
Abstract
Background
Measurement of plasma metanephrine, normetanephrine and 3-methoxytyramine is useful in the diagnosis of phaeochromocytomas, but many assays require a large volume of plasma due to poor assay sensitivity, and often require lengthy sample preparation. Our aim was to develop a method for measurement of plasma metanephrines using a small sample volume with minimal hands-on preparation.
Methods
Samples were deproteinised using 10 K spin filters prior to online solid phase extraction using a Waters Acquity UPLC Online SPE Manager (Waters, Manchester, UK) coupled to a Waters Xevo TQ-S mass spectrometer (Waters, Manchester, UK). The assay was validated and results compared to a previously published method.
Results
We achieved a limit of quantification of 37.5 pmol/L for metanephrine and 3-methoxytyramine and 75 pmol/L for normetanephrine using only 150 µL of sample. The assay was linear up to 30,000 pmol/L for all analytes and in a method comparison study results showed good agreement with a previously published LC-MS/MS assay.
Conclusions
We have developed a simple method for measurement of plasma metanephrine, normetanephrine and 3-methoxytyramine using only 150 µL of sample. There is minimal hands-on sample preparation required and the assay is suitable for routine use in a clinical laboratory.
Keywords
Introduction
Phaeochromocytomas and paragangliomas (PPGLs) are rare tumours of adrenal chromaffin cells or similar tissue in extra-adrenal paraganglia. Biochemical diagnosis of these tumours is important due to the non-specific nature of symptoms such as hypertension, palpitations, flushing and sweating. Analysis of plasma metanephrines is used as a first line test for the diagnosis of adrenal and extra-adrenal sympathetic PPGLs. 1
Plasma metanephrines are produced from catecholamines within chromaffin cells by the action of catechol-O-methyl transferase (COMT). Adrenaline is metabolized to metanephrine (MN), noradrenaline to normetanephrine (NMN) and dopamine to 3-methoxytyramine (3MT). Many of the current plasma metanephrine assays only measure MN and NMN, but there is evidence that measurement of 3MT gives important information on tumour location, the presence of underlying mutations of SDHB and SDHD genes and the likelihood of metastases, 2 with 3MT shown to be higher in patients with metastases compared to those without. It is advantageous to offer 3MT measurement as part of plasma metanephrine analysis, but this can be challenging due to the low picomolar concentrations found in the majority of samples.
Current methods for the measurement of plasma metanephrines include immunoassay,3,4 HPLC with electrochemical detection 5 and liquid chromatography tandem mass spectrometry (LC-MS/MS).6–10 Immunoassays do not offer the option of 3MT analysis, and HPLC with electrochemical detection suffers from interferences from common substances such as paracetamol, 11 so LC-MS/MS is increasingly the method of choice for metanephrine analysis.
LC-MS/MS analysis of analytes in complex matrices such as plasma often requires extensive sample preparation. Solid phase extraction is the most common sample preparation method for plasma metanephrines, with both offline7–10 and online 6 methods reported in the literature; however, 3MT was not measured in two of these assays,7,9 and some required high sample volumes for analysis.6,7
We wanted to develop an online solid phase extraction (SPE) method for the measurement of plasma metanephrines to decrease the hands-on preparation time for use in a busy clinical laboratory. The Waters Online SPE Manager (OSM), (Waters, Manchester, UK) is an online separation device designed for use with the very high back pressure encountered when running UPLC conditions. In particular, the extraction cartridges have been manufactured with thicker walls, to ensure a liquid tight junction under the higher pressures, but this reduces the amount of chromatographic packing material available for use. As a result of this fundamental design change, the preconditioning, washing and elution parameters of the OSM are uniquely different from those reported for similar systems designed for use under HPLC conditions. 6 We often receive low volume samples for plasma metanephrine analysis and wished to develop a method with minimal sample volume requirements with sufficient sensitivity to allow the quantification of MN, NMN and 3MT. We felt that the OSM combined with a highly sensitive mass spectrometer would allow us to develop such a method.
In this report, we describe the development and validation of a semi-automated online SPE-LC-MS/MS method for the measurement of MN, NMN and 3MT using only 150 µL of plasma.
Materials and methods
Calibrator and reagent preparation
Stock solutions were prepared by dissolving 10 mg of 3-methoxytyramine powder (Sigma-Aldrich, Poole, UK) in 0.1 mol/L hydrochloric acid. Catecholamine mix 2, consisting of 1 mg/mL of MN and NMN in methanol was used as the stock for MN and NMN (LGC standards, Middlesex, UK). Separate stocks were used for standards and quality control samples (QCs). The stocks were then diluted in phosphate buffered saline pH 7.4 (PBS; Sigma-Aldrich, Poole, UK) containing 0.1% (w/v) bovine serum albumin (BSA; Sigma, Poole, UK), to give standard concentrations of 0–30,000 pmol/L for MN and 3MT and 0–32,300 pmol/L for NMN and QC concentrations of 200, 1000 and 3000 pmol/L for MN and 3MT and 220, 1090 and 3270 pmol/L for NMN. Aliquots (200 µL) of these were stored at −80℃ for up to six months. A mixed internal standard of deuterated d3-metanephrine (α-d2 β-d1), d3-normetanephrine (α-d1, β-d2) and d4-3-methoxytyramine (α-d2, β-d2) (Medical Isotopes, New Hampshire, USA) was used as the internal standard at a working concentration of 2000 pmol/L in water. This was found to be stable at 4℃ for six months.
Sample preparation
Standards, QCs and patients’ samples (150 µL) were pipetted directly into a 10 K modified PES centrifugal filter (VWR, Leicestershire, UK). To this, 150 µL of working internal standard was added and the filter vortexed for 10 s. The filter was then centrifuged at 9962 g for 20 min, and the filtrate transferred into a well of a 96-deep well block (Porvair Sciences, Wrexham, UK). The block was heat sealed (Thermo, Hemel Hempstead, UK), and then centrifuged at 8000 g for 5 min.
Online solid phase extraction and liquid chromatography
The plate was transferred into a Waters Acquity UPLC system coupled to an Online SPE manager (OSM) (Waters, Manchester, UK). The OSM is under full software control and allows preconditioning, loading, washing and elution of the SPE cartridges. An Oasis weak cation exchange (WCX) 1 × 10 mm and 30 µm SPE cartridge (Waters, Manchester, UK) were placed in the right clamp and preconditioned twice with 200 µL of acetonitrile containing 2% (v/v) formic acid, followed by 250 µL of 80% acetonitrile: 20% 10 mmol/L ammonium formate pH 3.2 in water followed by 250 µL of 95% acetonitrile. The cartridge was then equilibrated with 250 µL water, and 100 µL of sample was loaded onto the cartridge via the Acquity autosampler with 250 µL of water. Unbound contaminants were removed by washing the cartridge with 200 µL of water followed by 200 µL of 95% acetonitrile. The cartridge was then automatically transferred to the left hand clamp for elution of the analytes from the cartridge directly onto the analytical column under the initial chromatographic conditions. The analytical column was an Atlantis Hilic Silica 2.1 × 50 mm, 3 µm column (Waters, Manchester, UK), coupled to a SecurityGuard Gemini C18 4 × 2 mm guard cartridge (Phenomenex, Macclesfield, UK). Whilst eluting the sample in the left clamp, the next sample is prepared in the right clamp, thus improving throughput.
Mobile phase A contained 100 mmol/L ammonium formate in deionized water adjusted to pH 3.2 with formic acid, and mobile phase B contained acetonitrile. Initial conditions were 5:95 (v/v) A:B. Metanephrines were eluted from the column using a gradient of 5–20% mobile phase A over 4.1 min. These conditions were held for 0.3 min before returning to initial conditions for a further 2.74 min to re-equilibrate the column. The flow rate was 0.3 mL/minute throughout and the total run time was 7.15 min.
Mass spectrometry
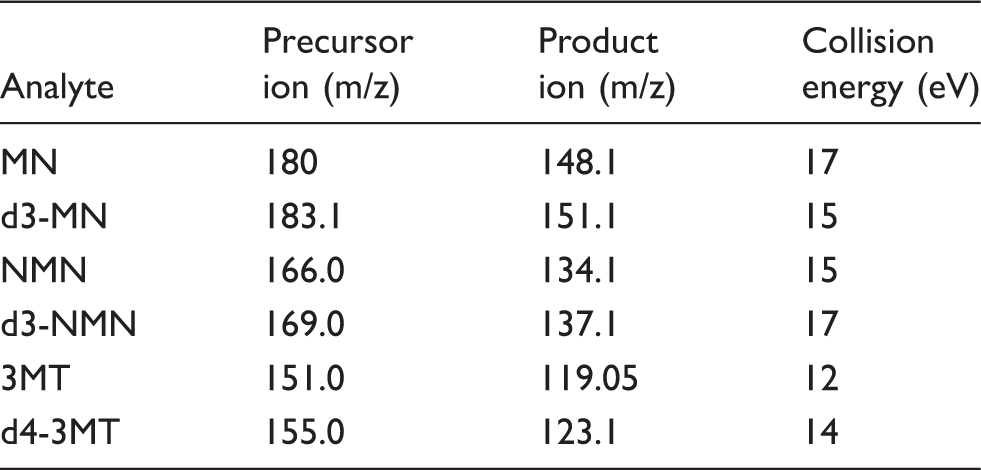
MRM Transitions and collision energies for each analyte.
Assay validation
Ion suppression
To investigate ion suppression, we infused a 2 nmol/L solution of d3-MN, d3-NMN and d4-3MT in water directly into the mass spectrometer via the Waters Intellistart™ Fluidics system to give a constant background signal. Plasma samples (n = 6) were prepared as detailed earlier and injected simultaneously via the autosampler. Ion suppression is seen as a reduction in background signal, and this is significant if it occurs where the compound of interest elutes, and although there is no official guidance on acceptable concentrations of ion suppression, for the purpose of this study we decided that a reduction in signal of less than 10% would be acceptable. In addition, plasma samples (n = 5) were doubly diluted with PBS/0.1% (v/v) BSA up to a 1:32 dilution to assess the linearity of the dilutions, as the compounds of interest would dilute non-linearly in the presence of ion suppression.
Lower limit of quantification
The lower limit of quantification (LLOQ) was in accordance with FDA Guidance. 12 Fifteen replicates of low, medium and high concentrations of MN, NMN and 3MT in PBS/0.1% BSA were analysed and the LLOQ was determined as the lowest concentration of each with % coefficient of variation (CV) <20 and accuracy within 20% of expected concentrations.
Imprecision and accuracy
To determine the intra-assay imprecision of the assay, the three QC samples were prepared and analysed 10 times within one batch. To determine the inter-assay imprecision, the three samples were analysed 15 times in different batches. The imprecision was deemed acceptable if the CV was <15%, and the accuracy was deemed acceptable if the mean value was within 15% of the weighed in value.
Linearity
The linearity of the method was evaluated by analysing calibrators containing MN and 3MT up to a concentration of 30,000 pmol/L and NMN up to 32,300 pmol/L. Calibration curves (n = 10) were produced using TargetLynx software, which plotted calibrator concentration values against the corresponding Online SPE-LC-MS/MS response (analyte peak height/stable isotope labelled analyte peak height). The assay was determined to be linear if the 1/× linear regression analysis produced an r2 >0.99.
Recovery
Three different concentrations of the three analytes (500, 1000 and 3000 pmol/L) were spiked onto six different plasma samples. Total recovery was calculated from measured compared to expected concentrations, and was deemed to be acceptable if between 80 and 120%. Extraction recovery was determined using the Advanced Method Development (AMD) feature in the MassLynx software. Briefly, two cartridges were placed in series, and a sample and washes were applied to the first cartridge. Any sample lost from the first cartridge (breakthrough analyte) was captured on the second cartridge. The second cartridge was then eluted, followed by the first, and the amount of analyte eluted was quantified using TargetLynx software. Extraction recovery was calculated as the peak height of the analyte recovered from cartridge 1 divided by the peak height of breakthrough analyte plus the peak height of the analyte eluted from cartridge 1.
Method comparison
Plasma samples (n = 206) were analysed by online SPE-LC-MS/MS and by LC-MS/MS with offline SPE. 10 All samples used were part of the prospective monoamine-producing tumor study (https://pmt-study.pressor.org) and were taken after an overnight fast and with the patient in the fully supine position for 30 min before blood sampling. Samples were initially analysed by offline SPE within one month of collection. They were then stored at −80℃ for a mean of 1.68 years, prior to shipping frozen in dry ice and storage for a maximum of 2 weeks before analysis by online SPE. Samples have been shown to be stable under these conditions. 13 All subjects provided informed consent under protocols approved by the local Ethics committees at each participating centre.
Diagnostic sensitivity and specificity
Receiver Operating Characteristic (ROC) analysis was carried out on 206 plasma samples to ascertain the diagnostic sensitivity and specificity of both online and offline SPE assays. Of the samples, 36 were from patients with confirmed PPGLs and 170 were from patients in whom PPGL had been excluded as a diagnosis.
Drug interference
It has previously been shown that isoproterenol and MDMA interfere with normetanephrine quantification, and isoetharine and MDA interfere with metanephrine. 14 To investigate this, compounds were spiked into water plus two plasma pools at concentrations at least three times the upper limit of the appropriate reference limit (isoproterenol 47 µmol/L, MDMA 5.2 µmol/L, isoetharine 42 µmol/L and MDA 5.6 µmol/L). The concentrations of metanephrine, normetanephrine and 3MT were calculated and compared to those in the pools spiked with an equivalent volume of water.
Statistical analysis
All statistical analyses were carried out using Analyse-it software (Analyse-it software Ltd, Leeds, UK) apart from ROC analysis which was done using the JMP Pro 10.0 software package (SAS, Cary, NC, USA).
Results
SPE and liquid chromatography
Chromatographic separation of 3MT, MN and NMN was achieved within 4 min (Figure 1) with a further 3 min required to wash and re-equilibrate the column, giving a total run time of 7.15 min. Each analyte co-eluted with the stable isotope labelled version.
Chromatogram of a plasma sample containing 149 pmol/L metanephrine, 345 pmol/L normetanephrine and 110 pmol/L 3-methoxytyramine. The internal standards d3-metanephrine, d3-normetanephrine and d4-3MT are also shown on the chromatogram.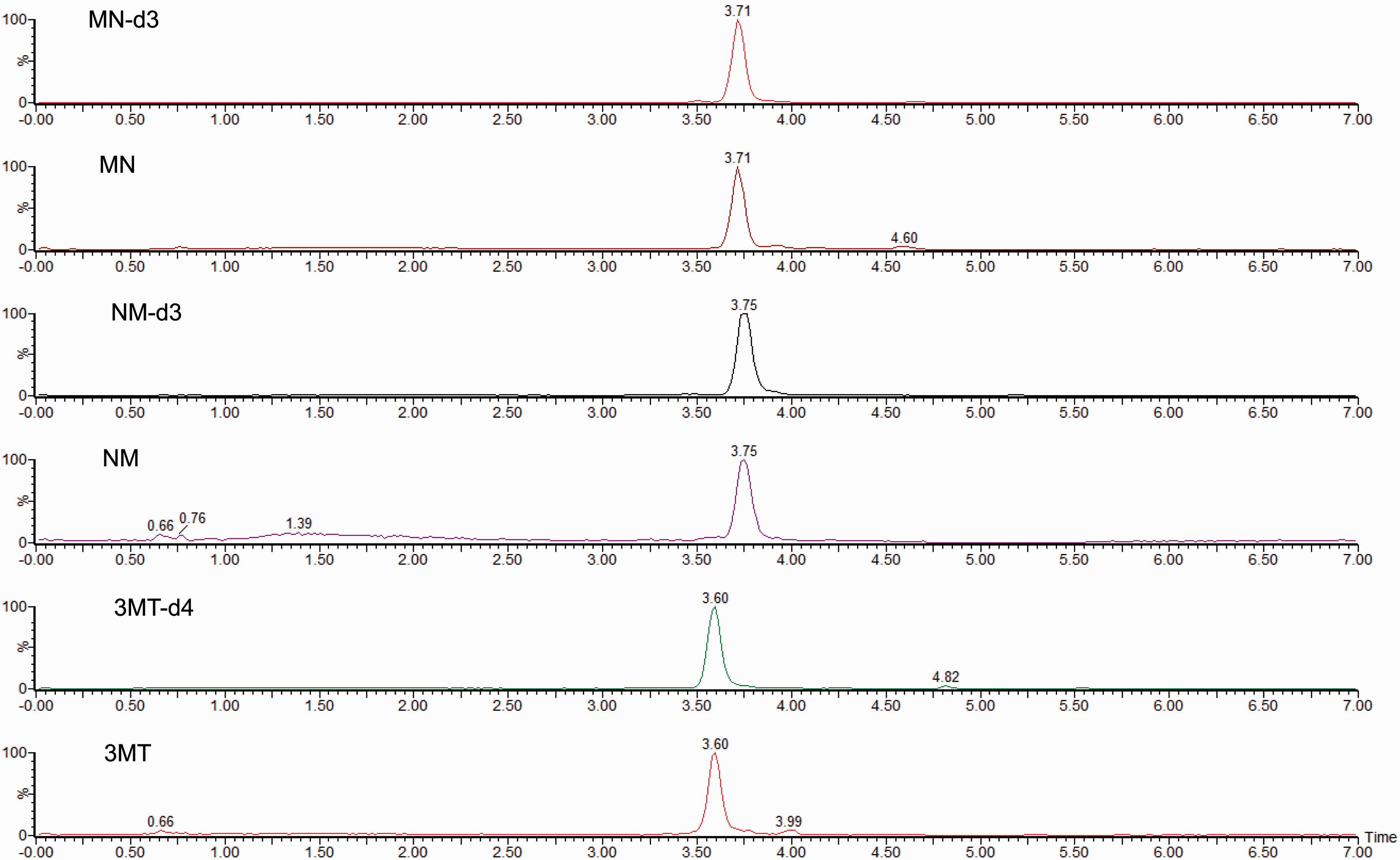
Ion suppression
There was no significant suppression of the background signal at the time of analyte elution for any of the three analytes. Ion enhancement occurs after 5 min, but this does not interfere with analyte quantification. We further investigated ion suppression by serially diluting patients’ samples in PBS/0.1% (v/v) BSA. The analyte:internal standard peak ratio was used to calculate the concentration of each dilution from a standard curve, and these concentrations were plotted against the expected values. Dilution linearity of five patients’ samples showed an r2 value >0.99 for all three analytes, indicating that ion suppression does not affect this assay.
Lower limit of quantification
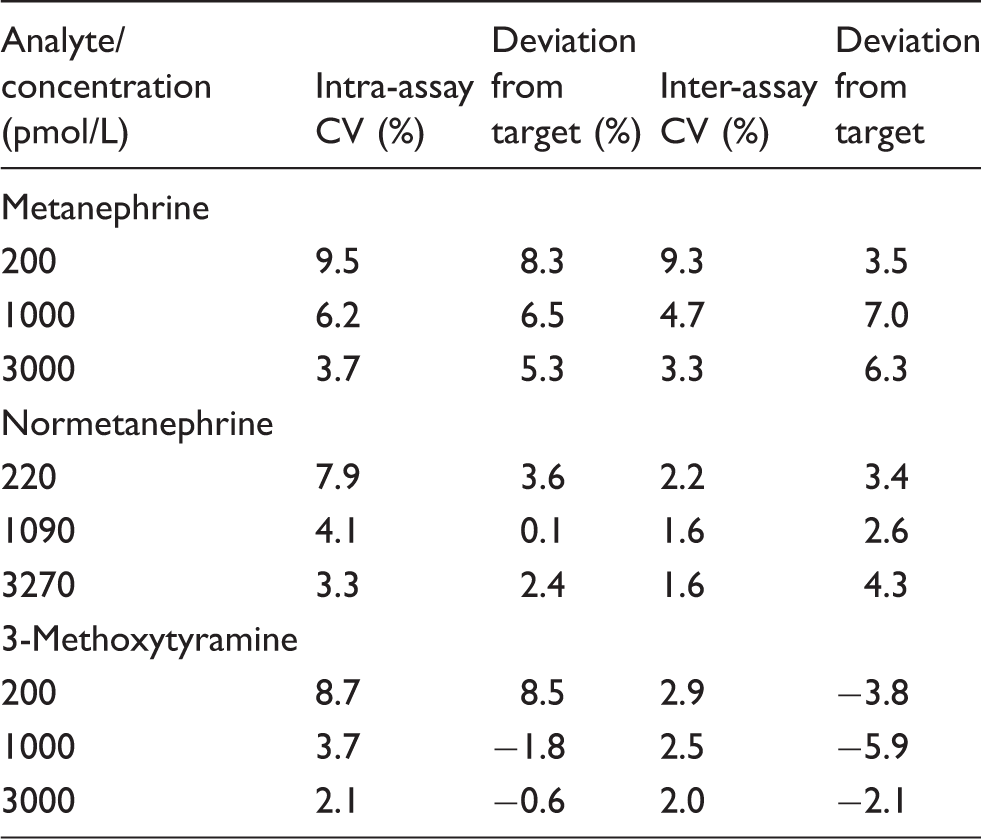
The lower limit of quantification for MN was 37.5 pmol/L, with a CV of 7% and a bias of +5.9%. The LLOQ for NMN and 3MT was 75 pmol/L, with CVs of 5.8 and 5.4% and deviation from the target value of −3.4 and +8.4%, respectively.
Imprecision
Intra- and inter-assay imprecision and deviation from target values.
Linearity
Standard curves were produced by plotting the analyte concentration on the x-axis and the response (analyte peak height/internal standard peak height) on the y-axis. The curve was linear up to 30,000 pmol/L for MN and 3MT and up to 32,300 pmol/L for NMN, with r2 values greater than 0.99 over multiple batches. A representative slope for MN was 0.9997 (SE 0.0002) with an intercept of 2.4604 (SE 2.0274), and the SE of the residuals was 4.1438. For NMN, a representative slope was 0.9971 with an intercept of 21.91 (SE 0.0014 and 19.09, respectively), and SE of the residuals was 39.03. A typical 3MT standard curve had a slope of 1.0053 (SE 0.0030) with an intercept of −36.95 (SE 38.88), and the SE of the residuals was 79.47.
Recovery
The mean total recovery of MN, NMN and 3MT was 98% (87–108%), 97% (86–107%) and 95% (80–109%), respectively, which were all within acceptable limits. The extraction recovery was 92% for MN, 94% for NMN and 97% for 3MT, indicating that losses from the cartridge are minimal.
Method comparison
MN, NMN and 3MT were measured in 206 samples by online SPE-LC-MS/MS and by a previously published LC-MS/MS method
10
with offline solid phase extraction. Sixteen of the 206 samples were below the LLOQ of our assay for MN and were excluded from the comparison. Bland-Altman analysis showed the online SPE method had a mean negative bias of 23% over a range of 37.5 pmol/L to >12000 pmol/L (Figure 2(a)). All the NMN results were within the measuring range, and Bland-Altman analysis showed a mean positive bias of 0.1% (Figure 2(b)). The NMN results ranged from 113 pmol/L to greater than 40,000 pmol/L. Of the 206 samples, 163 were below the measuring range of 3MT in our assay, leaving 43 for inclusion in the comparison, with the highest 3MT concentration >22,000 pmol/L. A mean negative bias of 12.4% was shown in the Bland-Altman analysis (Figure 2(c)).
Comparison between offline and online SPE methods for metanephrine, normetanephrine and 3-methoxytyramine. Panel (a) shows Bland-Altman analysis for metanephrine samples, and panels (b) and (c) show Bland-Altman analysis for normetanephrine and 3-methoxytyramine, respectively. The solid line represents the mean percentage difference between the two methods; dotted lines represent the confidence limits which are 1.96% SD.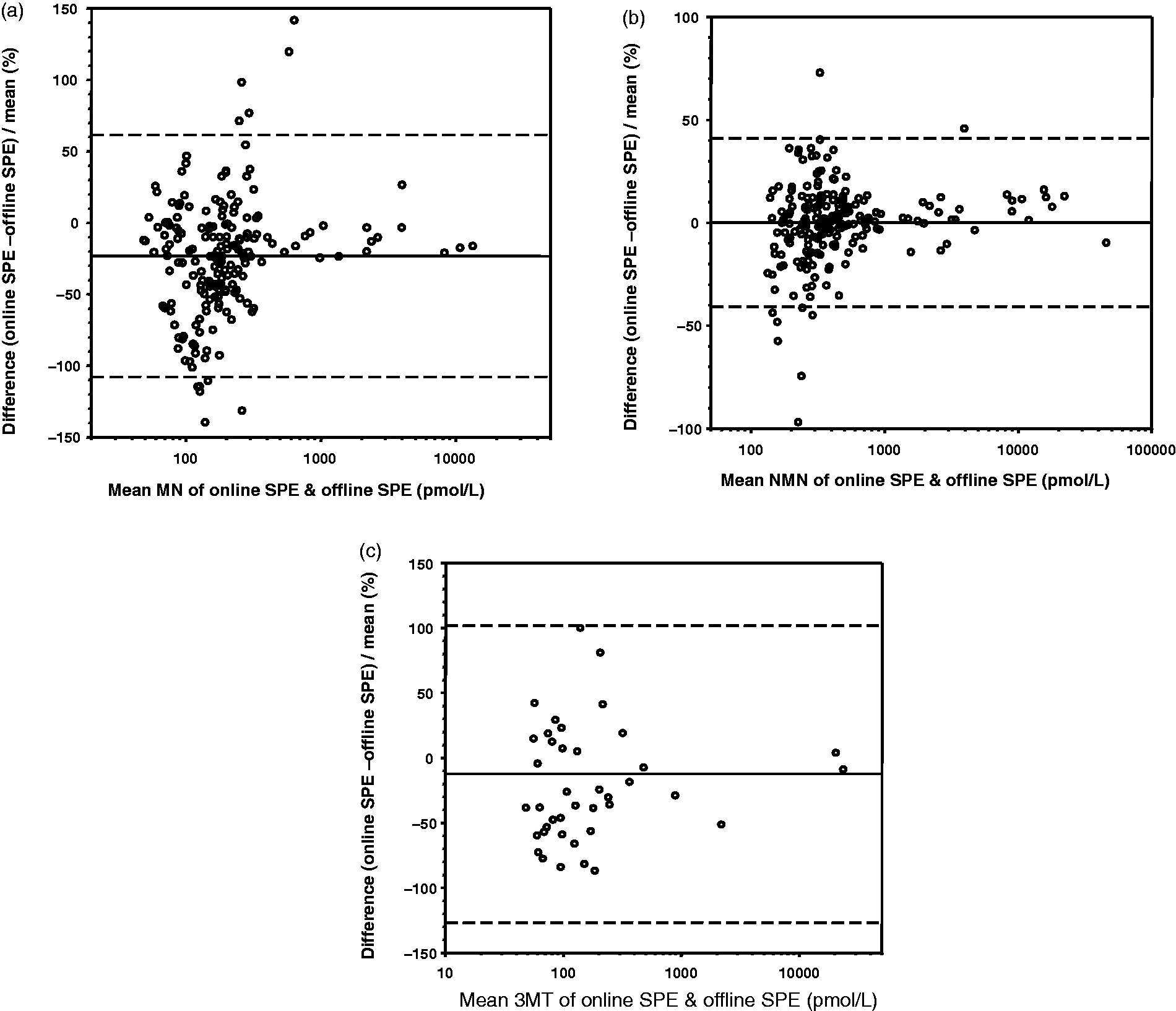
Diagnostic sensitivity and specificity
The diagnostic sensitivity and specificity was calculated for each assay using the cut-offs used in each laboratory (Table 3). The reference ranges used for online SPE were based on the work of Peaston et al.
8
with <510 pmol/L for MN, <1180 pmol/L for NMN and <180 pmol/L for 3MT. The reference ranges for the offline SPE method were 446 pmol/L for MN and 179 pmol/L for 3MT.
10
Age-adjusted reference ranges were used for NMN, as described by Eisenhofer et al.
15
ranging from 590 pmol/L for a 5-year-old to 1037 pmol/L for patients 60 years old and above. Using these cut-offs, the sensitivity of the online assay was 94.4%, whilst the offline assay had a sensitivity of 100%. The specificity of the two assays was similar at 96.5% for online SPE and 95.8% for offline SPE (Table 3). ROC analysis confirms the good diagnostic sensitivity and specificity of both assays, with an area under the curve (AUC) of 0.995 for offline SPE and 0.979 for online SPE (Figure 3).
Receiver Operating Characteristic (ROC) curve analysis for offline SPE and online SPE. The sensitivity and specificity of each assay are plotted for different cut-off points. The area under the curve is shown for each method. Diagnostic sensitivity and specificity of online and offline SPE methods. Note: The sensitivity and specificity were calculated using the cut-offs currently in use in the two laboratories.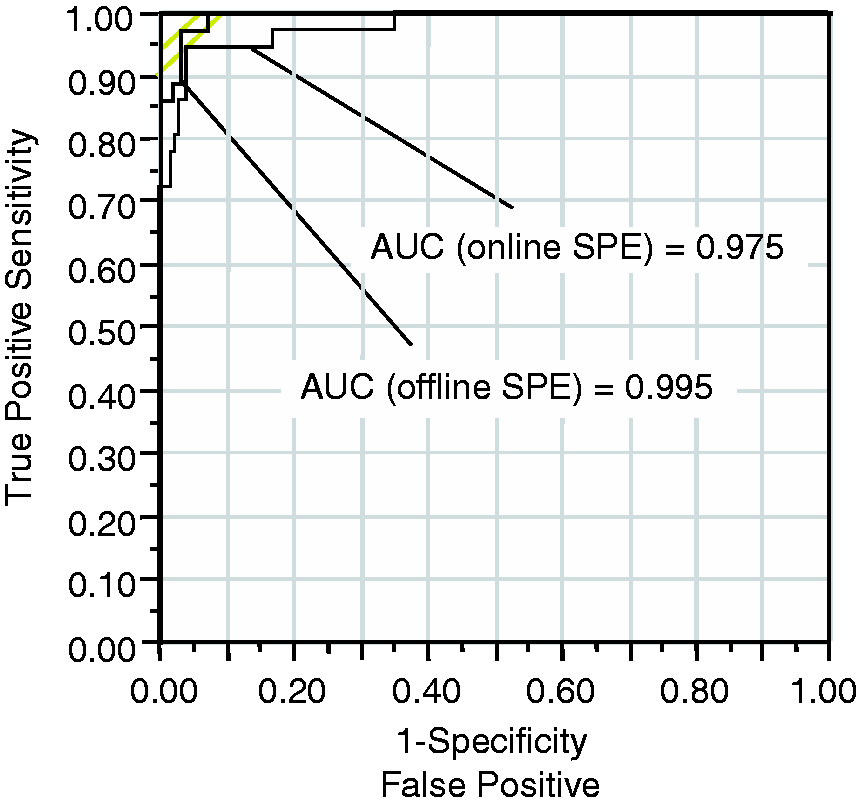

Drug interference
None of the drugs tested showed peaks in water at the retention times of MN, NMN or 3MT. In addition, the drugs did not affect the concentration of any of the three analytes in the plasma pools, indicating that interference from these drugs is negligible in this assay.
Discussion
We have developed a method for the measurement of MN, NMN and 3MT in plasma that is suitable for routine clinical use. The combination of centrifugal filters and online SPE for sample preparation is novel and require minimal hands-on preparation time, which is important in a busy clinical laboratory. The filtration is required in this assay as the OSM is coupled to an Acquity UPLC system, and the viscosity of plasma prepared without filtration would be too high for the narrow bore tubing of the UPLC system and would quickly cause blockages. We tried other methods to remove proteins such as acetonitrile precipitation and trichlorocacetic acid precipitation, but found that we did not get reproducible results using these techniques. We found that the recovery obtained from deproteinising the samples using the centrifugal filters was acceptable and consistent and therefore decided to validate the method using this sample preparation.
It has recently been reported that MN may cause ionic cross-talk in LC-MS/MS assays leading to overestimation of 3MT. 16 This is not a problem with this assay as the resolution between 3MT and MN is sufficient that cross-talk does not interfere in the quantification of 3MT (Figure 1).
The lower limits of quantification for MN, NMN and 3MT allow quantification of samples within published reference ranges, 8 and the linearity of the assay up to 30,000 pmol/L for MN and 3MT and 32,300 pmol/L for NMN allows quantification of the majority of raised samples. If we obtain a result higher than this, the dilution linearity experiment proved that it is valid to dilute the sample with PBS/0.1% (v/v) BSA to produce a concentration within the measuring range of the assay that can then be multiplied back up.
A method comparison against a published offline SPE LC-MS/MS method 10 was carried out using samples taken from patients who were enrolled in the prospective monoamine producing tumour study. The comparison between the two methods was acceptable, with a small negative bias observed with the online-SPE method for MN and 3MT and a small positive bias for NMN, and the diagnostic sensitivity and specificity for both the assays were shown to be very good. The largest differences were in very high results; this would not affect the diagnosis of any patient and may be due to calibration differences between the two assays. The samples were taken from patients after an overnight fast who had been in the supine position for 30 min. We currently use reference ranges derived from the work of Peaston et al. 8 which were based on samples taken from patients in the sitting position. This reference range was used as we carried out a comparison of our previous plasma metanephrine assay against the Peaston method, and our previous method compared well with our current method, with a mean bias of −3.2% for metanephrine and −4.1% for normetanephrine. It has been established that supine rest prior to blood sampling gives significantly lower results for the metanephrines compared to samples taken in the sitting position,17,18 and the difference between the cut-offs used may explain the false negative results with our method. Using the age-adjusted reference ranges for NMN and the supine cut-offs for MN and 3MT, 15 the sensitivity of our assay does improve to 97.2% (35 true positive, one false negative) in this supine, fasted population. This is at the expense of specificity, which falls to 91.3% (11 false positives, 115 true negatives); however, it may be argued that as plasma metanephrine analysis is used as a screening test for PPGL, the importance of not missing the diagnosis is such that the sensitivity should be maximized.
This work was carried out using samples that had been collected with ideal pre-venepuncture patient preparation, i.e. patient fully supine for 30 min. However, we are aware that the vast majority of samples that we receive are taken from patients in the sitting position and that the facilities for supine blood sampling may not be readily available in phlebotomy clinics. In such patients, therefore, the use of a fasting, supine reference range would significantly decrease the specificity of this test. Asking for a repeat sample collected under ideal conditions would increase the specificity, but as Därr et al. 18 suggest that there is a problem with this approach, as for a variety of reasons the majority of such patients do not get repeat metanephrine analysis undertaken. Därr et al. 18 also suggest that if supine samples cannot be taken, urine fractionated metanephrines should be used as an alternative test. However, the accurate collection of timed urine samples may also pose difficulties and we therefore suggest that the use of a ‘sitting position’ reference range for plasma metanephrines may represent a pragmatic alternative approach.
In conclusion, we have developed a method for plasma metanephrine analysis that is suitable for use in the clinical laboratory using a minimal amount of sample. We plan to carry out further work on appropriate cut-offs for our population to maximize the diagnostic sensitivity and specificity of the test in our routine patients’ samples.
Footnotes
Acknowledgements
Thanks are extended to members of Prospective Monoamine Tumor trial (Graeme Eisenhofer & Roland Därr, University Hospital Dresden at the TU Dresden, Dresden, Germany; Aleksander Prejbisz & Andrzej Januszewicz, Institute of Cardiology, Warsaw, Poland; Dirk Weismann & Martin Fassnacht University Hospital Würzburg, Würzburg, Germany; and Felix Beuschlein University Hospital Munich, Munich, Germany) for their contributions of patient materials.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
University Hospital of Dresden (EK189062010), Institute of Cardiology, Warsaw (1233-08112010), Medical University Hospital of Wurzburg (104/11), University Hospital of Munich (173-11).
Guarantor
JA.
Contributorship
BK conceived the study. The development and validation of the assay was performed by JA with aid from BK. MP provided the patients’ samples with corresponding ethical approval and carried out some of the statistical analysis. JA wrote the first draft of the paper, all authors reviewed and edited the manuscript and approved the final version.