection method.
Liquid chromatography tandem mass spectrometry method.
Defining the pretest probability of a PPGL in an individual patient is essential to the appropriate interpretation of results, which should not solely be based on the magnitude of the increase above the reference range. 5
Assurance when one evaluates the importance of a borderline result can be gained by collecting a repeat plasma or urine sample under standardized low-stress conditions, for example, collecting a fasting plasma sample in the supine position after 20 min rest and avoiding urine collections after exercise. 1 Darr et al. 49 recently reported that the collection of a plasma sample in a non-fasting ambulant population can give false-positive rates of up to 30% compared with a 5% false-positive rate in patients, where the samples were collected under supine and fasted conditions.
In contrast, Boote et al. 52 reported in a small study that diagnostic sensitivity and specificity were acceptable for PMets when a sample was collected in a seated position for detection of PPGL. However, it is the authors’ view that where practicable samples should be collected after fasting in the supine position.
Additional reassurance can be achieved by demonstrating similar patterns of NMA and MA in the plasma and urine, thus it is the authors’ recommendation in such situations that paired samples are collected. Moreover, patients should have any medication that may cause an analytical interference or result in a pharmacological increase in MAs (Table 3) modified and/or any underlying conditions managed optimally before re-testing.57–72
Genetic evaluation
Genes known to play a role in the development of PPGL (both hereditary and somatic) including associated syndromes and clinical and biochemical presentations.
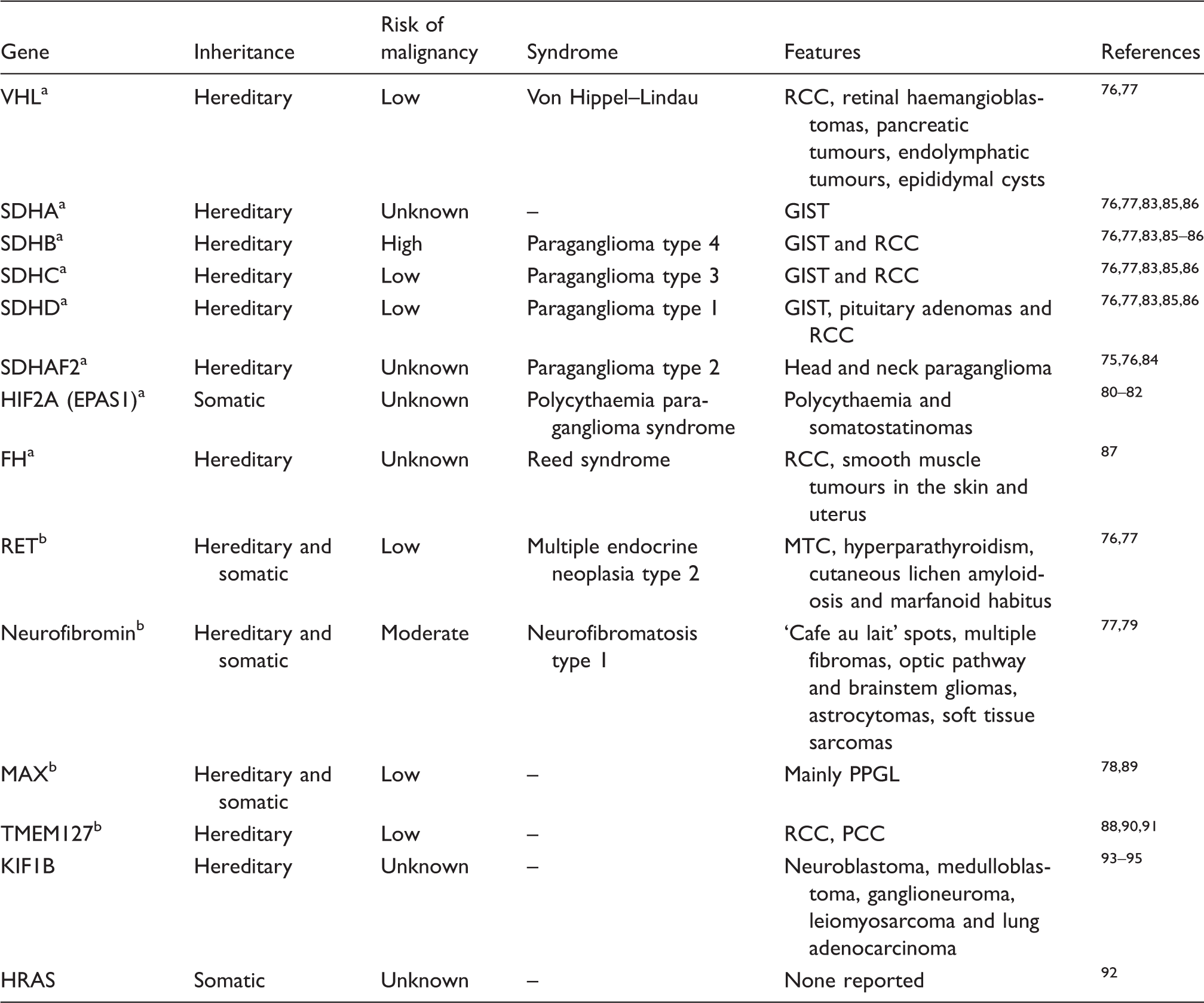
FH: fumarate hydratase; GISTs: gastrointestinal stromal tumours; HIF2: hypoxia-inducible factor 2; MAX: MYC-associated factor X; MTC: medullary thyroid carcinoma; RCC: renal cell carcinoma; MTC: medullary thyroid cancer; SDH: succinate dehydrogenase; TMEM127: transmembrane protein 127; VHL: von Hippel–lindau; RET: rearranged during transfection.
Cluster 1 mutation.
Cluster 2 mutation.
Mutations discovered to date cluster into two groups. Cluster one mutations encompass genes that are involved with the activation of the hypoxic response via specific transcription factors known as hypoxia-inducible factors (HIFs). Mutations in the genes responsible for regulation of these transcription factors can result in HIF pathways becoming constitutively active, leading to pseudohypoxic conditions that result in cell proliferation and angiogenesis.73,74
Cluster 2 mutations affect genes that are involved with kinase signalling pathways (notably MAPK and mTOR pathways). This leads to the activation of downstream targets that promote cell proliferation and survival.75–78
Genetic testing is relatively expensive; therefore, only the most likely mutations are tested for in a given patient. The decision on which mutations to test for is guided by clinical presentation, biochemical testing and imaging. The clinical presentation of type-1 neurofibromatosis (NF-1) is usually apparent so that genetic testing in not usually required. 79 Conversely, mutations in the SDHx gene can have major implications on prognosis so that knowledge of the specific mutation is important. The subunit SDHB is known to have a high likelihood of malignancy unlike mutations in SDHA, SDHC, SDHD and SDHAF2 subunits.76,77,83,85,86 However, with the increasing use and availability of next generation sequencing, the approach to genetic evaluation of patients with PPGL is likely to change from a less targeted approach looking for known mutations to one that will screen entire genes identifying both novel and known mutations.
The identification and understanding of the role of specific genetic mutations that predispose patients to PPGL increase the potential for the development of ‘personalized medicine’. An example of this is the mutations that have been identified in the rearranged during transfection (RET) gene. This gene is responsible for the multiple endocrine neoplasia 2 (MEN-2) phenotype and its mutations are grouped into one of four levels (A–D), with patients carrying level D mutations having the greatest likelihood of developing medullary thyroid carcinoma (MTC). 73
Syndromes associated with PPGL
MEN-2
MEN-2 is an autosomal dominant syndrome with RET proto-oncogene mutations associated with MTC, PPGL and hyperparathyroidism. 95 PPGL has a variable penetrance, depending on the familial kindred. Fifty per cent of patients with a mutation in RET will develop a PPGL at some point in their lives. 96 In only 25% of cases will it be the presenting disease, explaining why patients need to be screened lifelong for PPGL and certainly before any surgery. MTC carries almost 100% penetrance in these syndromes and can occur in patients under the age of five years depending on the codon affected. 97
Succinyl dehydrogenase syndrome
The SDHx family of nuclear genes (SDHA, SDHB, SDHC, SDHD) encodes for the subunits of the mitochondrial enzyme SDH. A fifth nuclear gene (SDHAF2 or SDH5) encodes a protein required for flavination of SDHA. Mutations in these genes are inherited in an autosomal-dominant manner and cause the development of single or multiple PPGL in the head and neck, mediastinum, abdomen and pelvis. The site of PPGL (dictated by the gene abnormality) indicates the likelihood of secretion and malignancy risk (34–97% in SDHB).1,98
Patients should be genetically screened for SDHx abnormalities in the case of head and neck paragangliomas, multiple PPGL (synchronous or metachronous), recurrent disease and early onset of PPGL (<45 years).1,99
Von Hipple–Lindau
Von Hipple–Lindau (VHL) is inherited in an autosomal-dominant manner in 80% of cases and is characterized by multiple vascular tumours in the eyes, brain and nervous system with benign and malignant tumours in sympathetic ganglia, kidneys, pancreas and adrenal glands. The VHL gene is a tumour suppressor gene. VHL is divided into subtypes depending on the risk of PPGL and renal cell carcinoma.100,101 Type 2 (A, B and C), characterized by missense mutations, is all high risk for PPGL. Prevalence of PPGL in this syndrome is 10–25%. 102 Mean age at diagnosis of PPGL in VHL is 30 years old. 103 Suggested VHL surveillance advises screening for PPGL from five years old. 101 Up to 20% of all PPGL are secondary to VHL, reinforcing the argument for genetic screening in young patients.
Neurofibromatosis type 1 (NF-1)
NF-1 is an uncommon autosomal-dominant disease that has a low risk (<1–2.9%) of PPGL. 104 However, PPGL can occur in the very young and there may be a higher risk of malignancy (7–12%).104,105 Genetic screening for PPGL in neurofibromatosis is not routinely performed, but the diagnosis should be considered in NF-1 patients with hypertension.
Takotsubo syndrome
Takotsubo syndrome is an acute cardiac entity characterized by a left ventricular wall motion abnormality with a circumferential extension beyond that expected from the coronary artery supply and resulting in left ventricular ballooning during systole. PPGL can be the causative stress. 106 In 80 reported cases, men constituted a higher proportion (30%) of PPGL-induced Takotsubo syndrome as compared with all cases (4.5%). Also patients with PPGL were significantly younger (19.8 years). 107 Patients present frequently with cardiac symptoms, including chest pain, palpitations and dyspnoea, but abdominal pain can be a warning symptom in these patients.
Imaging
Imaging is an important part of diagnosis and treatment planning for patients with PPGL. Imaging may be anatomical (important for surgical planning) or functional to assess sites of possible metastases and to evaluate possible treatment options in surgically unresectable or extensive disease.
Anatomical imaging
CT
Abdominal CT is often how PPGL are incidentally diagnosed. PPGL are thought to account for about 1.5–14% of adrenal incidentalomas.13–15 CT is a good method of the assessment of adrenal incidentalomas as benign adrenal cortical neoplasms will usually be fat filled and therefore have Hounsfield units (HUs) of <10 (with no contrast). Unfortunately, many CTs are performed with intravenous contrast, which makes the HU an unreliable assessment. CT can also be used to assess the absolute and relative washout of an adrenal lesion. An absolute percentage washout of greater than 60% or a relative percentage washout of over 40% indicates a benign lesion. PPGL do not typically have a benign appearance on CT with HU >10, and often appear suspicious and may be reported as malignant due to their heterogenous appearances due to their hypervascularity and central necrosis. However, combined with abnormal MAs, these indeterminate or suspicious features can be diagnosed as a PPGL. A CT may suggest malignancy based on gross invasion into other glands and vascular structures or the presence of abdominal metastases.
MRI
MRI may be used to further define the characteristics of an indeterminate adrenal lesion on CT. Typically, a PPGL will light up with a hyperintense ‘bright white’ signal in T2-weighted images. MRI is also used for follow-up for SDHx families to prevent the excess radiation exposure associated with whole body CT.
Functional imaging
Metaiodobenzylguanidine scintigraphy
Metaiodobenzylguanidine (MIBG) is an analogue of NAd and accumulates in the neurosecretory granules. It is combined with 123 iodine to visualize on single-photon emission computed tomography (SPECT) imaging or fused with SPECT-CT for improved anatomical information. Specificities for phaeochromocytoma range from 70 to 100% and 84 to 100% for paragangliomas, with sensitivities of 85–88% and 56–76%, respectively. 108 To aid differentiation of pathological from physiological uptake, a tumour/liver ratio may be used, as difficulties can arise with increased physiological uptake and in small tumours. There is a question whether all PPGL patients need an MIBG, but the authors’ unit still uses MIBG to assess for metastases and confirm localization. MIBG is more sensitive if performed while not on α-blockade.
Positron emission tomography computerized tomography
PPGL express somatostatin receptors and can be detected by somatostatin analogues when using positron emission tomography computerized tomography (PET-CT). While traditionally octreotide scintigraphy has been the second-line functional imaging after MIBG, more recently, dotanoc/dototate has been used. Dotanoc has an increased sensitivity to a greater number of somatostatin receptors than octreotide. 109 68Gallium dotanoc PET-CT has a sensitivity of 80–96%, specificity of 85% and 100% accuracy for malignant PPGL and corresponding metastases.109–111 These recent studies appear to show improved sensitivity compared with [ 18 F]-fluorodeoxyglucose (FDG) PET. This also opens the possibility of dotatate therapy in metastatic disease.
FDG PET-CT
[ 18 F]-FDG PET-CT relies on increased glucose metabolism in malignancy. While proven in many cancers, the role in PPGL is not clear. Studies have not clearly demonstrated a role for significantly differentiating between benign and malignant PPGL; there does, however, appear to be some evidence showing increased standardized uptake value max scores for PPGL in the SDHx syndromes 112 and in MIBG-negative PPGL patients. However, more recent data would suggest that 68 gallium dotanoc PET-CT has a higher sensitivity and increased sensitivity for individual metastatic lesions than [ 18 F]-FDG PET-CT. 111
Treatment
Treatment of PPGL relies on the expertise of endocrinologists, surgeons and anaesthetists. When performed within specialist teams and high volume units, these can be well controlled and safe operations. Surgery on unprepared patients with incidental PPGL can cause a crisis, resulting in significant hypertension, arrhythmias, stroke or death. The treatment of PPGL can be divided into medical, surgical and adjuvant treatment in malignancy. The treatment of a PPGL crisis is medical and supportive treatment of organ failure.
Medical treatment
Alpha-adrenergic receptor blockade is the mainstay of medical treatment for PPGL. Typically, phenoxybenzamine has been the agent of choice as a non-competitive and non-selective alpha-receptor antagonist. However, doxazosin can be used as a competitive, short-acting selective α1 receptor blocker. 113 There is some evidence that doxazosin may cause less postoperative hypotension. 114 Li et al. 114 demonstrated less intra- and postoperative variability in blood pressure with doxazosin compared with phenoxybenzamine.
During α-blockade, it is important that fluid balance is monitored to ensure that the vasodilation produced is being supported by adequate fluid intake. This is usually adequate orally, but is often supplemented by an intravenous infusion of crystalloid fluid the night before surgery. Once the α-blockade is complete with a satisfactory postural blood pressure drop of 20–40 mmHg, a reflex tachycardia may develop, and at this point, it is safe to introduce a β-blocker to reduce the risk of tachycardias and arrhythmias. However, the patient should be α-blocked before the introduction of a β-blocker. Alpha-blockers can also be given intravenously, for example phentolamine can be used in a PPGL crisis or immediately perioperatively. Calcium channel blockers can also be used, although less commonly.
Surgical treatment
Abdominal PPGL can be removed by an open adrenalectomy if large or malignant, laparoscopic adrenalectomy or retroperitoneoscopic (through the back) adrenalectomy for smaller lesions (usually <4 cm). A predominantly no touch technique is important, as pressure on the lesion causes a spike in secretion of catecholamine. Any PPGL surgery carries additional increased risks of stroke, myocardial infarction and death as compared with normal adrenal surgery due to the extremes of hypo- and hypertension that can occur in surgery despite α- and β-blockades. It is therefore essential that these operations are performed in specialist centres. If the lesion is suspicious of a malignant PPGL, then it is important to achieve clear margins in the surgical resection, and this may mean resection of surrounding structures if invaded by the tumour.
Postsurgical therapy
Routinely, no adjuvant therapy is required for a benign PPGL. The difficulty is predicting and defining malignancy in these tumours. Paragangliomas have a higher risk of being malignant than phaeochromocytoma. Surveillance imaging, the recurrence of symptoms or biochemically positive results, may reveal metastases or local recurrence and therefore confirm malignancy. Malignant tumours are rare and therefore adjuvant treatment is not well characterized. Adjuvant treatment is usually only employed when metastases are present and often requires a multimodal approach. The five-year survival rate for patients with metastatic disease is around 50%. 5 Review of a cohort of 90 patients in France with malignant PPGL assessed the natural history of PPGL and showed a progression-free survival of 46% at one year indicating that active surveillance of disease progression is a valid option, 115 especially when treatment options are limited. Malignant PPGL that are MIBG negative have a particularly poor prognosis; treatment options for these patients are poorly defined.
MIBG therapy
PPGL tumours and metastases that are positive on imaging for 123 I-MIBG can be treated with MIBG therapy in the form of 131 I-MIBG, which emits beta radiation. A number of studies have been published on this topic, but are not comparable due to study design making it difficult to draw reliable conclusions on the impact of 131 I-MIBG therapy. Yoshinaga et al. 116 prospectively recruited patients with metastatic neuroendocrine tumours for MIBG therapy. Forty-eight patients had malignant PPGL. Forty-five (94%) had previous treatment including surgery (79%), chemotherapy (50%) or standard radiotherapy (23%). Fifty-seven per cent of the patients received repeated MIBG therapy. Of the PPGL patients, 86% showed partial remission or stable disease by RECIST tumour size criteria: one partial regression, 54 stable disease and 10 progressive disease. In this study, no significant changes were seen in blood pressure at the first follow-up, but at the last follow-up, a significant reduction in systolic blood pressure was seen. Rutherford et al. 117 performed a retrospective review of 22 patients with PPGL assessed by World Health Organization (WHO) response criteria for symptomatic response, hormonal response and tumour size response. The patients were treated with surgery (86%), chemotherapy (32%) and radiotherapy (4%).
Nineteen per cent of patients showed complete or partial radiological response with 59% showing stable disease at six months. This study also demonstrated an improvement in hormone secretion and symptoms, but these factors had no significant effect on survival. A 68% five-year overall survival was seen in this study with a median survival of 17 years. A phase 2 prospective study showed 22% complete response or partial remission and 35% showed progressive disease at one year. Overall five-year survival was 64% with higher doses of MIBG. 118 Toxicity and side-effects of MIBG therapy can be significant and dose dependent. Side-effects are seen in 47–54% and include nausea, hypertension, abdominal pain, neutropaenia, hypothyroidism, ovarian failure and bone marrow suppression.116–118 Dotanoc therapy with 90 yttrium-dotanoc and 177lutetium-dotanoc can also be used for therapy based on uptake and success in neuroendocrine tumours.
Chemotherapy
Chemotherapy regimens that have been used for palliative chemotherapy include CVD (cyclophosphamide, vincristine and decarbazine) which gave a median progression-free survival of 5.4 months (2–26 months) 119 and on systematic review also reduced catecholamine secretion in 56% in combination therapy. 120 Temozolomide has been used as a single agent chemotherapy and associated with a tumour response and progression-free survival of 13.3 months in patients with SDHB mutations. 121 Chemotherapy is often used with other therapies and therefore it can be difficult to attribute which therapy is producing the effect. Many patients actually die from catecholamine excess, and hence the reduction in catecholamines is a significant benefit but combination with α-blockers and β-blockers will be required with a heavy tumour load and secretion.
Molecular-targeted therapy
Sunitinib is a tyrosine kinase inhibitor that is an effective antiangiogenic agent that targets vascular endothelial growth factor receptors 1 and 2 that are overexpressed in some metastatic PPGL. 122
Progression-free survival is similar to that achieved using chemotherapy at 4.1 months. 123 The PI3K/Akt/mTOR pathway is thought to be involved in the pathogenesis of metastatic PPGL. mTOR inhibitors have been shown to have antitumour effects on PPGL cells in animal models. 124 Trials are underway to further investigate the clinical applications.
Histological evaluation
Macroscopic features
Phaeochromocytoma are soft, encapsulated tumours with a yellow/brown to red cut surface (Figure 1(a)) and may weigh from just a few grams to up to 4 kg. Larger tumours can show areas of necrosis, cystic degeneration or haemorrhage. They usually have a thin rim of compressed adrenal cortex peripherally. If potassium dichromate is added to fresh tumour, it will turn dark brown (chromaffin reaction).
Pathological features observed in phaeochromocytoma. (a) Enlarged adrenal gland; (b) nested pattern (haematoxylin and eosin staining); (c) nuclear pleomorphism (haematoxylin and eosin staining staining); (d) sustentacular cells demonstrated by immunocytochemistry for S-100 protein.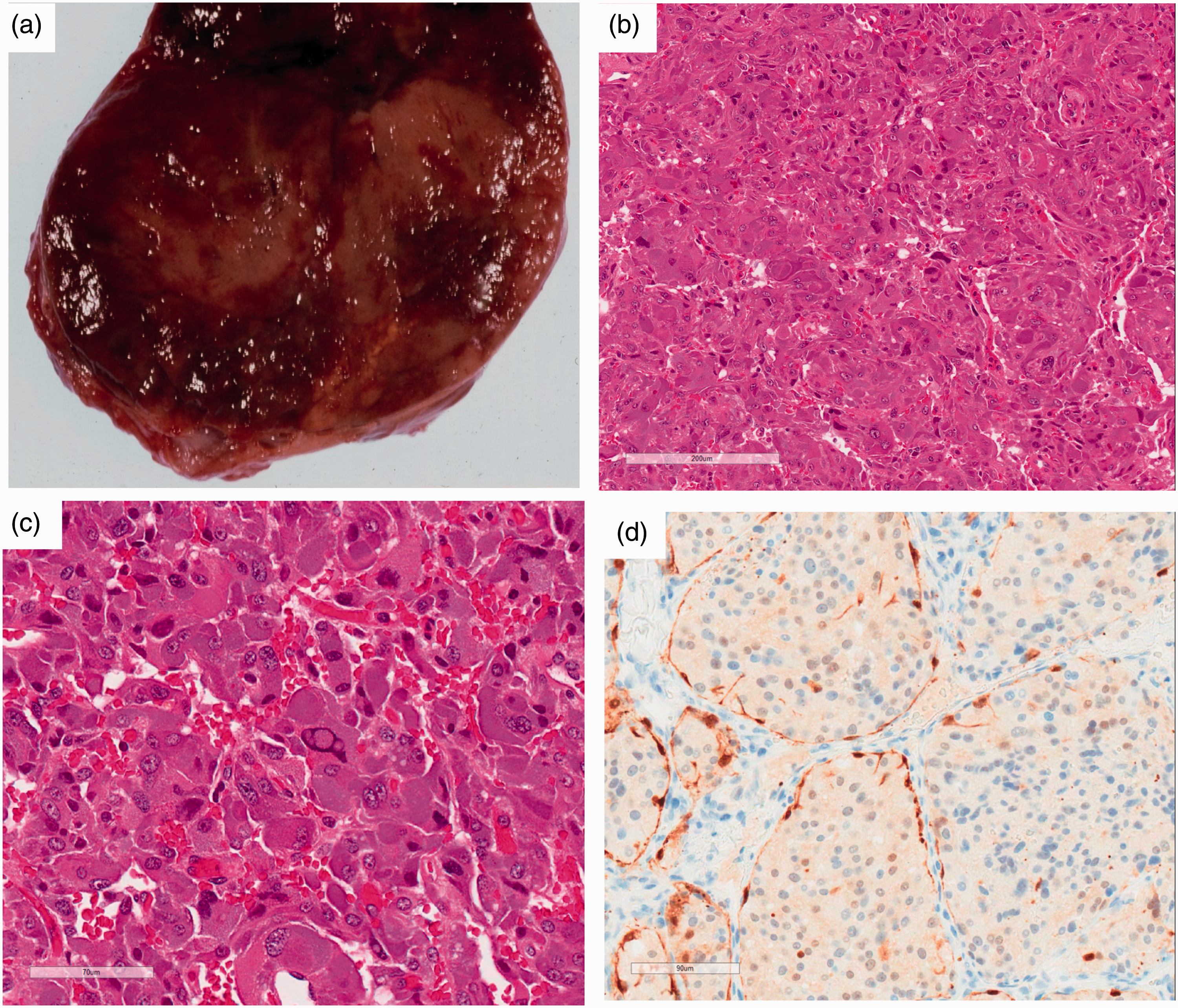
Microscopic features
Microscopically, the tumour cells have round to oval nuclei with a prominent nucleolus and may contain cytoplasmic inclusions. The cells have a finely granular basophilic or amphophilic cytoplasm and are usually arranged in nests (Zellballen pattern) surrounded by a rich vascular network (Figure 1(b)). Marked nuclear pleomorphism and hyperchromasia can be seen (Figure 1(c)), but similar to capsular and vascular invasion, this in itself is not a reliable indicator of malignancy. Mitoses are rarely found. Occasionally, the tumour cells may have an oncocytic appearance due to the presence of large numbers of mitochondria. Other features include ganglion-like cells, dystrophic calcification and melanin pigment, 125 and the presence of amyloid. 126 The nests are outlined by sustentacular cells, which provide structural support and are not visible with routine staining techniques, but can be highlighted with S-100 immunocytochemistry (Figure 1(d)).
PPGL can be associated with other tumours of the adrenal medulla, such as ganglioneuroma, ganglioneuroblastoma or neuroblastoma, and with other tumours such as adrenal cortical adenoma, spindle cell sarcoma 127 or neuroendocrine carcinoma. 128 Hyperplasia of the adjacent adrenal medulla can be an indication of MEN-2 syndromes, but it is difficult to identify and genetic screening is more accurate. 129
Immunohistochemical profile
PPGL express the neuroendocrine markers chromogranin, synaptophysin and neurone-specific enolase. They may express HMB45 (due to a close histogenic link with melanocytes 130 ) and more rarely cytokeratins and vimentin. S-100 highlights the sustentacular cells.
Behaviour
Phaeochromocytoma of the Adrenal gland Scoring Scale.
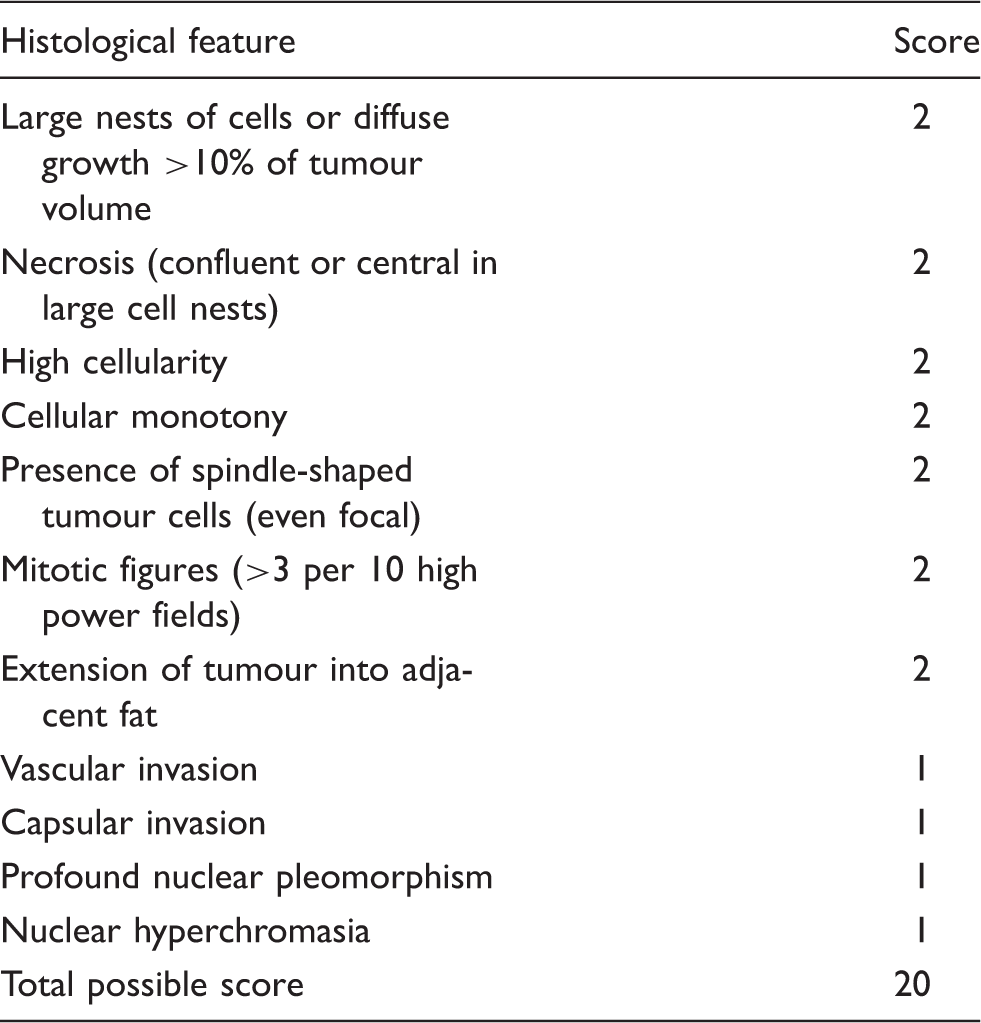
Grading system for Adrenal Phaeochromocytoma and Paraganglioma.

10 μm2: number of tumour cells in a square of a 10 µm observed under high power magnification (400×).
Follow-up
Guidance on the long-term follow-up of patients operated on for PPGL has been published by the European Society of Endocrinology. 135 In brief, this guidance recommends patients should be followed-up annually for 10 years to screen for local or metastatic recurrence or new tumours, unless they are high risk (i.e. young patient or those with genetic disease). If this is the case, patients should be followed annually for life.
From a biochemical perspective, it is proposed that PMets or UMets are measured two to six weeks postoperation in patients with elevated MAs preoperatively, and then annually to screen for local or metastatic recurrence or new tumours. If PMets or UMets remain elevated postoperatively, imaging should also be performed within three months following surgery.
In addition, it is proposed that plasma chromogranin A is measured preoperatively in patients with normal PMets or UMets (including 3-MT) and postoperatively when chromogranin A is raised preoperatively.
In biochemically inactive PPGL, imaging should be considered every one to two years to screen for local or metastatic recurrence or new tumours.
Genetic testing should be considered in all patients with histologically proven PPGL because over one-third of patients with PPGL have mutations in the susceptibility genes, and the presence of mutations has implications for the patient and family members. The extent of genetic testing is influenced by a number of factors including cost and the diagnostic algorithm employed for genetic testing. 83
Conclusion
PPGL is a rare disease but with significant clinical consequences. Advances in the biochemical diagnosis, genetic assessment and screening have seen significant developments in recent years. While surgical treatment is curative in many cases, the treatment of malignant disease remains a challenge.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
ASD and SLS.
Contributorship
All authors contributed to this review.