
Abstract
Objective
With a high-sensitivity troponin assay, it may be possible to exclude acute myocardial infarction with a single blood test on arrival in the emergency department by using a novel ‘rule out’ cut-off set at the limit of detection of the assay. We aimed to determine whether this can also be achieved using a contemporary sensitive troponin assay that does not meet ‘high-sensitivity’ criteria.
Methods
In a prospective diagnostic cohort study, we included patients presenting to the emergency department with suspected cardiac chest pain. For this secondary analysis, serum samples drawn on arrival were tested using a contemporary sensitive troponin I assay (s-cTnI; Siemens Ultra ADVIA Centaur, 99th percentile 40 ng/L, limit of detection 6 ng/L). Acute myocardial infarction was adjudicated by two independent investigators based on reference standard troponin testing ≥12 h after symptom onset.
Results
Of 414 participants, 70 (16.9%) had acute myocardial infarction and 205 (49.5%) had initial s-cTnI concentrations below the limit of detection. Using the limit of detection as a ‘rule out’ cut-off gave a sensitivity of 94.3% (95% CI 86.0–98.4%) for acute myocardial infarction. If only patients with s-cTnI below the limit of detection and no electrocardiogram ischaemia were considered to have acute myocardial infarction ‘ruled out’ (41.8% of the cohort, n = 174), sensitivity would rise to 97.1% (90.1–99.7%) and negative predictive value to 98.8% (95.9–99.9%).
Conclusions
Acute myocardial infarction cannot be excluded in patients with s-cTnI concentrations below the limit of detection using the contemporary sensitive assay evaluated. Future work with this assay should focus on serial sampling over 1–3 h and combination with clinical information and/or additional biomarkers.
Keywords
Background
The diagnosis of an acute myocardial infarction (AMI) hinges upon the detection of a rise and/or fall of cardiac troponin with at least one level above the 99th percentile of a healthy reference population. 1 With the increasing analytical sensitivity of modern assays, it is now possible to quantify troponin concentrations in apparently healthy individuals, including concentrations that are below the 99th percentile of a reference population.
For a troponin assay to be labelled as ‘high sensitivity’, it must (a) have adequate precision at the 99th percentile upper reference limit (defined as a coefficient of variation of <10%) and (b) detect troponin concentrations in over 50% of apparently healthy individuals. 2 The Roche high sensitivity troponin T (hs-cTnT; Elecsys 2010) and Abbott ARCHITECT high sensitivity troponin I are examples of assays that meet ‘high-sensitivity’ criteria. While the Siemens ADVIA Centaur troponin I assay also has adequate precision at the 99th percentile, it detects troponin concentrations in only 46% of apparently healthy individuals. 3 The assay has therefore been labelled as ‘contemporary sensitive’ rather than ‘high sensitivity’.
One of the key limitations of cardiac troponin as a biomarker of AMI is the time taken for circulating concentrations to rise after the onset of infarction. This means that patients who present to the emergency department (ED) with symptoms that are compatible with an acute coronary syndrome are routinely admitted to hospital for serial troponin testing over a number of hours.1,4,5 Ultimately, however, only a minority of those patients will be diagnosed with AMI.6,7 A strategy that would enable exclusion of that diagnosis in a proportion of patients at the time of initial presentation could reduce the need for serial sampling and therefore unnecessary hospital admissions.
Using hs-cTnT we have previously shown that it may be possible to exclude AMI in up to 27.7% of patients who had completely undetectable troponin concentrations (<3 ng/L, which is labelled as the ‘limit of blank’ of this assay). 8 In support of those findings and in a retrospective cohort of over 10,000 patients, Bandstein et al. 9 subsequently demonstrated that patients who have a normal electrocardiogram (ECG) and initial hs-cTnT concentrations below the limit of detection (LoD) of the assay (<5 ng/L) have extremely low probability of AMI, which may be considered to exclude the diagnosis. We do not yet know whether it would also be possible to exclude AMI in patients with initial troponin concentrations below the LoD of a ‘contemporary sensitive’ (rather than ‘high sensitivity’) assay such as the Siemens ADVIA Centaur troponin I Ultra.
Goals of this investigation
We sought to determine whether troponin concentrations below the LoD of a contemporary sensitive assay (Siemens ADVIA Centaur troponin I Ultra) could be used, either alone or in combination with the ECG, to exclude a diagnosis of AMI at the time of initial presentation to the ED.
Methods
Design, setting and selection of participants
In this secondary analysis from a prospective diagnostic cohort study, we included patients who presented to the ED at Stepping Hill Hospital, Stockport, United Kingdom, with chest pain that the treating physician suspected might be cardiac. We excluded patients whose symptoms occurred >24 h prior to presentation, those with another medical condition necessitating hospital admission, patients with renal failure requiring dialysis, those with suspected myocardial contusion secondary to trauma, pregnant women, non-English speakers, prisoners and those for whom all means of follow up would be impossible. All participants provided written informed consent, and ethical approval was obtained from the Research Ethics Committee North West–Cheshire (reference 09/H1014/74). From data collected in this cohort study, we have already published an external validation of the Manchester Acute Coronary Syndromes clinical decision rule, an evaluation of the diagnostic performance of the clinical judgement of emergency physicians and an analysis of the diagnostic value of pain severity.10–12
Methods and measurements
Clinical data were recorded at the time of presentation to the ED. ECGs were interpreted contemporaneously by the treating emergency physician, and the presence or absence of changes consistent with acute myocardial ischaemia (in the treating physician’s opinion) was recorded in the case report form. All participants also underwent venepuncture at the time of initial presentation. Troponin T (cTnT, Roche Diagnostics Elecsys 4th generation, 99th percentile 10 ng/L, coefficient of variation <10% at 30 ng/L) was measured on arrival and at least 12 h after symptom onset, which formed the reference standard biomarker for AMI.
Serum samples drawn at the time of presentation and 12 h after symptom onset were frozen at −70 ℃ pending later testing for sensitive troponin I (s-cTnI; Siemens troponin I Ultra, ADVIA Centaur). The 99th percentile of this assay is reported by the manufacturer as 40 ng/L. The lowest concentration, at which the coefficient of variation is <10%, is 30 ng/L. The functional sensitivity of the assay (defined as the minimum troponin concentration with a coefficient of variation <20%) is 17 ng/L, and the LoD is 6 ng/L.
Follow up
Patients were followed up after 30 days by (a) checking mortality status using a national database (the National Health Service Strategic Tracing Service database), (b) review of medical records and (c) either telephone, home visit or review in clinic. If it was not possible to contact the patient directly, their general practitioner (GP) was contacted. Follow-up was only considered valid in this event if the patient had been in contact with their GP and if sufficient information was available to determine ED attendances, hospital admissions, investigations and episodes of chest pain. If a patient had attended another hospital during the follow up period, copies of relevant records were obtained from that hospital.
Outcomes
The primary outcome for this analysis was a diagnosis of AMI. The diagnosis of AMI was adjudicated by two independent investigators with reference to clinical, laboratory and imaging data but blinded to s-cTnI concentrations. Discrepancies were to be resolved by discussion although none occurred. AMI was defined in accordance with the third universal definition 13 and required a rise and/or fall of cTnT with at least one level above the 99th percentile (10 ng/L) and evidence of a rise and/or fall of at least 20 ng/L based on the analytical characteristics of the assay.
As a secondary outcome, we also evaluated major adverse cardiac events (MACE) within 30 days, defined as AMI (including prevalent AMI diagnosed at the initial presentation), death (all cause) or the need for coronary revascularisation (including the detection of a new coronary stenosis >50% of the lumen of a major epicardial vessel, as reported by the responsible interventional cardiologist).
Sensitivity analysis
Recognising that our reference standard biomarker for AMI was troponin T, which may have different diagnostic performance to the troponin I assay being investigated, we also evaluated the diagnostic performance of the presentation s-cTnI concentration for predicting a subsequent s-cTnI rise to above the 99th percentile (40 ng/L) measured at least 12 h from symptom onset. To avoid incorporation bias, patients who presented to the ED more than 12 h after symptom onset were excluded from this analysis.
Statistical analysis
All statistical analyses were undertaken in SPSS version 20.0 (SPSS Inc, Chicago, Illinois) and/or MedCalc version 12.4.0.0 (Mariakerke, Belgium). Baseline characteristics were summarised using descriptive statistics. To evaluate diagnostic performance, we calculated sensitivity, specificity, positive predictive values and negative predictive values together with 95% CI. We evaluated the overall diagnostic performance and also calculated the area under the receiver operating characteristic (ROC) curve.
Results
In total, 414 patients, who presented between April and July 2010, were eligible for inclusion in this analysis. All patients completed follow up at 30 days. In total, 70 (16.9%) patients had AMI and 86 (20.8%) patients developed MACE within 30 days (Figure 1). The baseline characteristics of participants are shown in Table 1. A comparison of the baseline characteristics of included patients with those who were excluded because no serum sample was available is shown in the Appendix (Supplementary Table 1). Included patients had a higher overall prevalence of angina but there were no other significant differences. In total, 16.4% (n = 68) patients had initial s-cTnI concentrations above the 99th percentile and 49.5% (n = 205) had concentrations below the LoD of the assay (<6 ng/L); 41.8% (n = 174) patients had both s-cTnI concentrations <6 ng/L and no evidence of ECG ischaemia.
Participant flow diagram. Baseline characteristics of included patients. AMI: acute myocardial infarction. Numbers relate to frequencies with percentages in parentheses unless otherwise stated.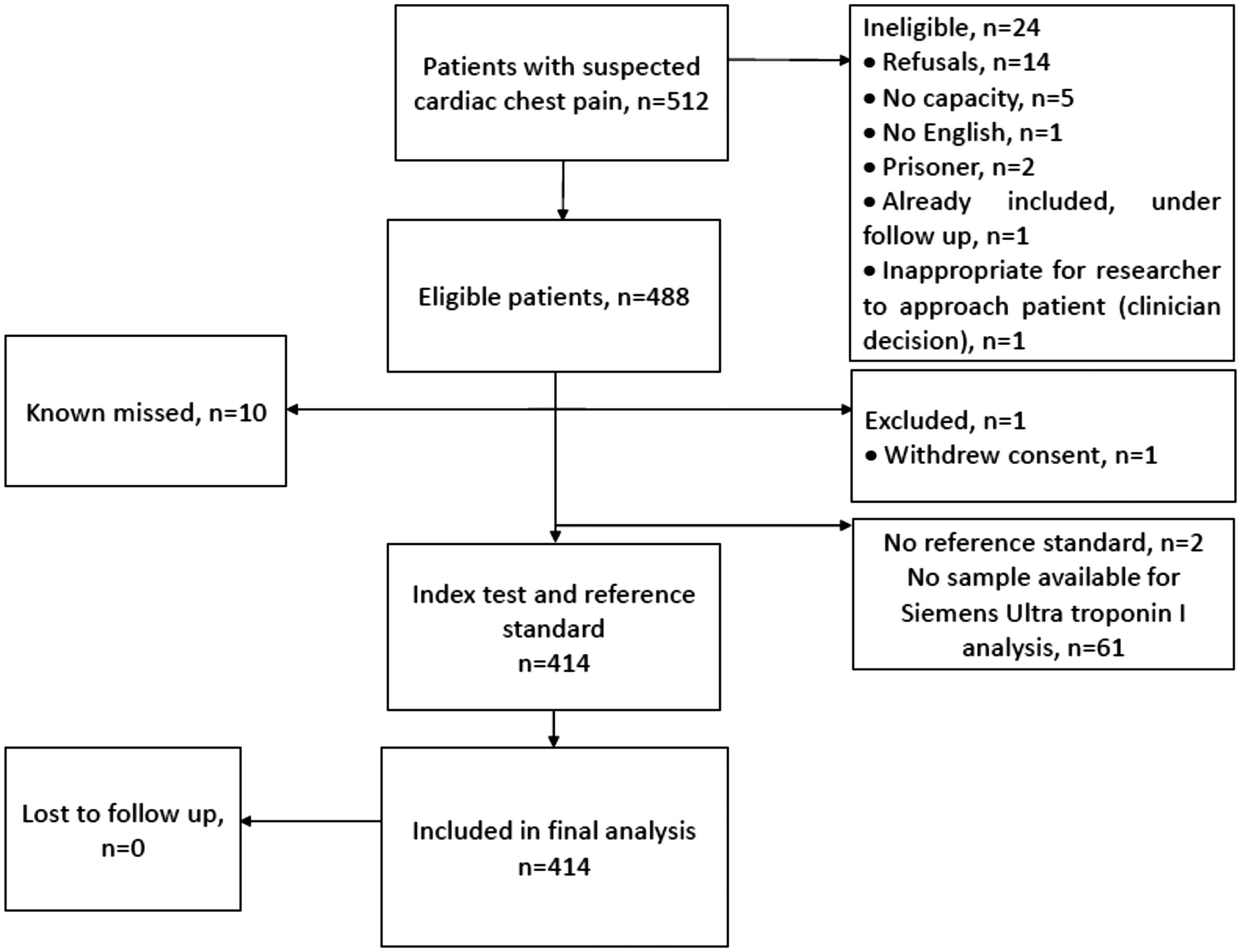
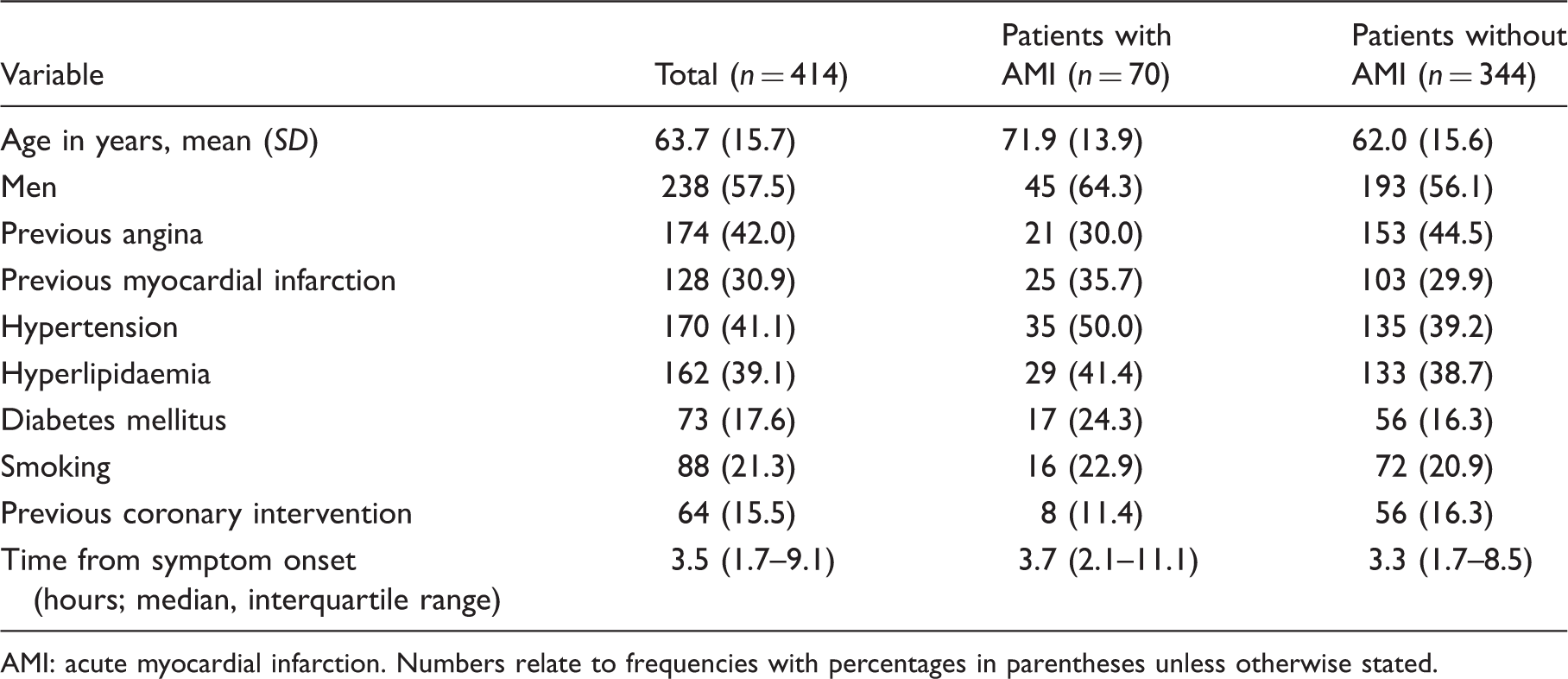
Diagnostic performance of the Siemens sensitive troponin I (s-cTnI) assay for diagnosing AMI at ‘rule out’ cut-offs studied.
PPV: positive negative values; NPV: negative predictive values.
Patients could be ‘ruled out’ only if they have a s-cTnI concentration below this threshold and no evidence of ECG ischaemia at the time of presentation to the ED.
If only patients with s-cTnI below the LoD (<6 ng/L) and no evidence of ECG ischaemia were considered to have the diagnosis ‘ruled out’, sensitivity would rise to 97.1% (95% CI 90.1–99.7%) and negative predictive value to 98.8% (95% CI 95.9–99.9%). Thus, this strategy would miss 2.9% (n = 2) of AMIs and would mean that discharged patients have a 1.2% posttest probability of AMI. The findings were similar among patients whose time from symptom onset was both <6 h and ≥6 h (see Supplementary Tables 2 and 3, available online).
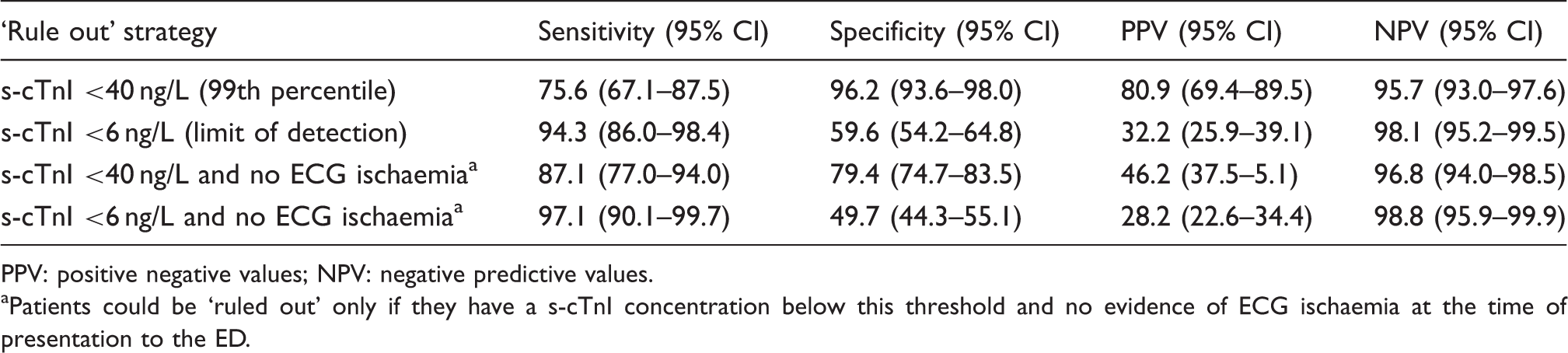
Diagnostic performance of the Siemens troponin I (s-cTnI) assay for MACE at ‘rule out’ cut-offs studied.
PPV: positive negative values; NPV: negative predictive values.
aPatients could be ‘ruled out’ only if they have a s-cTnI concentration below this threshold and no evidence of ECG ischaemia at the time of presentation to the ED.
Sensitivity analyses
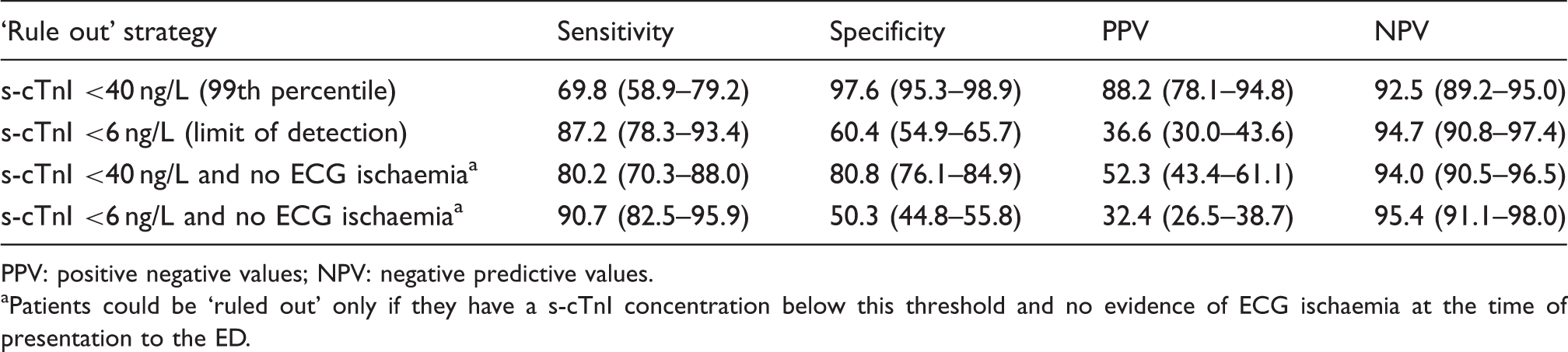
Diagnostic performance of Siemens sensitive troponin I (s-cTnI) concentrations at presentation for predicting any subsequent troponin I rise to above the 99th percentile at 12 h following symptom onset.
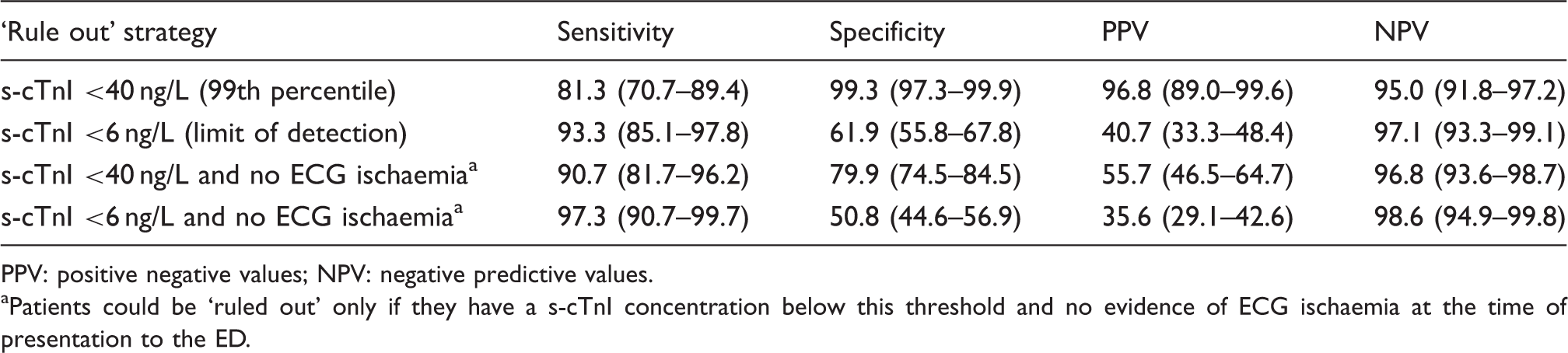
PPV: positive negative values; NPV: negative predictive values.
aPatients could be ‘ruled out’ only if they have a s-cTnI concentration below this threshold and no evidence of ECG ischaemia at the time of presentation to the ED.
Of the two patients who were missed by this strategy, one had presented within 1 h of symptom onset. That patient had a s-cTnI concentration of 342 ng/L at 12 h. The patient also had a normal c-TnT (<10 ng/L) at admission, which rose to 50 ng/L at 12 h, and underwent percutaneous coronary intervention to a stenosed left anterior descending artery on an inpatient basis. The other patient who was missed by this strategy also presented 1 h after symptom onset and had a s-cTnI concentration of 68 ng/L at 12 h. At the time of arrival, this patient did have an elevated cTnT of 40 ng/L.
Discussion
These findings suggest that the contemporary sensitive troponin I assay we have evaluated (Siemens ADVIA Centaur troponin I Ultra) has insufficient diagnostic sensitivity and negative predictive value to exclude AMI at the time of presentation to the ED using cut-offs below the 99th percentile. Among the 41.8% of patients who had initial s-cTnI concentrations below the LoD and no evidence of ECG ischaemia, the posttest probabilities of AMI and MACE were found to be 1.2% and 4.6%, respectively. While this may be useful for risk stratification and to assist with triage to an appropriate level of inpatient care, this level of diagnostic accuracy is unlikely to be considered sufficient for clinicians to consider this a safe ‘rule out’ strategy. 14
Using a high sensitivity troponin T assay (Roche Diagnostics 5th generation Elecsys, 99th percentile 14 ng/L, coefficient of variation <10% at 12 ng/L), previous work has shown that AMI could be excluded with very high negative predictive value in patients who have completely undetectable concentrations (below the limit of blank of the assay, <3 ng/L) at the time of initial presentation.8,15 A further retrospective evaluation of over 14,000 patients suggested that patients with initial concentrations below the LoD of the assay (5 ng/L) and no ECG evidence of ischaemia have a very low probability of AMI. 9
The troponin I assay that we have evaluated meets precision criteria for a high sensitivity assay (coefficient of variation <10% at the 99th percentile) but does not detect troponin concentrations in >50% of healthy individuals. Thus, it is labelled as a ‘contemporary sensitive’ assay rather than as having ‘high sensitivity’. 2 The diagnostic performance of this assay at the conventional 99th percentile cut-off has been shown to be similar to high sensitivity assays. 16 With serial testing over 3 h, it may be possible to exclude AMI with high diagnostic sensitivity using this troponin I assay. 6 In this research, we determined whether the detection of extremely low troponin concentrations by this assay at the time of arrival in the ED could be used immediately to exclude AMI in a proportion of patients. We found that the diagnostic performance of this assay at the conventional 99th percentile cut-off is comparable to that reported by Collinson et al. 17 In a cohort of 850 patients, that group reported a sensitivity of 73.0%. Keller et al. 6 reported a higher sensitivity of 90.2%. However, our findings suggest that sensitivity remains suboptimal to exclude the diagnosis of AMI even at the unconventional cut-off set at the LoD of the assay.
Limitations
This study does have some important limitations. First, of the 477 patients who participated in this study in total, only 414 patients had serum aliquots available for this analysis. This raises the possibility of selection bias, although the baseline characteristics of those included were not significantly different to those without available serum samples. Second, our reference standard for AMI was a troponin T assay at ≥12 h. A small proportion of patients with a rise and/or fall of troponin T may not have a detectable rise and/or fall of troponin I (and vice versa), which could have led to underestimates of diagnostic performance given that we were evaluating a contemporary sensitive troponin I assay. To address this, we included a sensitivity analysis evaluating the diagnostic performance of initial troponin I concentrations for predicting subsequent troponin I elevations, which demonstrates that the conclusions drawn would remain unchanged. While it is important to recognise these limitations, it is therefore unlikely that they have affected our findings.
Finally, it is important to consider the precision of the assay at such low troponin concentrations. The functional sensitivity of the Siemens ADVIA Centaur troponin I Ultra assay, at which the coefficient of variation is below 20%, is 17 ng/L. Because of concerns about precision below that concentration, some laboratories would not report lower concentrations. The precision of the assay may have contributed to our findings and presents a further barrier to clinical implementation of a ‘rule out’ cut-off set at the LoD of this assay. It is important, also, to recognise that samples in this study were analysed in batches, which could have reduced the impact of precision on our findings.
Conclusions
In summary, troponin I concentrations measured using a contemporary sensitive assay cannot be used to exclude AMI at the time of presentation to the ED, even among patients who have concentrations below the LoD of the assay and a normal ECG. Further research should focus on (a) the combination of troponin concentrations with additional clinical information and/or alternative biomarkers and (b) the earliest time point at which AMI can be safely excluded with serial sampling.
Footnotes
Acknowledgements
We acknowledge the support of the Manchester Academic Health Science Centre, the Manchester Biomedical Research Centre and the National Institute for Health Research (United Kingdom). We also acknowledge the support of all the staff in the Emergency Department and Biochemistry Department at Stepping Hill Hospital who assisted with this work, in particular Sister Joanna Jarvis.
Declaration of conflicting interests
For the purposes of this research, reagents were donated to the research team without charge by Siemens Diagnostics. For additional analyses from this cohort study (not reported here), Roche Diagnostics and Abbott Laboratories also donated reagents without charge. Dr Body has undertaken other research using reagents that were donated without charge by Roche Diagnostics, Siemens Diagnostics, Abbott Laboratories, Alere Diagnostics and Randox Laboratories. Dr Body has accepted travel and accommodation for conferences from Roche Diagnostics and Randox Laboratories. Dr Body is a principal investigator for a study sponsored by Roche Diagnostics. Dr Lewis has accepted travel and accommodation to lecture for Randox Laboratories.
Funding
This study was funded by a grant from the United Kingdom College of Emergency Medicine and was supported by: (a) fellowship funding (an Academic Clinical Lectureship for the first author) from the United Kingdom National Institute for Health Research (NIHR), (b) by the NIHR Clinical Research Network (UK CRN 8376) and (c) reagents donated for the purposes of the research by Siemens.
Ethical approval
This study was approved by the Research Ethics Committee North West–Cheshire (reference 09/H1014/74).
Guarantor
RB.
Contributorship
RB conceived the study. RB, GB and PL were involved in protocol development. RB gained ethical approval, supervised/undertook patient recruitment and data analysis. RB and SC adjudicated patient outcomes. RB wrote the first draft of the manuscript, and the analyses were critically reviewed by all authors. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.