
Abstract
The porphyrias are disorders of haem biosynthesis which present with acute neurovisceral attacks or disorders of sun-exposed skin. Acute attacks occur mainly in adults and comprise severe abdominal pain, nausea, vomiting, autonomic disturbance, central nervous system involvement and peripheral motor neuropathy. Cutaneous porphyrias can be acute or chronic presenting at various ages. Timely diagnosis depends on clinical suspicion leading to referral of appropriate samples for screening by reliable biochemical methods. All samples should be protected from light.
Investigation for an acute attack:
• Porphobilinogen (PBG) quantitation in a random urine sample collected during symptoms. Urine concentration must be assessed by measuring creatinine, and a repeat requested if urine creatinine <2 mmol/L.
• Urgent porphobilinogen testing should be available within 24 h of sample receipt at the local laboratory. Urine porphyrin excretion (TUP) should subsequently be measured on this urine.
• Urine porphobilinogen should be measured using a validated quantitative ion-exchange resin-based method or LC-MS.
• Increased urine porphobilinogen excretion requires confirmatory testing and clinical advice from the National Acute Porphyria Service.
• Identification of individual acute porphyrias requires analysis of urine, plasma and faecal porphyrins.
Investigation for cutaneous porphyria:
• An EDTA blood sample for plasma porphyrin fluorescence emission spectroscopy and random urine sample for TUP.
• Whole blood for porphyrin analysis is essential to identify protoporphyria.
• Faeces need only be collected, if first-line tests are positive or if clinical symptoms persist.
Investigation for latent porphyria or family history:
• Contact a specialist porphyria laboratory for advice. Clinical, family details are usually required.
Introduction
The porphyrias are a group of rare, mostly inherited metabolic disorders caused by deficiency of specific enzymes in the haem biosynthetic pathway, or in one porphyria a gain of enzyme function (Figure 1).1,2 Each disorder leads to particular patterns of accumulation of porphyrin precursors, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) and porphyrin intermediates. The porphyrias may cause acute neurovisceral attacks or light-sensitive skin disease or both. In practice, the porphyrias have very diverse manifestations, can present in many different settings including emergency care and affect patients at almost any age (see Table 1). Diagnosis is challenging and critically dependent on timely and appropriate biochemical testing.3,4 Clinicians are often unfamiliar with required samples and tests, and reliant on laboratory advice to guide investigation.
The haem synthesis pathway. XLEPP: X-linked erythropoietic protoporphyria. The main types of porphyria.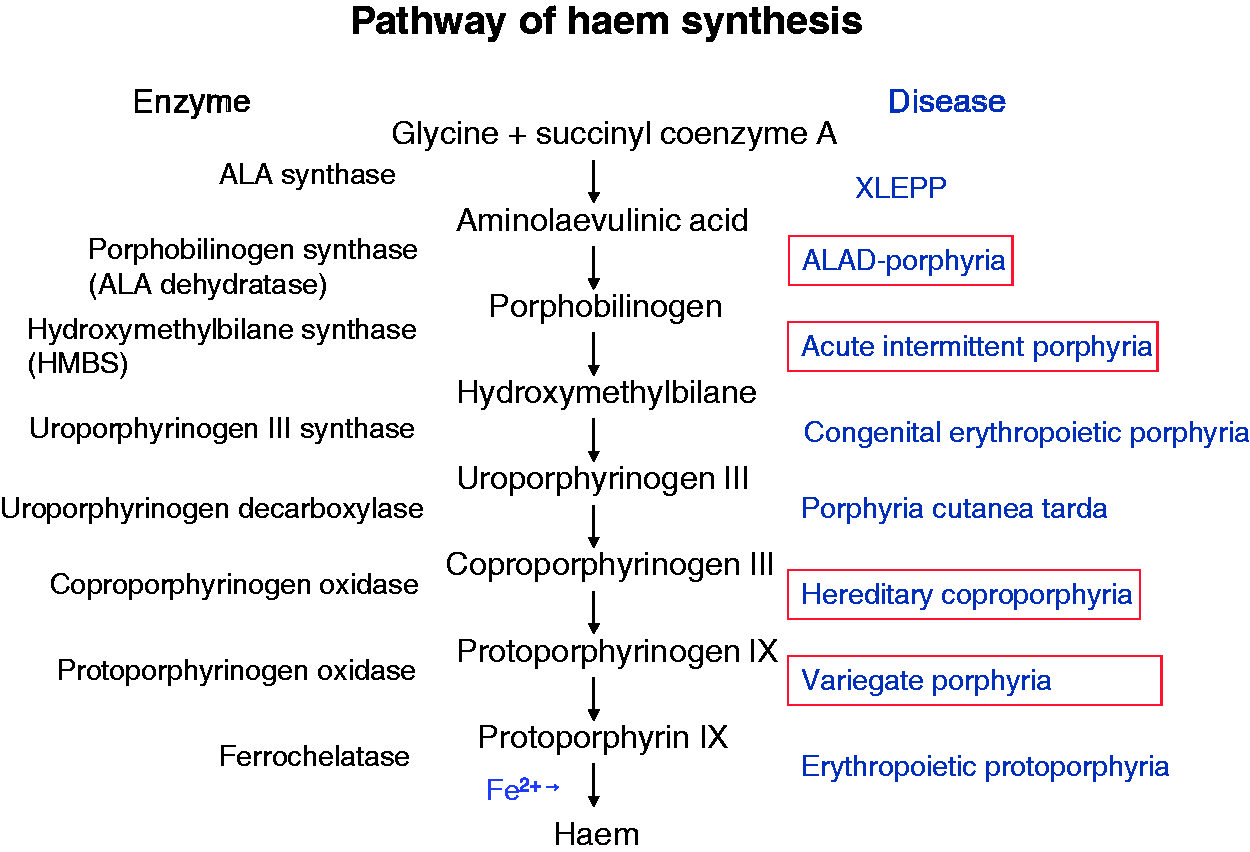
Acute porphyrias
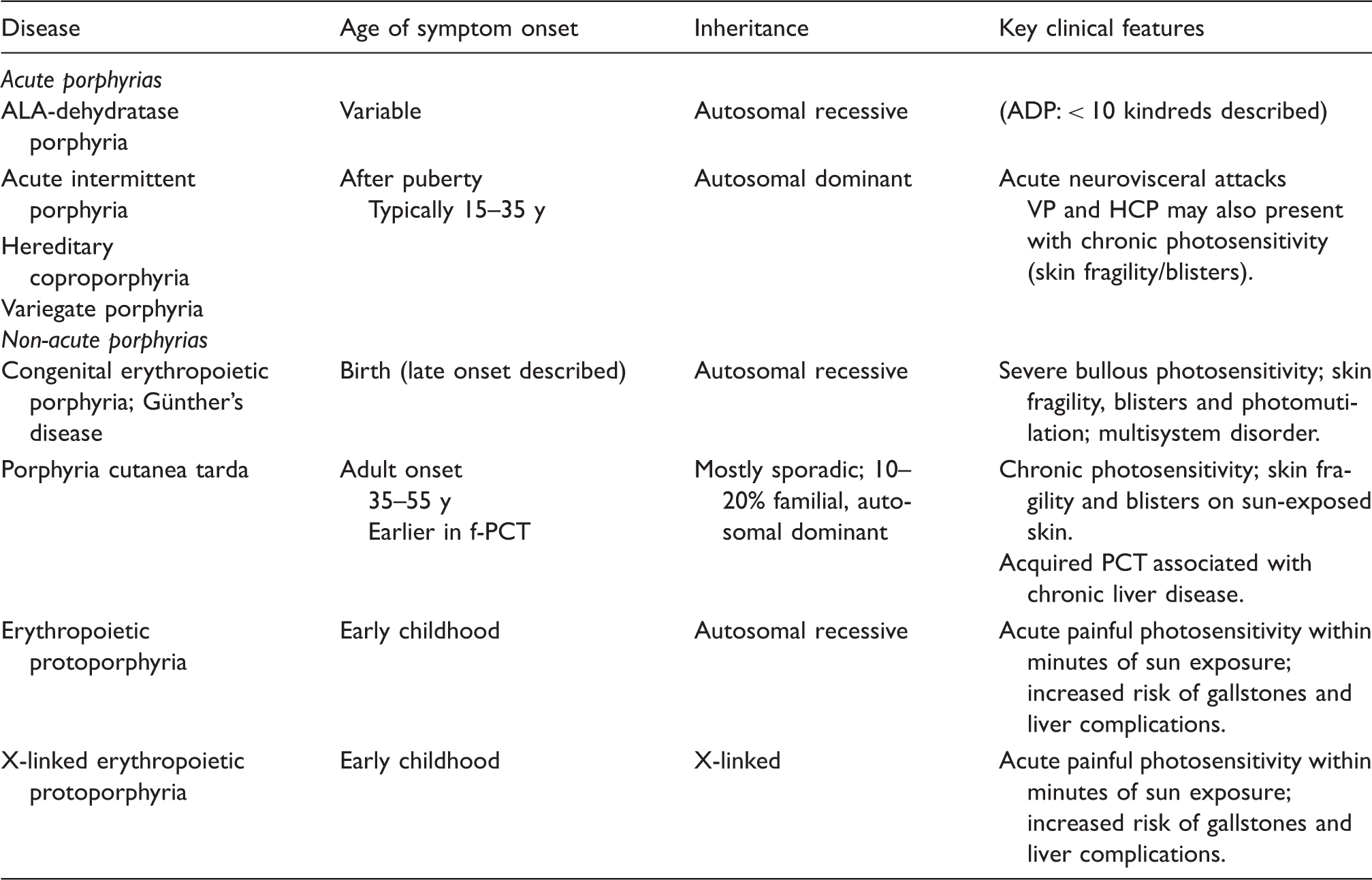
Four disorders cause potentially life-threatening acute neurovisceral attacks: acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP) and aminolevulinic acid dehydratase porphyria (ADP). AIP, VP and HCP are autosomal dominantly inherited. ADP is an exceedingly rare autosomal recessive disease. 5 Acute porphyria attacks almost never occur before puberty and affect a minority of porphyria patients (10–20%).1,2,6 Drugs, alcohol, infection, reduced calorie intake and stress are recognized factors in precipitating acute attacks. Acute attacks are more common in young females and may be related to endogenous hormone cycles, tending to occur premenstrually. Acute attacks are characterized by severe, poorly localized abdominal pain (also felt in the lower back and thighs), nausea, vomiting, constipation, hypertension and tachycardia. These features are attributable to autonomic neuropathy. Hyponatraemia is frequently present. Severe attacks may progress to a predominantly motor neuropathy and encephalopathy with weakness, paralysis, and psychiatric and behavioural disturbance. Sensory neuropathy may also occur. It is well described that an acute porphyria attack may mimic other disorders, for example, Guillain-Barré syndrome (an acute, ascending polyneuropathy). 6 An important clinical practice point is that in patients with advanced polyneuropathic or encephalopathic features, the symptoms of abdominal pain may be less prominent or overlooked. The only specific treatment available is to provide the end product of the haem biosynthetic pathway as intravenous haem, either as haem arginate (Normosang®) in Europe or haemin (Panhematin®) in the United States of America. There is evidence that this treatment is most effective if given early.7,8 Unfortunately, diagnostic delay is common and contributes to a poorer outcome, with prolonged requirement for intensive care and rehabilitation.
In the UK, there is now a National Acute Porphyria Service (NAPS) that provides 24 h access to expert advice on treatment of acute attacks, determining the need for haem arginate treatment and follow-up of all patients with a new acute attack in specialist porphyria clinics 9 (see Appendix 2).
Cutaneous porphyrias
Cutaneous porphyrias present with two distinct patterns of photosensitivity. 10
Skin fragility and blisters in sun-exposed areas are characteristic of disorders that cause accumulation of relatively water-soluble porphyrins in the upper dermis. This type of chronic photosensitivity is seen in congenital erythropoietic porphyria (CEP; also known as Gunther’s disease), 11 porphyria cutanea tarda (PCT) and may also be present in a proportion of patients with two of the acute porphyrias, VP and HCP. PCT is the commonest porphyria and is distinctive because it is mainly an acquired condition, presenting in middle age, usually on a background of chronic liver disease (although usually that liver disease is unrecognized until after investigations prompted by the diagnosis of PCT).
CEP is an extremely rare, severe multisystem disease, usually presenting soon after birth because of the finding of red urine. It causes lifelong skin fragility and blistering photosensitivity which may result in progressive scarring and photomutilation unless light avoidance measures are absolutely rigorous.
Erythropoietic protoporphyria (EPP) and X-linked erythropoietic protoporphyria (XLEPP) 12 cause acute pain in the skin after only minutes of sun exposure, due to photoactivation of accumulated hydrophobic protoporphyrin in lipid membranes, especially in endothelial cells of dermal capillaries. EPP and XLEPP typically present in infancy and childhood though there may be long delays in diagnosis. The intensely distressing episodes may be described as prickly, burning or itching pain, often lasting about three days once started and only eased by applying cold. When specifically asked many patients will have noted some associated swelling, especially of the backs of the hands, but often at the time of clinical examination the skin appears normal. These conditions may not be recognized for years because of the unusual sensation described and the frequent lack of clinical signs.
Diagnostic approach
Porphyrin analysis based on presenting symptoms.

ADP: ALA-dehydratase deficiency porphyria; AIP: acute intermittent porphyria; CEP: congenital erythropoietic porphyria; PCT: porphyria cutanea tarda; HCP: hereditary coproporphyria; VP: variegate porphyria; EPP: erythropoietic protoporphyria; XLEPP: X-linked dominant protoporphyria.
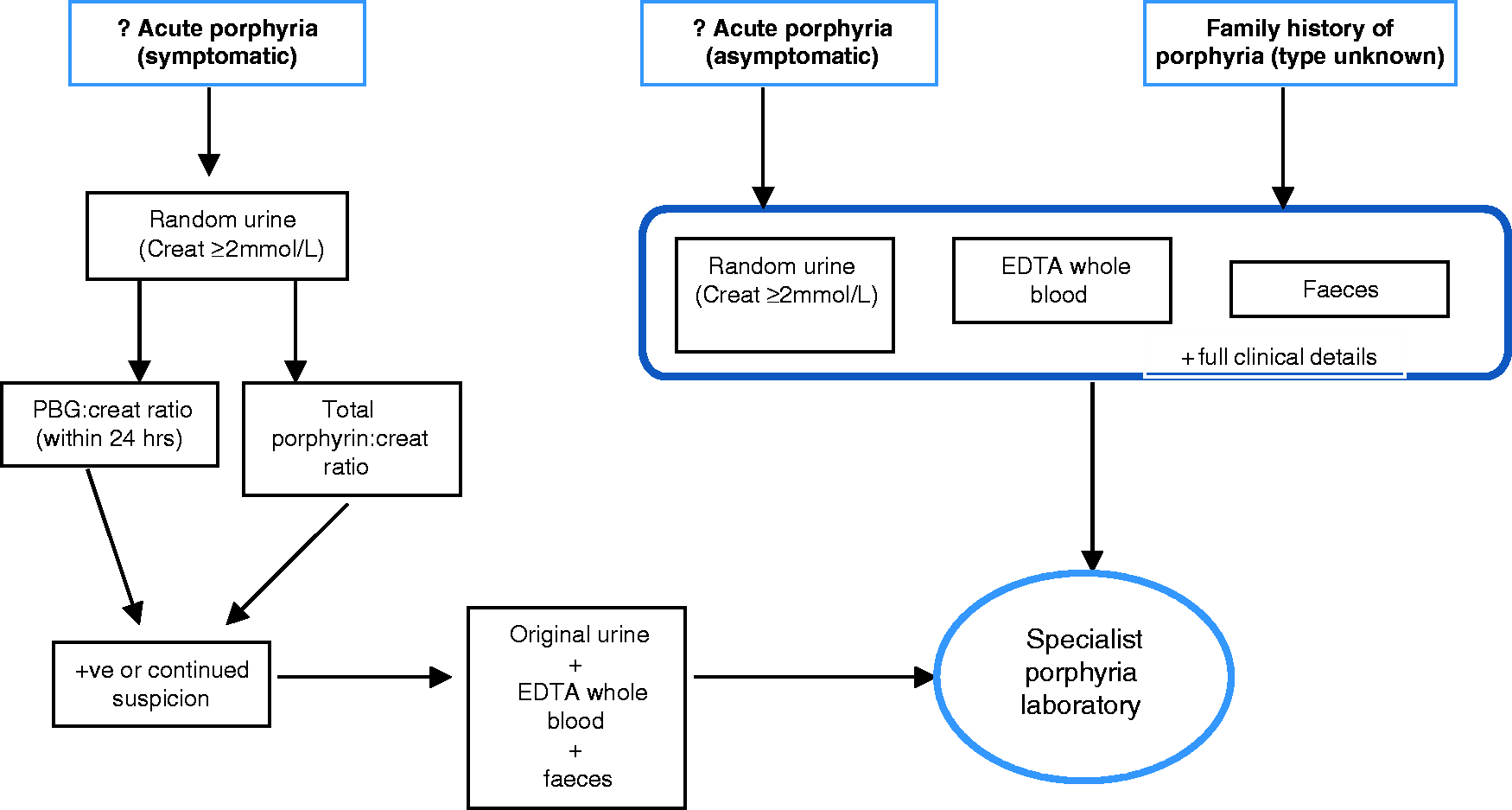
Algorithm for the investigation of acute porphyria or history of porphyria type unknown.
As there are no published evaluations of the positive and negative predictive values of a plasma and urine porphyrin measurement as first-line tests for cutaneous porphyrias, we have reviewed data from the five BIPNET specialist laboratories over the past five years in order to evaluate this diagnostic approach. We reviewed 457 patients in whom an unequivocal new diagnosis of PCT had been made on detailed porphyrin analysis (plasma porphyrin, urine porphyrin ± faecal porphyrin). All patients had a positive plasma porphyrin scan, with a fluorescence emission maximum between 617 and 623 nm and increased TUP with high-pressure liquid chromatography (HPLC) fractionation of the urines showing increased uroporphyrin and heptacarboxylic porphyrin excretion. Similarly, review of the EPP diagnoses in the Cardiff laboratory over the past 20 years confirmed that a positive plasma porphyrin fluorescence emission scan accompanied the finding of increased erythrocyte protoporphyrin in all 424 patients. The diagnostic sensitivity of plasma porphyrin scans in active VP has previously been established and has not been revaluated here. 15 Review of HCP patients is more problematic due to small number of cases and difficulty in establishing the presenting features accurately for referred samples. Most HCP patients present with acute symptoms 16 which are accompanied by increased urine PBG excretion. 3 Cutaneous presentation alone is very rare and usually associated with hepatobiliary dysfunction. 16 As with other cutaneous porphyrias the skin symptoms in HCP are due to photosensitization by circulating porphyrins and although this would be expected to be confirmed by a positive plasma porphyrin screen the paucity of data means that the predictive value of this single test cannot be accurately evaluated.
Pitfalls in laboratory diagnosis of cutaneous porphyria generally result from failure to send the correct sample(s) or to provide adequate clinical details to allow appropriate investigation and/or interpretation of results. Increased TUP may be incorrectly interpreted by clinicians as confirming a clinical diagnosis of PCT in a patient with blistering photosensitivity unless followed up by further appropriate testing (Figure 3).
The confirmation of the type of porphyria requires sophisticated methods of analysis that are available in specialist porphyria laboratories with expertise in interpretation and adherence to internal and external quality assurance (EQA) of the assays. 17 This may require faecal porphyrin analysis to distinguish between different types of porphyria. We would strongly encourage all laboratories that are making specific porphyria diagnoses to participate in a specialist network such as BIPNET, which can be contacted via details on their website. Once porphyria has been diagnosed referral to a porphyria or metabolic clinic for clinical advice and management is recommended. Similarly investigations for the inheritance of porphyria in asymptomatic family members are usually carried out in specialist porphyria laboratories and/or molecular genetics departments in conjunction with counselling from geneticists and porphyria specialists.
Samples and sample handling
A fresh early morning urine sample (∼10–20 mL) protected from light and collected preferably when the patient has symptoms is recommended. The collection of a 24-h urine specimen unnecessarily delays analysis and may give misleading results due to incomplete collection and inappropriate storage. Quantitative urine measurements (PBG and TUP) should always be expressed as a ratio to urine creatinine concentration. However, the results from dilute urine samples (urine creatinine <2.0 mmol/L) should be interpreted with caution (e.g. urines from young children or patients in intensive care) as they may give misleading ratios calculated from analytical data close to the detection limits of the methods used. Wherever possible, it is recommended that a repeat, preferably early morning, sample should be requested if urine has a creatinine <2.0 mmol/L. This statement applies to both qualitative and quantitative measurements of TUP and PBG.
Blood (5 mL) should be collected preferably into a blood collection tube with EDTA anticoagulant, although heparinized whole blood is acceptable for a plasma fluorescence scan. A stool sample (5–10 g) can be collected into an approved container for the measurement of stool porphyrins. Recommended porphyrin analysis based on presenting symptoms is shown in Table 2.
Sample stability
Investigations performed at the Cardiff centre have shown that porphyrins in urine, faeces and whole blood are stable for up to four days if stored protected from light at 4℃, and for up to two days if stored at room temperature. Porphyrins in separated plasma are less stable and exposure to ambient light at room temperature for 6 h can result in a 50% loss. The stability of PBG in urine was investigated at both Cardiff and Salford and the results show that a decrease of up to 37% can occur if the sample is exposed to light at room temperature for one day and up to 14% even if light protected. Freeze–thawing of urine samples, particularly repeated cycles, can also result in a marked decrease in PBG concentration and a corresponding increase in TUP.
All samples should be protected from light by wrapping labelled containers in black plastic or tin foil immediately after collection and transported to the receiving laboratory as soon as possible. Samples must remain light protected until the point of analysis. In the event that samples are to be referred on to a specialist porphyria laboratory, every effort should be made to ensure that they are despatched within 24 h of receipt to prevent the degradation of porphyrins. Properly light-protected samples may be sent by first class post. If samples are received prior to a weekend or public holiday, faecal samples can be frozen, light protected until despatch but urine and blood samples should be stored at 4℃ also light protected. Haemolysed blood is unsuitable for plasma porphyrin analysis.
Staffing and service quality
The methods for porphyrin analysis available in the laboratory should be determined by the workload, technical expertise and the availability of appropriate equipment for the analysis. Although porphyrias are suspected far more frequently than they are diagnosed, porphyrin tests are infrequent analyses in most general clinical laboratories. UK laboratories are currently accredited to at least CPA standard (www.ukas.com) and most are preparing for UKAS accreditation under an internationally recognized standard ISO 15189 which requires demonstration of individual competencies. This is likely to pose problems for laboratories where low sample numbers are analysed. ISO standards also necessitate use of specific internal quality assurance (IQC) material and participation in appropriate EQA schemes both of which may be disproportionately expensive relative to the numbers of samples assayed. Where a laboratory has no access to necessary equipment or there is insufficient workload to ensure that training is maintained and competency achieved, we suggest that laboratories liaise either with other local laboratories within their network or a specialist porphyria laboratory for porphyria testing.
Quality control
Internal quality control
It is essential to monitor performance of the methods used with commercially available IQC material where available. Lyphochek® Urine Control Level 2 manufactured by Bio-Rad Laboratories, Inc. can be used for TUP methods or ClinChek® I and II manufactured by Recipe® Chemical and Instruments and supplied by Waters Limited for TUP, ALA and PBG. Urine controls and calibrator are also available from Chromsystems®. There is no commercially available IQC material for total faecal porphyrin, quantitative erythrocyte porphyrin or plasma porphyrin.
External quality assessment schemes
The Welsh External Quality Assessment Scheme (WEQAS, www.weqas.com) have an EQA Scheme for TUP, urine PBG (qualitative and quantitative) and qualitative plasma porphyrins. This scheme is run bi-monthly with three specimens. The Royal College of Pathologists of Australasia (RCPA, www.rcpaqap.com.au) Chemical Pathology Quality Assurance Program run a bi-monthly porphyrin scheme for urine, faeces, whole blood, quantitative plasma analysis and HPLC fractionation of urine and faeces. The RCPA also runs a separate scheme for clinical interpretation of a case and biochemical results. A third scheme run by the European Porphyria Network (EPNET) has two distributions a year with samples from a patient with a clinical case for interpretation and biochemical analysis of all available porphyrin assays including genetic analysis. These last two schemes are more suitable for specialist porphyria laboratories offering an extensive array of porphyrin tests.
Equipment
Quantitative urine PBG and TUP measurements require a UV-Vis scanning spectrophotometer, accurate and precise pipettes and a refrigerator or cold room for storage of the reagents at 4℃. A −20℃ freezer is required for storage of plasma and faeces, if analysis is delayed for more than 24 h. The equipment used for porphyrin analysis should be maintained and serviced annually to ensure it is in working order. Porphyrin fluorescence detection requires a red-sensitive photomultiplier tube for both the fluorimeter and the HPLC fluorometric detector in order to perform plasma porphyrin scans, total faecal porphyrin, red cell-free protoporphyrin and HPLC fractionation of urine and faecal porphyrins. These investigations will therefore generally need to be referred to a local network or a specialist porphyria laboratory that has the appropriate equipment (Appendix 2).
Methodology
Effective investigation of porphyrias must take into account the clinical symptoms and every effort should be made to collect samples while symptoms are present. Distinguishing the cutaneous from the acute porphyrias is a useful approach for decisions on which tests to perform. Laboratories should offer quantitative PBG excretion to test for acute porphyrias and, in the absence of a plasma sample, TUP excretion to detect an acute porphyria where there has been a delay in sample collection and urine PBG excretion has returned to normal (VP and HCP). The investigation of photosensitive porphyria requires blood analysis using a scanning fluorimeter fitted with a red-sensitive photomultiplier and should be performed at a laboratory with appropriately trained staff. Although HPLC fractionation of faecal porphyrins is essential for the differential diagnosis of VP and HCP, total faecal porphyrin analysis is not an essential test for the primary screening for porphyrias. False-positives can result from increased faecal porphyrins due to bacterial degradation of haem in the gut to dicarboxylic porphyrins. The haem may be dietary in origin or pathological.
The methods used for the measurement of PBG and TUP that are currently used by our laboratories are described in Appendix 1. The withdrawal from the market of the Trace® PBG kit by Thermo Scientific has left a gap in the methodology for rapid urinary PBG analysis. Previously widely used qualitative methods such as the Watson-Schwartz test 18 the Rimington method 19 and the Hoesch test 20 are all simple procedures with a lower detection limit of approximately 12 μmol/L. However, these methods have been criticized for low sensitivity and poor specificity21,22 and EQA data from WEQAS showed that at a concentration of 10.7 μmol/L (upper reference limit) the percentage assessed as positive was 40% vs. 80% positive using the Trace® PBG kit (WEQAS porphyrin guide). We recommend that in the absence of a suitable semi-quantitative PBG screening method, all PBG analyses should be performed using fully quantitative ion exchange resin based methods as outlined in Appendix 1, which have lower detection limits of 2.5 μmol/L.
The full quantitation of PBG in urine takes more time to perform two levels of quality control and a patient sample than the semi-quantitative Trace® PBG kit: approximately 75 min vs. 30 min. However, the argument for the use of full PBG quantitation is based both on the relative insensitivity of the Trace® PBG kit and economics. The Trace® PBG kit was expensive, typically costing £57 for the analysis of two IQC levels and a patient sample and considerably more if performed outside normal working hours. The most widely used commercially available kit for PBG measurement is available from BioRad Laboratories Limited and consumable costs for two IQC and a test sample are approximately £19. The authors have as yet no experience with a PBG kit quite recently brought on to the market by Recipe chemical and instruments company and distributed in the United Kingdom by Waters Limited. This kit is cheaper at £9 for two IQC and a patient sample but further work on assessing its suitability is required.
Role of specialist porphyria laboratories
There is evidence from Welsh and European EQA schemes that certain analyses, in particular red cell protoporphyrin quantitation, plasma fluorescence emission scanning and faecal porphyrin measurement are performed to a variable standard. 17 It is therefore questionable whether these tests should be performed outside those specialist centres that have an adequate workload to maintain analytical expertise.
Specialist porphyria laboratories are linked to porphyria clinics and clinical specialists are available for clinical and technical advice. A specialist porphyria laboratory is defined by EPNET as having the ability to distinguish between all types of porphyria, perform a defined set of porphyrin assays, provide test result interpretation, offer clinical advice and participate in EQA schemes. 17
It is important to refer specimens for confirmatory quantitative porphyrin analysis when urine PBG and/or porphyrin testing is positive in a suspected acute porphyria or when a TUP is positive in a suspected bullous porphyria. It is also important to send blood, urine and faecal samples to a specialist laboratory when screening tests are negative, but clinical suspicion remains high. It can be extremely difficult to rule out porphyria in asymptomatic patients who present with either a family history or previous personal diagnosis with the exception of VP where detection of the protein complexed porphyrin by spectrofluorimetric scan is clinically sensitive and specific. 23 Asymptomatic family members of patients with inherited porphyria may show no biochemical abnormalities and may be falsely reassured by normal biochemical results. In these cases, and for children before puberty, molecular genetic analysis is required and should be accessed through referral to a clinical genetics service or a specialist porphyria clinic (Appendix 2).
Although many patients with previous acute porphyria attacks (particularly those with AIP) continue to show biochemical abnormalities for many years even when clinically well, many will eventually become completely latent. 14
Other specific situations that require specialist expertise include: investigation of pseudoporphyria and hyperbilirubinaemia. Pseudoporphyria is a photodistributed bullous disorder with the clinical and histological features of PCT but without any underlying increase in porphyrin production. It can be caused by medications, chronic renal failure or dialysis, excessive sun exposure and UVA radiation. 24 The hereditary hyperbilirubinaemias may also have abnormal urine coproporphyrin excretion and the relative proportions of the coproporphyrin isomers can help in the differential diagnosis. In Dubin–Johnson Syndrome, the coproporphyrin I isomer is increased to greater than 80% of the total coproporphyrin. Rotor syndrome will show an increase in coproporphyrin I with normal concentrations of coproporphyrin III, and in Gilbert’s disease an increase in both the coproporphyrin I and III isomers is found. 25
Evidence of increased porphyrin production and excretion can occur in other disease states and is not always indicative of porphyria. It is important to recognize these secondary causes of porphyrin overproduction as a lack of awareness can lead to misdiagnosis of porphyria.
Porphyrinuria is a term used to describe increased urine porphyrin excretion not caused by porphyria. It is usually associated with liver dysfunction, various drugs and toxins metabolized by the liver, most frequently excess alcohol consumption and rarely lead poisoning. Urine and faecal porphyrin analysis by HPLC fractionation of the porphyrin isomers is required to distinguish between secondary causes of porphyrinuria and porphyria.
Scope of the guidelines
The guidelines recommend best methodological practice for general clinical laboratories to provide safe, cost-effective initial testing for suspected porphyria and algorithms for onward referral of appropriate samples to specialist porphyria laboratories. The guidelines represent the consensus of clinicians and scientists in the British and Irish Porphyria Network (BIPNET; www.bipnet.org.uk) whose laboratories meet the quality standards for specialist laboratories within the European Porphyria Network (EPNET; www.porphyria-europe.org).
Footnotes
Acknowledgements
The authors would like to thank Orphan Europe for their support for BIPNET meetings, WEQAS for EQA data, Dr Robert Dawe of The Photobiology Unit, Ninewells Hospital, Dundee, for his valuable contribution to the revision of the text and Professor George Elder, Emeritus Professor, University of Cardiff for his invaluable advice and support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Orphan Europe has provided financial support for BIPNET meetings and the set-up costs for the BIPNET website.
Ethical approval
Not applicable.
Guarantor
JW and MB.
Contributorship
JW conceived the idea for the guidelines and co-wrote the first draft with JM. All the authors are members of either the laboratory or clinical subgroups of the British and Irish Porphyria Network (BIPNET) and all contributed to the development of the final version.