
Abstract
Familial lipoprotein lipase (LPL) deficiency (FLLD) is a rare autosomal recessive genetic disorder caused by homozygous or compound heterozygous mutations in the LPL gene. FLLD individuals usually express an impaired or non-functional LPL enzyme with low or absent triglyceride (TG) hydrolysis activity causing severe hypertriglyceridaemia. Here we report a case of FLLD in a 29-year-old man, who initially presented with eruptive cutaneous xanthomata, elevated plasma TG concentration but no other co-morbidities. Subsequent genetic testing of the patient revealed compound heterozygosity of a novel duplication (p.R44Kfs*4) leading to a premature stop codon in exon 2 and a known mutation (N291S) in exon 5 of the LPL gene. Further biochemical analysis of the patient's postheparin plasma confirmed a reduction of total lipase activity compared with his heterozygous father carrying the common N291S mutation and to a healthy control. Also the patient showed increased (1.85-fold) activity of hepatic lipase (HL), indicating a functional link between HL and LPL. In summary, we report a case of FLLD caused by compound heterozygosity of a new duplication and a common mutation in the LPL gene, resulting in residual LPL activity. With such mutations, individuals may not receive a diagnosis before classical FLLD symptoms appear later in adulthood. Nevertheless, early diagnosis and lipid-lowering treatment may favour a reduced risk of premature cardiovascular disease or acute pancreatitis in such individuals.
Introduction
Familial lipoprotein lipase (LPL) deficiency (FLLD) is a rare autosomal recessive genetic disorder, causing low or absent LPL activity and leading to severe hypertriglyceridemia due to impaired clearance of chylomicrons from the plasma. 1 The disease is often diagnosed in childhood and clinically manifested by elevated plasma triglyceride (TG), episodes of abdominal pain, recurrent acute pancreatitis, eruptive cutaneous xanthomata and hepatosplenomegaly. 1
FLLD is caused by homozygous or compound heterozygous mutations in the LPL gene that impair the function of the enzyme. 1 LPL is the key enzyme regulating the catabolism of TG-rich lipoproteins in blood and making free fatty acids available for tissue uptake and subsequent utilization or storage. 2,3 The active enzyme is a non-covalent dimer of the 448-amino acid LPL monomer secreted primarily by adipose tissues and skeletal muscle. 2 LPL contains a catalytic centre, which is activated by apolipoprotein (apo) C-II. 4 Secreted LPL binds to heparin sulphate proteoglycans on the luminal surface of the endothelium, from where it can sequester and hydrolyze the TG content of chylomicrons and very low-density lipoprotein (VLDL) particles in the blood. 4,5 Administration of intravenous heparin displaces LPL and hepatic lipase (HL) into the circulation, allowing measurement of lipase activity and mass in postheparin plasma to obtain functional information regarding genotype/phenotype relation. 6
The LPL gene spans a region of 30 kb on chromosome 8p22 and consists of 10 exons. 7 Currently 161 different LPL mutations have been reported in the Human Gene Mutation Database (HGMD). 8 The current spectrum of reported mutations includes missense/nonsense, splicing, regulatory, small deletions, small insertions, small indels (deleted/inserted bases), gross deletions, gross insertions/duplications and one case of a complex rearrangement. The majority of these LPL mutations comprise missense mutations which mostly map to exons 5 and 6. 8
Many reported cases of FLLD include infants or children who presents with aforementioned symptoms, leading to an early diagnosis and treatment. 9–11 At present it is still a subject of debate whether FLLD is associated with increased risk of developing cardiovascular disease or not. 11–13 Nevertheless, it is still of importance to identify, diagnose and treat hyperlipidaemic individuals who do not present with symptoms before adulthood due to less severe LPL gene mutations. 12
Here, we present a case of a compound heterozygous 29-year-old man presenting with eruptive cutaneous xanthomata and found to be a carrier of a novel duplication (p.R44Kfs*4) leading to a premature stop codon in exon 2 and a known mutation (N291S) in exon 5 of the LPL gene.
Methods
DNA analysis
From peripheral blood leukocytes DNA was extracted using a Maxwell 16 Blood DNA Purification Kit (Promega Corporation, Madison, WI, USA). The LPL gene comprises 10 coding exons, of which the last exon contains a single nucleotide of the stop codon followed by the entire 3′-untranslated region. For screening the nine coding regions including exon–intron boundaries were amplified by polymerase chain reaction (PCR) using Taq DNA polymerase (Sigma) and intron specific primers (sequences available upon request). PCR products were subsequently bidirectionally sequenced on an ABI 3730 XL DNA Analyser (Applied Biosystems, Foster City, CA, USA).
Biochemical analyses
For postheparin lipase activity measurements, the proband, his father and a control person (overnight fasting) were given an intravenous dose of heparin (60 U/kg body mass). Plasma samples were drawn before and 15 min after heparin administration and stored in aliquots at −80℃.
Total and hepatic lipase activity was determined using a Confluolip assay kit (PROGEN Biotechnik GmbH, Heidelberg, Germany). In brief, samples were diluted 100× in substrate buffer in a total volume of 600 µL in a heated (37℃) quartz cuvette with continuous stirring. Kinetic measurements were performed using a LS 55 fluorometer (Perkin Elmer, Waltham, MA, US) set to 342 nm for excitation and 400 nm for emission. Lipase activity was calculated and expressed in pmol/mL/min. HL was selectively measured by performing the assay under LPL-inhibiting buffer conditions. Lipase activities are presented in Table 1 as a mean of three independent measurements.
Plasma total cholesterol, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol and TG, gamma-glutamyltransferase (GGT), alanintransaminase (ALT) and glucose were all determined by enzymatic colorimetric tests on an Architect c16000 system (Abbott Diagnostics, Lake Forest, IL, USA). Thyroid stimulating hormone (TSH) was determined by chemiluminescent microparticle immunoassay (CMIA) on an Architect i2000 system (Abbott Diagnostics). Haemoglobin A1c (HbA1c) was measured by cation exchange chromatography using Tosoh G7 (Medinor A/S, Brøndby, Denmark).
According to biochemical method validation data, the elevated concentration of TG in the probands plasma did not cause interference in any of the assays used.
Case report
Clinical analysis of the proband and his parents.

ND: not determined; TSH: thyroid stimulating hormone; GGT: gamma-glutamyltransferase; ALT: alanine transaminase; Hba1C: glycated (glycosylated) haemoglobin.
Lipase assay variation is expressed as percentage of standard deviation.
Age-dependent reference values are given in parentheses.
*Percent postheparin lipase activity compared with an individual with a normal (WT) genotype.
†Percent postheparin hepatic lipase activity compared with WT.
Genetic screening for mutations in nine coding regions including exon–intron boundaries revealed two heterozygous LPL mutations in the patients DNA (Figure 1(a)): One mutation (c.953A > G) in exon 6 was the common N291S (also N318S, new nomenclature, Figure 1(a), Table 1) encoded by exon 5. The other mutation was a novel LPL duplication c.128dupT in exon 2 resulting in a frame shift leading to a premature stop codon (p.R44Kfs*4; Figure 1(a) and Table 1). The novel mutation predicts a truncated gene product having a size of only 10.3% of the wildtype (WT) LPL monomer. In order to confirm compound heterozygosity of the two recessive alleles, the probands parents were subjected to genetic testing as well. The father was found heterozygous of the known N291S mutation in exon 5, whereas the mother was heterozygous of the novel duplication c.128dupT in exon 2 (Figure 1(a), Table 1).
Mutational analysis in the proband and his parents. (a) Combined chromatogram of DNA sequence analysis of LPL gene mutations and pedigree of the family. Black symbols represent the N291S mutation and grey symbols represent the novel R44Kfs*4 mutation. Question mark indicates no sample obtained for genetic analysis. (b) Photograph of the lipidaemic plasma phenotype observed in the proband but not in his heterozygous father (N291S) or a normal control.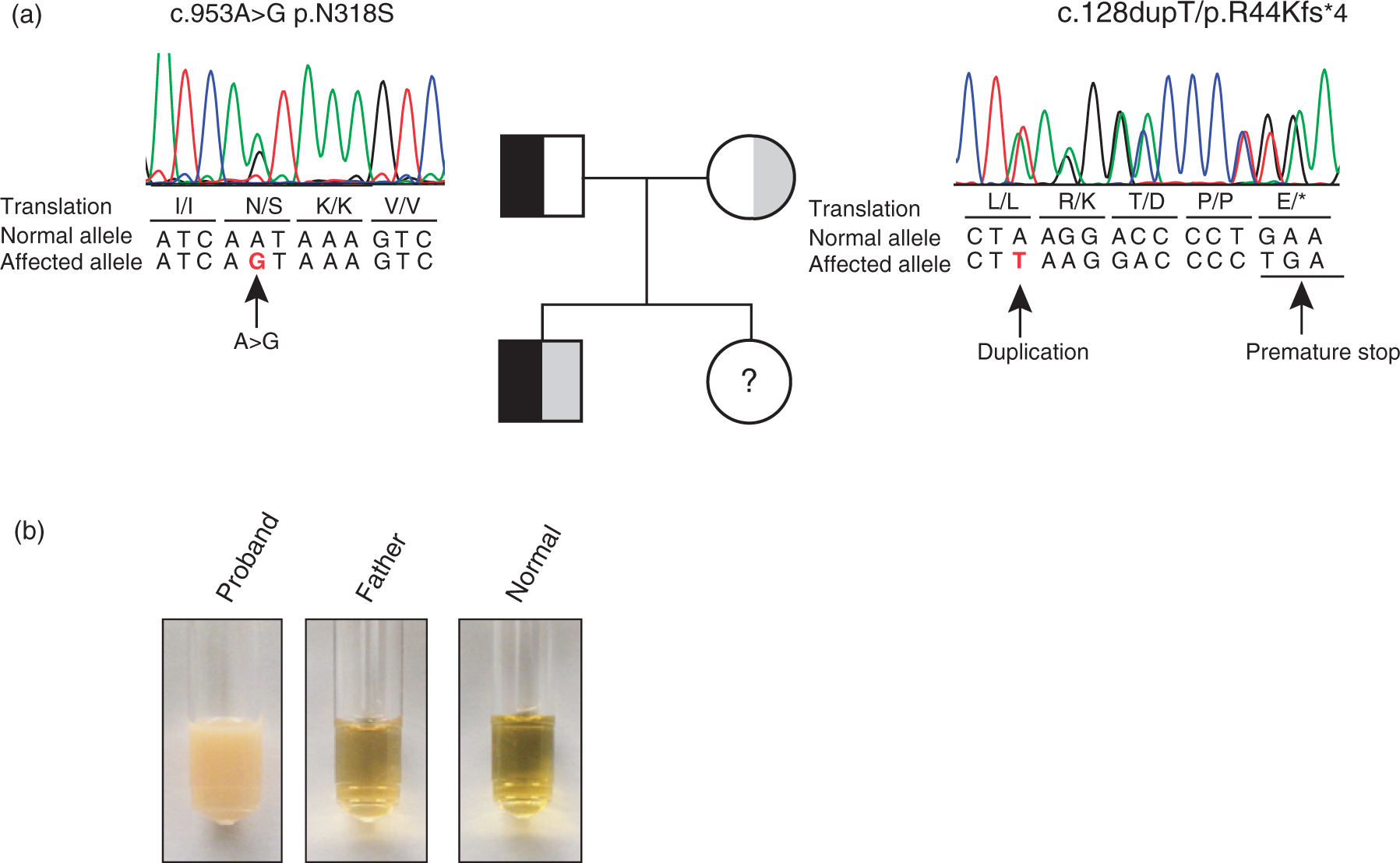
To gain further functional insight into the lipolytic capacity of the proband, postheparin total lipase and HL activity was determined. We noted that the plasma of the fasting proband appeared milky compared with his father and a healthy individual (Figure 1(b)). As expected, the level of total lipase activity of the probands postheparin plasma was reduced to (73.2 [7.0%]) of healthy control (WT). The total lipase activity of his father was less reduced (84.4 [5.4%]) of WT. The hepatic lipase contributes significantly (≈1/3) to the total lipase activity in postheparin plasma. 14 Thus, we also measured HL activity alone by inhibiting LPL in conditions of high ionic strength and pH 8.8. Interestingly, the probands plasma contained elevated HL activity (184.6 [20.2%]) compared with WT and to his father (107.0 [5.4%]), indicating a postheparin LPL activity much lower than 73% of WT. Thus, our biochemical analyses were compatible with the compound heterozygotic genotype of the proband which predicts one loss-of-function allele and one allele with reduced activity.
Discussion
Here we present a case of FLLD in a 29-year-old man, who initially presented with eruptive cutaneous xanthomata and elevated plasma TG concentration. Given the relatively late presentation and absence of pancreatitis, the case represents a relatively mild rather than a classic case of FLLD. The patient was found to be compound heterozygous for two mutations in the LPL gene and we established that the known LPL mutation N291S was inherited from his father, while the mother gave rise to a second mutated allele. The latter was a duplication in exon 2 (c.128dupT) that is novel according to HGMD.
The novel LPL duplication c.128dupT in exon 2 predicts a frame shift (p.R44Kfs*4) leading to a premature stop codon (Figure 1(a) and Table 1). The encoded small truncated gene product is without expected enzymatic function; thus, we anticipated that LPL activity of the compound heterozygous proband would be less than half the amount of WT. Although we did not selectively inhibit HL to obtain LPL-specific activity determinations, we showed that postheparin HL activity makes up a larger fraction of postheparin total lipase activity in the proband compared with his father (N291S) or WT (Table 1). This suggests that LPL activity is significantly more reduced compared with the determined postheparin total lipase activity (73.2 [7.0%]) in good agreement with the compound heterozygous genotype of the proband. We therefore suggest that the R44Kfs*4 mutation adds to the number of disease-causing LPL loss-of-function mutations.
The N291S allele has carrier frequencies ranging from 1 to 7 percent according to population-based studies 15 and are present in approximately 50% of all cases with LPL mutations in our local population (unpublished data). It has been shown that otherwise healthy carriers of N291S have significantly elevated mean plasma triacylglycerol concentrations over non-carriers. 16 Moreover, normolipidaemic carriers have a significantly greater postprandial chylomicron response to fat load test, indicating that the expression of the mutation is influenced by environmental factors such as diet. 14 Another study established a link between LPL and atherosclerotic disease by showing that the N291S mutation is significantly associated with reduced HDL cholesterol levels in men with premature atherosclerosis. 17 At the molecular level, the N291S residue is not part of any domain known to be involved in triacylglycerol hydrolysis and the mutant enzyme is not defective in secretion or heparin binding in transfected COS1 cells. 18 Nevertheless, independent studies have shown that the N291S mutation reduce LPL activity. 17,18 Consistent with this, our postheparin analyses of the father showed reduced total lipase activity and normal HL activity compared with WT (Table 1). It has been noted that increased postheparin HL activity measurements often are elevated in hyperlipidaemic patients. 19 This is also the case in this report (Table 1). We speculate that the mechanism by which HL activity increase in hyperlipidaemic and FLLD individuals involves HDL, since HL mobilization and activity is controlled by electrostatic-dependent association of HL with HDL. 20,21
Several new lipase-modulating proteins have been identified that could play a regulatory role in maintaining a high plasma lipolytic capacity in individuals carrying mutations in the LPL system. 13 These include glycosylphosphatidylinositol-anchored HDL-binding membrane protein 1 (GPIHBP1), 22 angiopoietin-like proteins (Angptls) 23 and lipase maturation factor 1 (LMF1). 24 The latter plays a role in intracellular maturation of secreted lipases that assemble into homodimers in the endoplasmic reticulum, 24,25 whereas GPIHBP1 and angptls act stimulatory and inhibitory, respectively, on lipase activity during transendothelial transport. 26–28 Clearly, future genetic and biochemical studies including further genetic testing of individuals presenting with dyslipidaemia awaits to fully elucidate the role of lipases and lipase-modulating proteins in normal physiology and diseases.
In conclusion, we report on case of compound heterozygous LPL deficiency due to a known N291S mutation and a novel duplication in exon 2 (c.128dupT) predicting a frame shift (p.R44Kfs*4) leading to a premature stop codon. Postheparin plasma analyses of the proband, his father and a healthy control accordingly confirmed a decreased total lipase activity of the proband. Of interest, we also observed an increase in the postheparin HL activity of the proband, a finding of which clearly requires further investigation.
Acknowledgement
We would like to acknowledge Klaus Brusgaard for setting up the LPL gene analysis, Bjarne Rasmussen, Marianne Overgaard and Ragnhild Larsen for excellent technical assistance.
Declaration of conflicting interests
The authors declare that they do not have any conflict of interest.
Funding
This research was funded by Odense University Hospital.
Ethical approval
Not applicable.
Guarantor
MO.
Contributorship
MO conducted the genetic analysis, performed biochemical analyses and wrote the manuscript. MO, CLB and DS researched the case history. CLB conducted blood sampling for postheparin analyses. DS was the patient`s genetic counsellor. SF and MN contributed to data interpretation and writing of the manuscript. All authors approved the final draft.