
Abstract
Background
One of the main challenges in the measurement of glucagon is the premise that it is unstable in human plasma. Traditionally, protease inhibitors have been used to prevent its degradation; however, their use is controversial. Here, we investigated the optimal method of sample collection for glucagon, with measurement by liquid chromatography tandem mass spectrometry (LC-MS/MS) and two commercially available immunoassays.
Methods
Blood from healthy fasting volunteers (n = 10) was processed under a variety of preanalytical conditions including collection in EDTA vs. lithium heparin tubes and the addition of aprotinin and/or a dipeptidyl-peptidase IV (DPPIV) inhibitor. Additionally, the effect of freeze thaw was assessed. Plasma glucagon concentrations were measured by LC-MS/MS and two commercially available immunoassays (HTRF® sandwich immunoassay, Cisbio and Milliplex MAP Human Metabolic Hormone Panel, Merck Millipore).
Results
A systematic bias of Milliplex > LC-MS/MS > HTRF was noted and plasma glucagon concentrations were significantly different between methods (Milliplex vs. LC-MS/MS P < 0.01; Milliplex vs. HTRF P < 0.0001; LC-MS/MS vs. HTRF P < 0.001). The addition of aprotinin, DPPIV inhibitor or a combination of aprotinin and DPPIV inhibitor had no effect on plasma glucagon concentrations when compared to ‘non-stabilized’ samples or each other. Whether samples were taken in EDTA tubes or lithium heparin tubes made no difference to plasma glucagon concentrations. These findings were consistent for all three methods. Plasma glucagon concentrations were not significantly different after two freeze–thaw cycles (performed on samples in EDTA tubes containing aprotinin and DPPIV inhibitor).
Conclusions
This study demonstrates that glucagon is stable in both EDTA and lithium heparin tubes when stored at −80℃. Furthermore, the addition of aprotinin and DPPIV inhibitors is unnecessary.
Introduction
Glucagon has an important physiological role in energy homeostasis as well as cardiovascular regulation. Furthermore, it is a biomarker for diseases such as diabetes and neuroendocrine tumours. One of the main challenges in the measurement of glucagon is the premise that it is unstable in human plasma. Studies indicate that the circulating serine protease dipeptidyl-peptidase IV (DPPIV) is the primary enzyme for glucagon degradation in blood. In order to counteract degradation, inhibitors such as the serine protease inhibitor aprotinin 1 or the proprietary P800 inhibitor cocktail are often added to samples to stabilize them. 2 However, their use is controversial as other studies have found these to be ineffective in preventing degradation.3,4 Here, we investigated the optimal method of sample collection for glucagon, with measurement by liquid chromatography tandem mass spectrometry (LC-MS/MS). Additionally, measurement was also made by two commercially available immunoassays in order to ascertain whether peptide degradation could cause a positive bias as immunoassays could potentially cross-react with inactive fragments.
Methods
A 20 ml blood sample was collected from 10 fasting healthy volunteers (West London National Research Ethics Committee, ref. 11/LO/1782). Immediately after collection, 2 mL samples were decanted into various collection tube types: (1) K3 EDTA anticoagulant (ref 368860 – Becton Dickinson Vacutainer System, NJ, USA) with the addition of aprotinin (Bayer, Newbury, UK; final concentration 250 kallikrein inhibitor units/mL) and a DPPIV inhibitor (Ile-Pro-Ile, Sigma-Aldrich, Dorset, UK; final concentration 10 µg/mL); (2) K3 EDTA anticoagulant with the addition of aprotinin; (3) K3 EDTA anticoagulant with the addition of a DPPIV inhibitor; (4) K3 EDTA anticoagulant alone; (5) lithium heparin (ref 367883 – Becton Dickinson Vacutainer System, NJ, USA) with the addition of aprotinin and a DPPIV inhibitor; (6) lithium heparin with the addition of aprotinin; (7) lithium heparin with the addition of a DPPIV inhibitor and (8) lithium heparin alone. Following collection, tubes were placed on ice and centrifuged at 2300 × g for 10 min to obtain plasma, which was stored at −80℃ for up to nine months. Samples were analysed on ice within 4.25 h of defrosting. Two further aliquots, collected in K3 EDTA tubes with aprotinin and DPPIV inhibitor, underwent one and two further freeze–thaw cycles, respectively.
Glucagon concentrations were measured by LC-MS/MS based on a method previously described. 5 This consisted of a Waters Acquity ultra-high performance liquid chromatography UPLC system (Waters Corporation, MA, USA) coupled to an AB SCIEX 5500 QTRAP (Applied Biosystems/MDS SCIEX, Ontario, Canada) with an electrospray ion source (intra-assay CVs were ≤11.9% at 4, 7 and 13 pmol/L). Secondly, glucagon was assayed by a Homogenous Time Resolved Fluorescence (HTRF®) sandwich immunoassay (Cisbio, Codolet, France). Thirdly, we used the Milliplex MAP Human Metabolic Hormone Panel which involves a sandwich immunoassay with fluorescence detection (Merck Millipore, Darmstadt, Germany). For both immunoassays, intra-assay CVs were < 10% across the working range. Differences between tube types and methods were analysed by two-way analysis of variance with correction for repeated measurements. P < 0.05 was considered statistically significant.
Results
Glucagon concentrations as measured by liquid chromatography tandem mass spectrometry (LC-MS/MS), Milliplex immunoassay (Milliplex) and Cisbio HTRF immunoassay (HTRF) in various tube types.
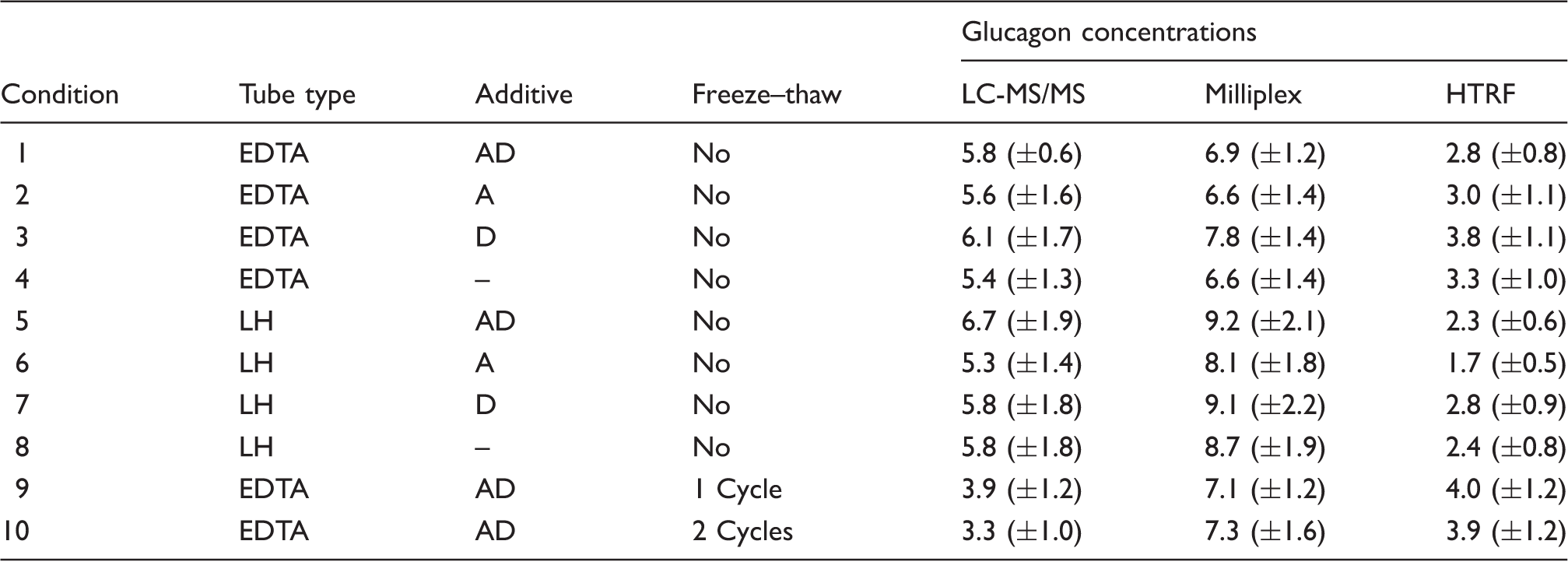
EDTA: K3 EDTA anticoagulant; LH: lithium heparin tube; A: aprotinin added; D: DPPIV inhibitor added (mean plasma glucagon concentration (pmol/L) ± SEM, n = 10).
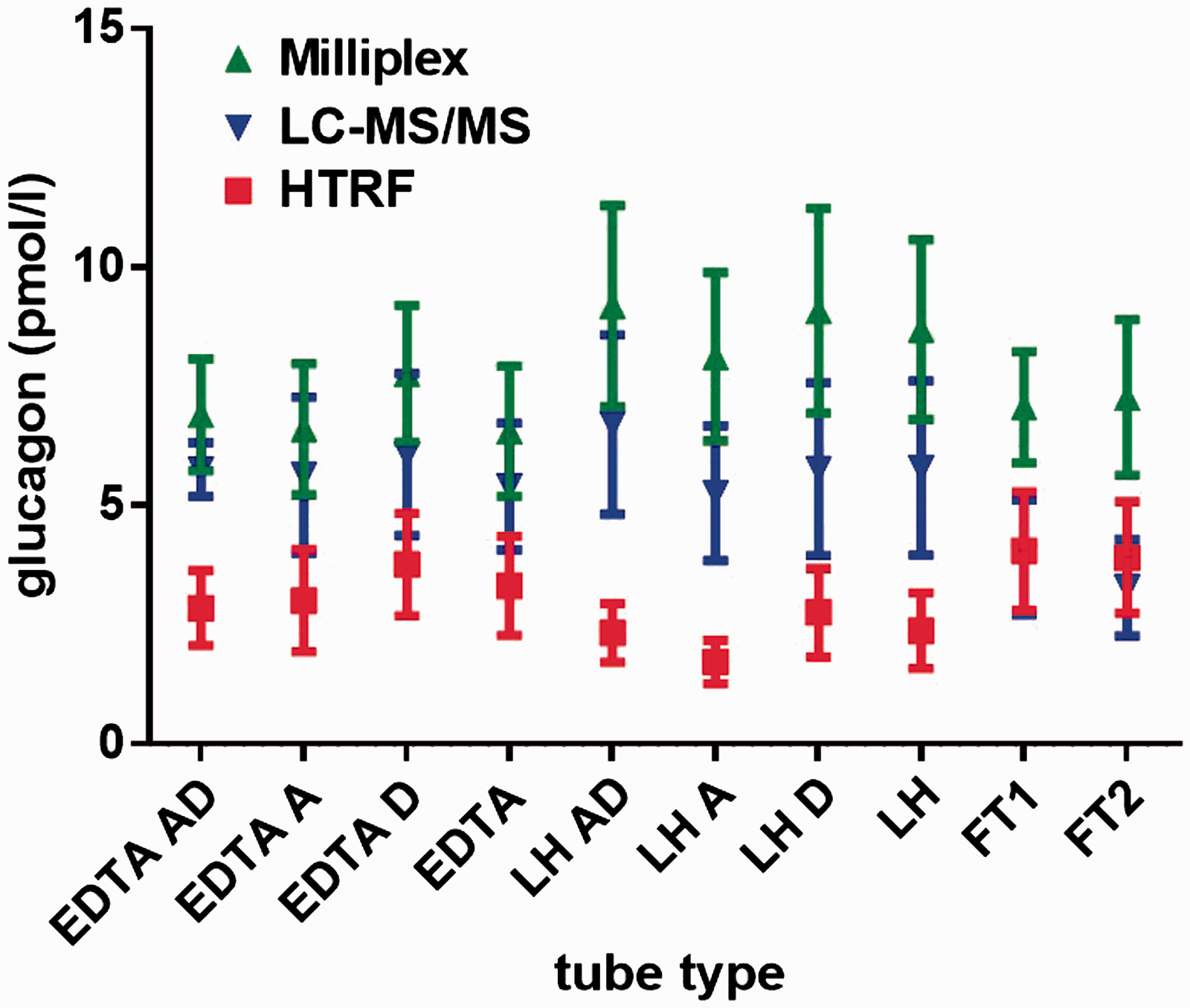
Glucagon concentrations as measured by Milliplex immunoassay (Milliplex), Cisbio HTRF immunoassay (HTRF) and liquid chromatography tandem mass spectrometry (LC-MS/MS) in various tube types.
Importantly, glucagon concentrations were not statistically different when compared with the various tube types. The addition of aprotinin, DPPIV inhibitor or a combination of aprotinin and DPPIV inhibitor had no effect on plasma glucagon concentrations when compared to ‘non-stabilized’ samples or each other. Whether samples were taken in EDTA tubes or lithium heparin tubes made no difference to plasma glucagon concentrations. These findings were consistent for all three methods. The LC-MS/MS data suggested a trend towards lower concentrations following freeze–thaw cycles, but no significant difference was observed using the immunoassays.
Discussion
This paper describes, for the first time, the preanalytical stability of glucagon, as measured by LC/MS-MS and two different immunoassays. Consistent with Emmen et al., 3 we find that aprotinin has no effect on endogenous glucagon stability. Consistent with Wewer Albrechtsen et al., 4 we find that glucagon stability is unaffected by DPPIV inhibition and freeze–thaw when measured using immunoassay. Wewer Albrechtsen et al. 4 did not measure endogenous glucagon, rather, samples were spiked in vitro, so the work described here measuring endogenous concentrations of glucagon provides robust confirmation of Wewer Albrechtsen et al.’s findings. An explanation for the discrepant findings in historical studies of the effect of stabilizers on glucagon stability may be related to the varying specificity of the antibodies used in the glucagon immunoassays resulting in cross-reactivity with glucagon-like peptides, where DPPIV inhibition is more important.
The LC-MS/MS data suggested a trend towards lower concentrations following freeze–thaw cycles, as reported previously, 5 but no significant difference was observed using the immunoassays. This finding for LC-MS/MS is consistent with previous data where after four freeze–thaw cycles, glucagon was approximately 40–50% compared to a non-freeze-thaw sample when measured by LC-MS/MS. 5 It is possible that freeze–thaw increases glucagon aggregation, leading to glucagon precipitating out during LC-MS/MS extraction and thus resulting in artefactually lower concentrations. This requires further investigation.
A marked systematic bias is noted between the three assays. Bak et al. 6 previously compared seven commercially available immunoassays for glucagon measurement and found striking differences in recovery between the assays. These differences may be explained by poor standardization of reference materials, varying concentrations of antibody cross-reactivity, different matrix effects or the presence of factors in the plasma, causing non-specific interference. LC-MS/MS in theory circumvents some of these issues, particularly the potential for cross-reactivity with similar compounds or inactive degradation fragments leading to inaccurate quantitation.
As we were primarily interested in the physiological values of glucagon in clinical use, this study examined fasting glucagon concentrations in normal human volunteers. Further study may include basal and stimulated glucagon concentrations (e.g. post-arginine) or hyperglucagonaemic patients with type 2 diabetes or glucagonoma.
In conclusion, glucagon is stable in both EDTA and lithium heparin tubes when centrifuged immediately and stored at −80℃. Importantly, the addition of aprotinin and DPPIV inhibitors is unnecessary. We have confirmed this by three separate analytical methods.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The Section of Endocrinology and Investigative Medicine is funded by grants from the MRC, BBSRC, NIHR, an Integrative Mammalian Biology (IMB) Capacity Building Award, an FP7-HEALTH-2009-241592 EuroCHIP grant and is supported by the NIHR Imperial Biomedical Research Centre Funding Scheme. The views expressed are those of the authors and not necessarily those of the above-mentioned funders, the NHS, the NIHR or the Department of Health. The research presented here was supported by the NIHR Clinical Research Facility at Imperial College Healthcare NHS Trust. JC is the recipient of a Wellcome Trust Clinical Research Training Fellowship. BJ is the recipient of a Medical Research Council Clinical Research Training Fellowship. TMT is supported by grants from the MRC and the Wellcome Trust. SRB is supported by an NIHR Senior Investigator Award and the MRC.
Ethical approval
West London National Research Ethics Committee approved (Ref. 11/LO/1782).
Guarantor
JC.
Contributorship
JC conceived the study. JC, BJ, JH, RK, CC, SB and TT were involved in the study design and data analysis. JC wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.