
Abstract
Background
Extraction followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis is the method of choice when it comes to the accurate quantification of 25-OH-vitamin D in blood samples. It is generally assumed that the addition of exogenous internal standard allows for the determination of the endogenous analyte concentration. In this study we investigated the extraction properties of endogenous and exogenous 25-OH-vitamin D.
Methods
Eight samples were used for the evaluation of the extraction procedure and 59 patients’ samples for a method comparison. The methanol-to-sample ratio (v/v) and the sample-to-hexane ratio (v/v) were varied and the LC-MS/MS signals of endogenous 25-OH-vitamin D3, spiked 25-OH-vitamin D2 and internal standard of the extracts recorded. The optimized ‘in-house’ LC-MS/MS assay was compared to two automated chemiluminescence immunoassays from DiaSorin and Abbott.
Results
Mathematical analysis of the data revealed a differential extraction of endogenous 25-OH-vitamin D3, spiked 25-OH-vitamin D2 and non-equilibrated internal standard. Exogenous 25-OH-vitamin D can be measured accurately if a definite methanol-to-sample ratio is used. Endogenous 25-OH-vitamin D is affected by critical quantification issues due to a differential slope in the extraction profile. The actual 25-OH-vitamin D concentration can be one-third above the measured extractable concentration. Results confirm that the ‘in-house’ LC-MS/MS assay provides reproducible 25-OH-vitamin D results.
Conclusions
Discordant concentrations of 25-OH-vitamin D from LC-MS/MS assays can be caused by selection of suboptimal extraction conditions. Furthermore, a different sample pretreatment or solvent extraction system may result in a different dissociation and extraction yield of endogenous 25-OH-vitamin D and therefore contribute to variations of LC-MS/MS results.
Introduction
25-OH-vitamin D is a problematic analyte due to its tight binding (Ka = 1 × 108 to 1.3 × 109 M)1 to vitamin D binding protein (DBP). Dietary and synthesized 25-OH-vitamin D which originates within the blood is defined as endogenous. It is predominantly bound to DBP. Exogenous 25-OH-vitamin D, i.e. internal standard or spiked 25-OH-vitamin D, has been added to a serum sample after the blood sample has been taken. A major proportion of these molecules might be subjected to different binding characteristics, e.g. non-specific adsorption to albumin.
Automated chemiluminescence immunoassays are the predominant applications for the quantification of 25-OH-vitamin D in clinical laboratories due to factors such as high throughput, non-extracting sample analysis and huge array of automated menus. In recent years LC-MS/MS methods with preceding extractions are becoming the method of choice due to superior accuracy and reliability 2 while still encompassing the ability to sample a large menu of analytes.
Comparisons of LC-MS/MS assays with chemiluminescence immunoassays for 25-OH-vitamin D have been reported over the past few years.3–7 Those studies illustrate a high ‘variability’ which we define as mean of the absolute value of inaccuracy and ‘bias’ between LC-MS/MS assays and chemiluminescence immunoassays. We distinguish the ‘sample specific bias’ which is calculated as the mean of the inaccuracy of individual samples and the ‘proportional bias’ revealed from regression analysis. In the latest vitamin D External Quality Assessment Scheme (DEQAS) review (2012), LC-MS/MS methods generally revealed a positive bias compared to automated analyser methods. 8
Laboratory studies using LC-MS/MS methods and common extraction procedures highlighted that calibration is a major cause of interlaboratory bias of 25-OH-Vitamin D results. 9 The opposite conclusion has been drawn from an interlaboratory comparison of 15 laboratories carrying out LC-MS/MS testosterone assays which employed a wider variety of extraction methods. 10 Chromatographic and mass spectrometric interferences have also been discussed 11 as a possible source of ‘discordance’, which we define as variability and/or bias.
Recent candidate reference methods for 25-OH-vitamin D did not consider differential extraction12,13 and Carter 14 reported that a requirement for non-extraction 25-OH-Vitamin D assays (chemiluminescence immunoassays) was that the analyte is totally displaced from its binding protein but we uphold that this is also imperative for extraction-based assays. Hejboer et al. 15 concluded that not all of the 25-OH-vitamin D is displaced from the DBP in sample treatment procedures of common automated immunoassays.
The key for minimizing variability and bias between different methods is knowledge of the true recovery of an analyte within a sample treatment procedure. For this reason we define three different concentrations within this study: the total, the extractable and the extracted 25-OH-vitamin D concentration. In the past, it was generally supposed that the total and extractable concentrations are identical in extraction-based assays and that the internal standard (IS) in LC-MS/MS assays corrects for procedural losses of analytes. Thus, the extractable concentration could easily be deduced from the extracted concentration. However, if the extraction of endogenous 25-OH-vitamin D and IS is different, then the evaluation procedure needs to be addressed.
In this study we investigate the extraction of endogenous and exogenous 25-OH-vitamin D from serum samples and discuss consequences for quantification with regard to differential extraction characteristics. Furthermore, the concentration of extractable 25-OH-vitamin D determined by LC-MS/MS is compared to concentrations obtained from two common automated chemiluminescence immunoassays from DiaSorin and Abbott.
Materials and methods
Study design
The impact of different sample storage conditions on the stability of analytes and analyte–matrix interactions was examined as basis for the study. The primary goal was to carry out experimental series of different extraction conditions and evaluate extraction efficiencies and the extractable concentration for endogenous, natural 25-OH-vitamin D3, exogenous, spiked 25-OH-vitamin D2 and IS. The results of the extractable concentration obtained from the mathematical analysis of the extraction experiments were compared to results from the routine LC-MS/MS assay, the LIAISON assay and gravimetric determinations.
The secondary part of the study comprises complementary information from a comparison of the routine LC-MS/MS assay with two automated chemiluminescence immunoassays, which pinpoints at imprecision, inaccuracy and bias of the methods. Upstream extraction was added to the LIAISON assay to look at the extraction efficiency of the sample treatment procedure within the LIAISON assay. The comparisons were completed by an evaluation of the different methods with a set of external quality control samples.
Samples
Eight blood samples from haemochromatosis patients, 40 serum samples and 22 pooled serum samples preselected from routine analysis, and 20 DEQAS samples have been collected for the study. Blood donations from haemochromatosis patients have been used to produce five spiked 25-OH-vitamin D2 serum samples (HCNO01–HCNO05), two grossly haemolysed serum samples (HCHL01, HCHL02) and one turbid serum sample (HCTU01). Five spiked samples were used for storage experiments. Extraction experiments were carried out with a total of eight serum samples, two spiked samples, two haemolysed and one turbid sample and three pooled samples (PONO1–PONO03). Forty preselected samples and 19 pooled samples were used for comparative analysis of the ‘in-house’ LC-MS/MS assay and the automated LIAISON and ARCHITECT assays. Samples were selected in order to achieve an equal distribution over the relevant clinical range of 25-OH-vitamin D concentrations. The set of 19 pooled serum samples was additionally used for the upstream extraction before the LIAISON assay. All samples were split into nine aliquots and kept frozen at −20℃ until analysis. Four sets of five DEQAS samples (October 2012 to July 2013) were used to assess our ‘in-house’ LC-MS/MS assay against an external quality assurance programme.
Sample stability
Five samples were split into two aliquots. One aliquot of each sample was spiked with 25-OH-vitamin D2. Variable amounts of 25-OH-Vitamin D2 (10 g/L in 70% methanol) were added to different samples, mixed and incubated for 1 h at room temperature. Then, each aliquot was further split into three series of six aliquots. The first series was kept frozen at −20℃ until analysis. The second series underwent 11 freeze–thaw cycles with intervallic storage at room temperature for 5 h. The third series was kept at room temperature for 11 days and was then stored at −20℃ until analysis. The spiked samples were measured by the LC-MS/MS assay and the LIAISON assay in triplicates; the non-spiked aliquots were measured by the LIAISON assay in triplicates.
Routine LC-MS/MS assay
For ‘in-house’ LC-MS/MS, 150 µL of 70% (v/v) methanol containing deuterated [2H6] 25-OH-vitamin D3 was added to 60 µL of serum or plasma and mixed for 2 min, to dissociate 25-OH-vitamin D from DBP, prior to extraction with 1.5 mL of hexane. Following vortexing for 2 min the tubes were centrifuged, 10 min at 3220 g, and 1 mL of hexane phase was removed into borosilicate glass tubes and dried at 37℃ under moderate airflow. Dried extracts were reconstituted in 300 µL 70% (v/v) methanol.
A methanol gradient on an ACE C8 column (50 × 2.1 mm i.d., 3 µm particle size) (ACE, UK) at 40℃ with a flow rate of 0.3 mL/min was applied. A Shimadzu HPLC system (Shimadzu Corp., Japan) was coupled to an AB SCIEX 4000 triple quadrupole mass spectrometer (AB SCIEX, USA) and was operated under conditions similar to those described previously. 16 Positive ion transitions (m/z) were 401.3/383.4 for 25-OH-vitamin D3, 413.3/355.4 for 25-OH-vitamin D2 and 407.3/389.4 for deuterated 25-OH-vitamin D3 as IS. Linear calibration curves of analyte-to-IS ratios of signal areas were used to quantify analyte concentrations. The analyte-to-IS ratio of the samples accounts for separation loss, differences in extraction yields between samples and possible variations in ionization due to matrix effects.
Extraction experiments
A variation of the above extraction conditions was carried out to allow for the determination of the extractable 25-OH-vitamin D concentration. In a first experimental series, the 70% methanol-to-sample ratio was altered while the sample volume of 60 µL and a hexane volume of 1.5 mL were kept constant. The volume of added 70% methanol which contains IS in the above concentration was varied from 30 to 300 µL in 10 experiments.
In a second experimental series, the sample-to-hexane ratio was altered while the hexane volume was 1.5 mL and the 70% methanol-to-sample ratio was 2.5. The sample volume was increased from 20 to 200 µL in 10 experiments. This series was repeated at constant total volume. The above sample volumes were used, but the volume of hexane was varied to achieve a constant total volume of 2 mL.
Absolute signal areas were recorded for these extraction experiments and linear calibration curves for 25-OH-vitamin D3, 25-OH-vitamin D2 and IS were used to calculate extracted concentrations. The extracted concentrations were corrected for differences in volumes (IS volume in first extraction series, sample volume in second series), separation loss (because only a part of the hexane volume is transferred) and dilution factors (between volume of extract and volume of sample or IS). LC-MS/MS intensity fluctuations of the extracted samples were low. Ion suppression of the extracts was found to be negligible.
The curve fit data evaluation which resulted in the extractable concentration was performed with SigmaPlot 11 (Systat Software Inc., USA).
Automated analysers
Two automated chemiluminescence immunoassays were used. All samples were measured in triplicates with the LIAISON 25-OH-vitamin D TOTAL assay of DiaSorin (2012). The model LIAISON® (DiaSorin, Italy) and all recommended supply materials were used according to the DiaSorin instructions. 25-OH-vitamin D2 concentrations were calculated as a difference of the concentrations of the spiked and the non-spiked samples.
The samples were additionally analysed with the current release of the ARCHITECT 25-OH-vitamin D assay on the ARCHITECT ci8200 analyser (Abbott Laboratories, USA).
The sample treatment procedure of the LIAISON analyser was surveyed by an upstream extraction procedure. For this purpose the extraction procedure of the routine LC-MS/MS assay was slightly modified: 100 µL of sample and 250 µL of 70% methanol were mixed, and 1.5 mL of hexane was added. The reconstitution was done in 250 µL of 0.05 M phosphate buffer at pH = 7.0 containing 9.0 g/L NaCl and 1.0 g/L Bovine Serum Albumin (BSA). This alteration was required to achieve a complete set of sample results within the calibration range of the LIAISON analyser. Final concentrations were obtained by correcting the LIAISON analyser results of the extracts for separation loss, partitioning between hexane and methanol/aqueous phase, and dilution.
The imprecision of the LC-MS/MS, the LIAISON and the ARCHITECT assay was determined from triplicate measurements. The coefficient of variation (CV) was averaged for 59 serum samples, 40 of which were run at five different assays (interassay imprecision), 19 samples were measured within a single assay (intraassay imprecision).
Results
Sample storage
Exaggerated freeze–thaw cycles (11 times) had no effects on the 25-OH-vitamin D concentration. However, a slight increase in 25-OH-vitamin D3 concentration of 11 ± 5% (standard deviation [SD]) was recorded with the LC-MS/MS assay after prolonged storage at room temperature on the bench in diffuse light (11 days). No differences were found with the LIAISON assay.
Vitamin D − protein dissociation
The extracted concentration of the 25-OH-vitamin D varied with the 70% methanol-to-sample ratio. A typical extraction profile for the analytes and the IS of sample HCNO02 is shown in Figure 1.
25-OH-vitamin D extraction profile of sample HCNO02 at constant sample volume (60 µL) and constant hexane volume (1500 µL). 25-OH-vitamin D signal integrals are converted to absolute extracted concentrations. This sample contained 100 nmol/L of IS and 26 nmol/L of 25-OH-Vitamin D2 based on gravimetric and volumetric determination.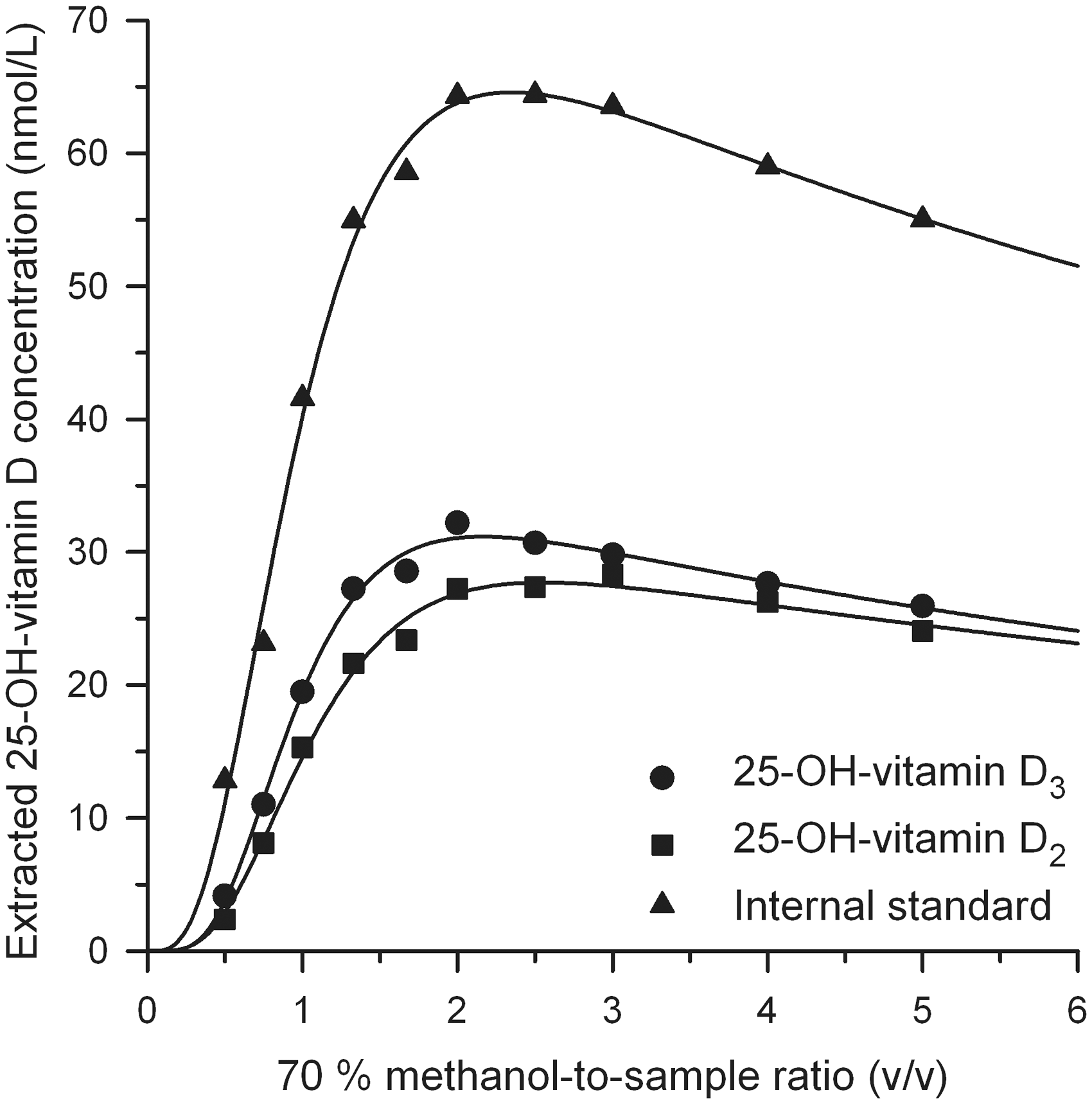
The extracted concentration c can be empirically described by
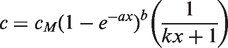
The extracted concentration increases to a maximum and then decreases at increasing 70% methanol-to-sample ratio due to the increasing (methanol:sample)-to-hexane ratio. The ascending part of equation (1) represents an asymmetric sigmoidal function which describes the dissociation or desorption characteristics with parameter
Equation (1) provided very good curve fitting results (mean R2 = 0.9887) covering 10 data points for a total of 18 curves of eight serum samples.
The extracted 25-OH-vitamin D concentration peaked at a 70% methanol-to-sample ratio of 2.63 (mean value of 12 curve fits) for five normal serum samples. The ratio was 2.52 for endogenous 25-OH-vitamin D3, 2.65 for IS and 2.85 for spiked 25-OH-vitamin D2.
Differential extraction of endogenous and exogenous 25-OH-vitamin D
The profiles of the extracted concentration show different maximum levels for different analytes/IS (Figure 1) and different samples. Comparative analysis of the components (analytes and IS) requires scaling of the profiles to the same height. Therefore, the ‘absolute extracted 25-OH-vitamin D concentration’ of each individual extraction profile was transformed into a ‘relative extracted 25-OH-vitamin D yield’ (Figure 2). The relative extracted 25-OH-vitamin D yield was scaled to 100% at the maximum of the curve fit. Then, the arithmetic mean of five normal samples at each 70% methanol-to-sample ratio was calculated for 25-OH-vitamin D3 and IS, and of two samples for 25-OH-vitamin D2. A new curve fit was performed for each component and the effective mean curve fit was rescaled to 100% relative extracted 25-OH-vitamin D yield at the curve maximum (see Figure 2).
Mean relative ‘extracted’ yield for 25-OH vitamin D3, IS and 25-OH-vitamin D2 of five samples (three samples PONO01–PONO03, two samples HCNO01–HCNO02 and the latter two were spiked with 25-OH-vitamin D2) as a function of the 70% methanol-to-sample ratio (v/v).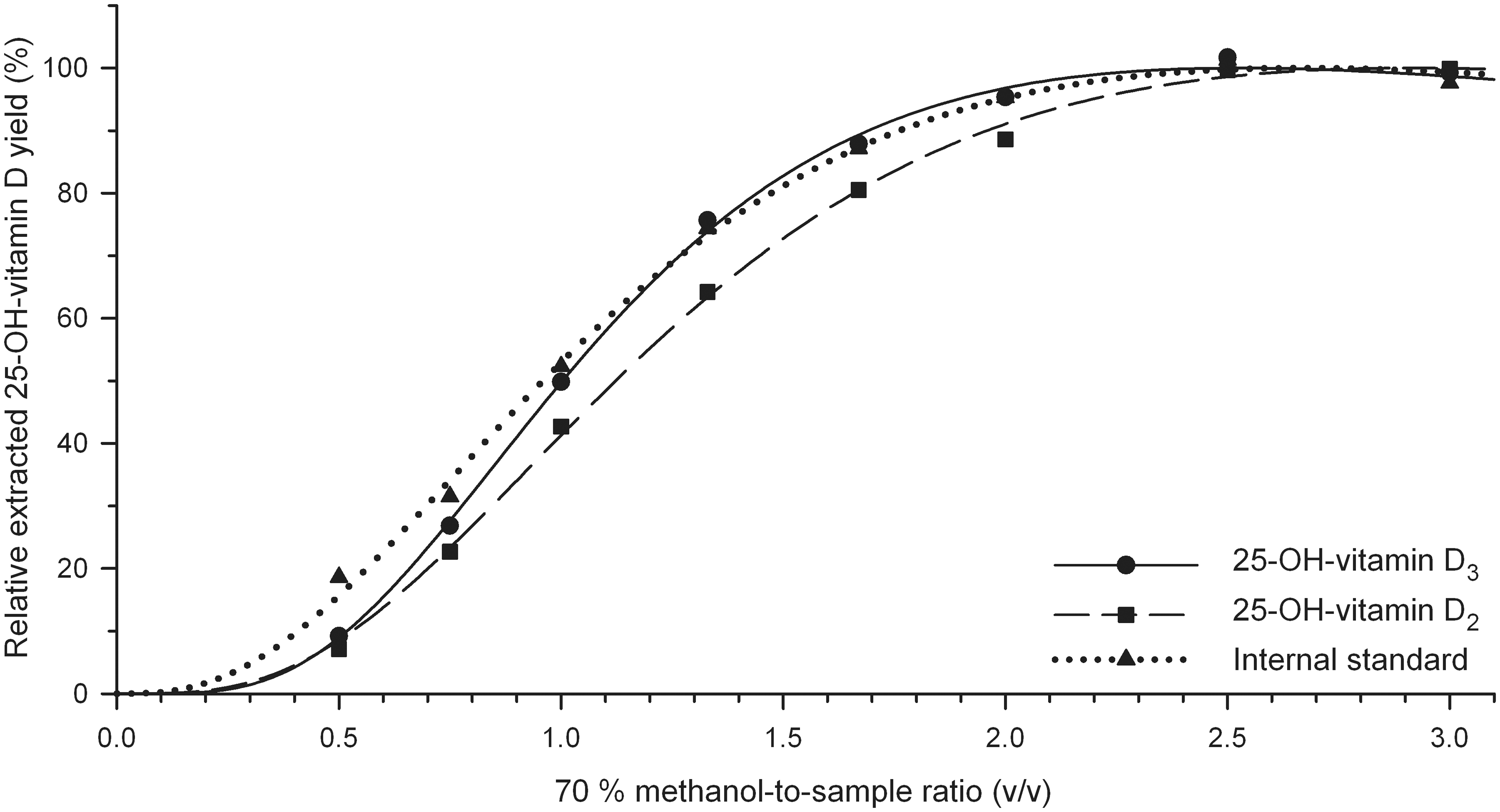
There are two clear differences of the curves.
The curves show a distinct parallel shift between the spiked 25-OH-vitamin D2 data (squares, dashed line) and the IS data (triangles, dotted line). The extraction of the equilibrated 25-OH-vitamin D2 required a higher 70% methanol-to-sample-ratio compared to non-equilibrated IS. The curve of endogenous 25-OH-vitamin D3 (continuous line) shows a steeper slope of 1.93 compared to the curves of the IS (1.42) and spiked 25-OH-vitamin D2 (1.50). The mean difference in slope between endogenous 25-OH-vitamin D3 and exogenous IS is 0.50 ± 0.08 (SD) for all eight samples.
In a wide range of the 70% methanol-to-sample ratio up to a value of 2.5, the ratio of 25-OH-vitamin D3 to IS that is used for quantification in the routine LC-MS/MS assay is varying. Haemolysed and turbid samples had an increased slope parameter of 2.79 and 2.80 for 25-OH-vitamin D3, respectively, and 2.29 and 2.35 for the IS, respectively.
The above experiments can be summarized that equilibration of an exogenous component causes a parallel shift of the extraction profile, exogenous origin is responsible for the difference in slope compared to the endogenous analyte and matrix conditions determine the absolute value of the slope of the endogenous analyte. Overall, our data analysis revealed an optimum 70% methanol-to-sample ratio of 2.5 for the three analysed components and a variety of sample matrices.
Determination of the extractable 25-OH-vitamin D concentration
In the previous sections the optimum extraction conditions for the dissociation of 25-OH-vitamin D and DBP were identified. In this section extraction efficiencies for those conditions are determined. For this purpose, the ‘extractable’ 25-OH-vitamin D concentration needs to be extrapolated from measured ‘extracted’ 25-OH-vitamin D concentrations.
A set of experiments at a constant 70% methanol-to-sample ratio of 2.5 was conducted where the sample-to-hexane ratio was varied. ‘Extracted’ concentrations of 25-OH-vitamin D were fitted with equation (2)
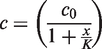
In an analogous way to the previous section, the curve fit of the absolute ‘extracted’ concentration was transformed to a relative ‘extracted’ 25-OH-vitamin D yield and scaled to 100% at a sample-to-hexane ratio of 0.04, the same ratio which is used in the extraction procedure of the routine assay. The curve fits showed no distinct difference between the components for five normal samples. Therefore, the arithmetic mean at each sample-to-hexane ratio of five samples was calculated for all analytes and IS (12 data sets). The newly performed mean curve fit for all components was rescaled to 100% relative ‘extractable’ 25-OH-vitamin D yield at x = 0 (see Figure 3).
Mean relative ‘extractable’ yield for 25-OH-vitamin D3, 25-OH-vitamin D2 and IS from Figure 2 as a function of the sample-to-hexane ratio (v/v) at constant 70% methanol-to-sample ratio (2.5) and constant hexane volume (1.5 mL). Extrapolation of the rational curve fit to a ratio of zero results in the ‘extractable’ yield for 25-OH-vitamin D.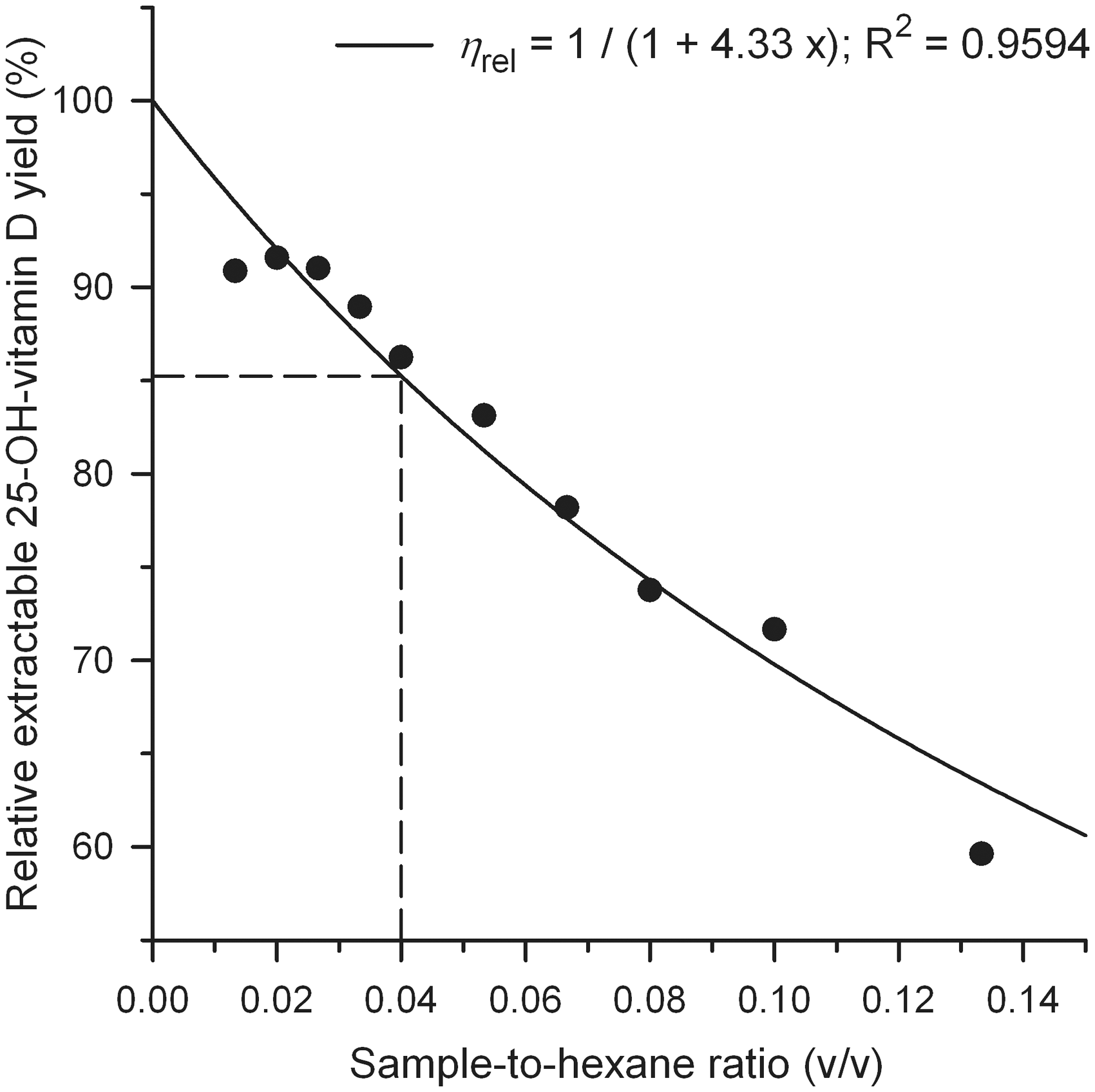
For the curve fit in Figure 3, equation (2) was modified to
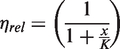
Haemolytic and turbid samples show a larger curvature of 0.45 and 0.35, respectively, for 25-OH-vitamin D3 and the IS. This difference in K values is consistent with differences of the parameter k of equation (1). The extraction experiments at a very low and high sample-to-hexane ratio exhibit a high error due to a poor signal-to-noise ratio and inaccurate loss calculation.
The determination of the curvature was not affected by potentially different mixing properties caused by different total volumes in the tubes. The total volume was kept constant for the extraction series of two samples and the sample-to-hexane ratio was varied in a similar range as earlier. No difference in the curvature was found (data not shown).
Concordant results from curve fit analysis and the routine LC-MS/MS assay
The curve fit procedure revealed the ‘extractable’ concentration of endogenous 25-OH-vitamin D3, spiked 25-OH-vitamin D2 and IS from the extraction series. In this section those extrapolated concentrations are compared to concentrations obtained by the routine LC-MS/MS assay, the LIAISON assay, and the gravimetric and volumetric determination of spiked 25-OH-vitamin D2 and IS.
Comparison of 25-OH-vitamin D3 (a) and 25-OH-vitamin D2 (b) concentrations obtained from curve fit analysis of the extraction data (equation (2)), the ‘in-house’ LC-MS/MS assay (cLC-MS/MS) and the DiaSorin LIAISON assay (cLIAISON) and from the added amount of 25-OH-vitamin D2 (cspiked).
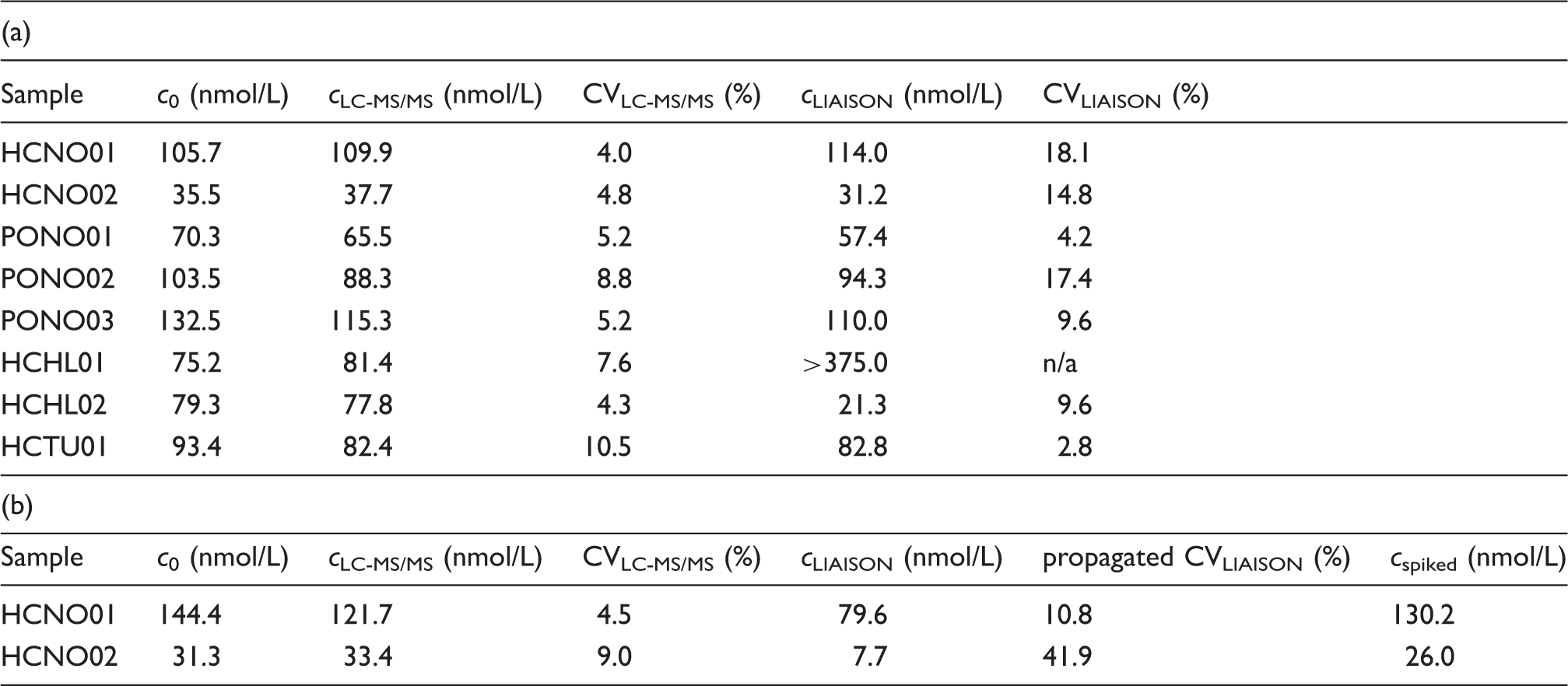
The curve fit results agree with the concentrations obtained from our ‘in-house’ LC-MS/MS assay within the limits of experimental error (sample specific bias: +5%). The 25-OH-vitamin D3 results of the LIAISON analyser are similar to the ‘in-house’ LC-MS/MS assay for six samples (sample specific bias: −4%). However, both haemolysed samples show a major inaccuracy on the LIAISON analyser yielding a 25-OH-vitamin D3 concentration of 21 nmol/L for sample HCHL02 and a concentration above the calibration range (>375 nmol/L) for sample HCHL01 compared to 78 and 81 nmol/L, respectively, of the LC-MS/MS assay.
In Table 1(b) the 25-OH-vitamin D2 results are compiled. The curve fit results are 11 and 20% higher than the concentration that is expected by gravimetric and volumetric measurement of the added amount of 25-OH-vitamin D2. The LIAISON results are well below those gravimetric/volumetric concentrations.
The curve fit analysis for the IS reveals a mean concentration of 95.2 ± 9.1 nmol/L (SD) for eight samples. This is concordant with the added concentration of IS of 100 nmol/L.
Assessment of the ‘in-house’ LC-MS/MS assay and automated chemiluminescence immunoassays
Regression analysis for 59 serum samples of the two automated analyser methods against the ‘in-house’ LC-MS/MS method is shown in Figure 4.
Regression analysis of 25-OH-vitamin D concentrations from 59 serum samples of the assays LIAISON (a) and ARCHITECT (b) versus LC-MS/MS.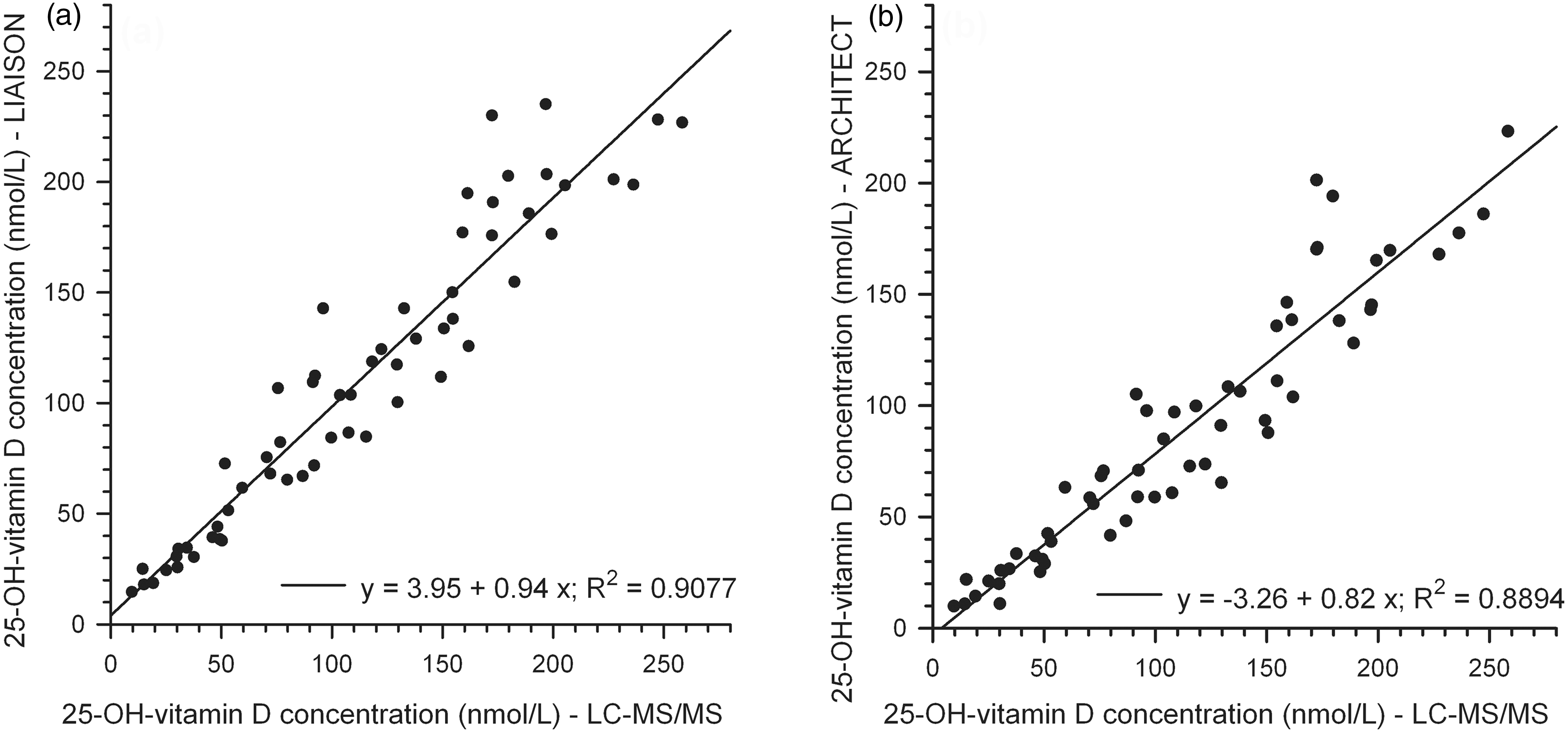
The slopes of 0.94 and 0.82 indicate a proportional negative bias of 6% for the LIAISON assay (Figure 4(a)) versus the LC-MS/MS assay and of 18% for the ARCHITECT assay (Figure 4(b)), respectively. The sample specific bias for 59 samples averages to +0.5% for the LIAISON assay versus the LC-MS/MS assay and to −21.4% for the ARCHITECT assay. The variability for the LIAISON data is high (15.4%) as is the imprecision of the LIAISON data (%CV = 8.3) compared to a %CV = 3.6 of the ARCHITECT data.
The sample-specific bias of our ARCHITECT data was in good agreement with the data received from corresponding mean DEQAS results of 20 samples (−19.3%). The DEQAS data between the mean LIAISON results and our LC-MS/MS results revealed a much lower sample specific bias (−26.1%) than our ‘in-house’ comparison. The mean DEQAS LC-MS/MS results had a negative proportional bias of 16% compared to our ‘in-house’ LC-MS/MS results (2.96 + 0.84 x; R2 = 0.9619) with a sample-specific bias of −11.3%.
Improved correlation of the LIAISON assay by the use of extracts
The high inaccuracy of the LIAISON data of individual samples, the high imprecision of the LIAISON data and the inconsistency in the comparisons of the LIAISON assay and the ‘in-house’ LC-MS/MS assay between our serum sample results and the DEQAS results cause concern and are the topic of the final experimental investigation within this study.
Nineteen pooled serum samples which were examined within the previous section were extracted based on the LC-MS/MS assay extraction procedure. The dried extracts were reconstituted in buffer, and the values obtained from the LIAISON analyser were used to calculate the ostensible concentration in the serum samples taking into account procedural losses and dilutions within the extraction procedure.
In Figure 5 the LIAISON data of extracts (continuous line) and serum samples (circles, dashed line), both in singlicates, are plotted against the LC-MS/MS means of triplicate measurements.
Regression analysis of 25-OH-vitamin D concentrations from 19 extracts (continuous line) and the corresponding non-extracted serum samples (dashed line) of the LIAISON assay against the LC-MS/MS assay. The data points of the extracts are corrected for procedural losses during extraction.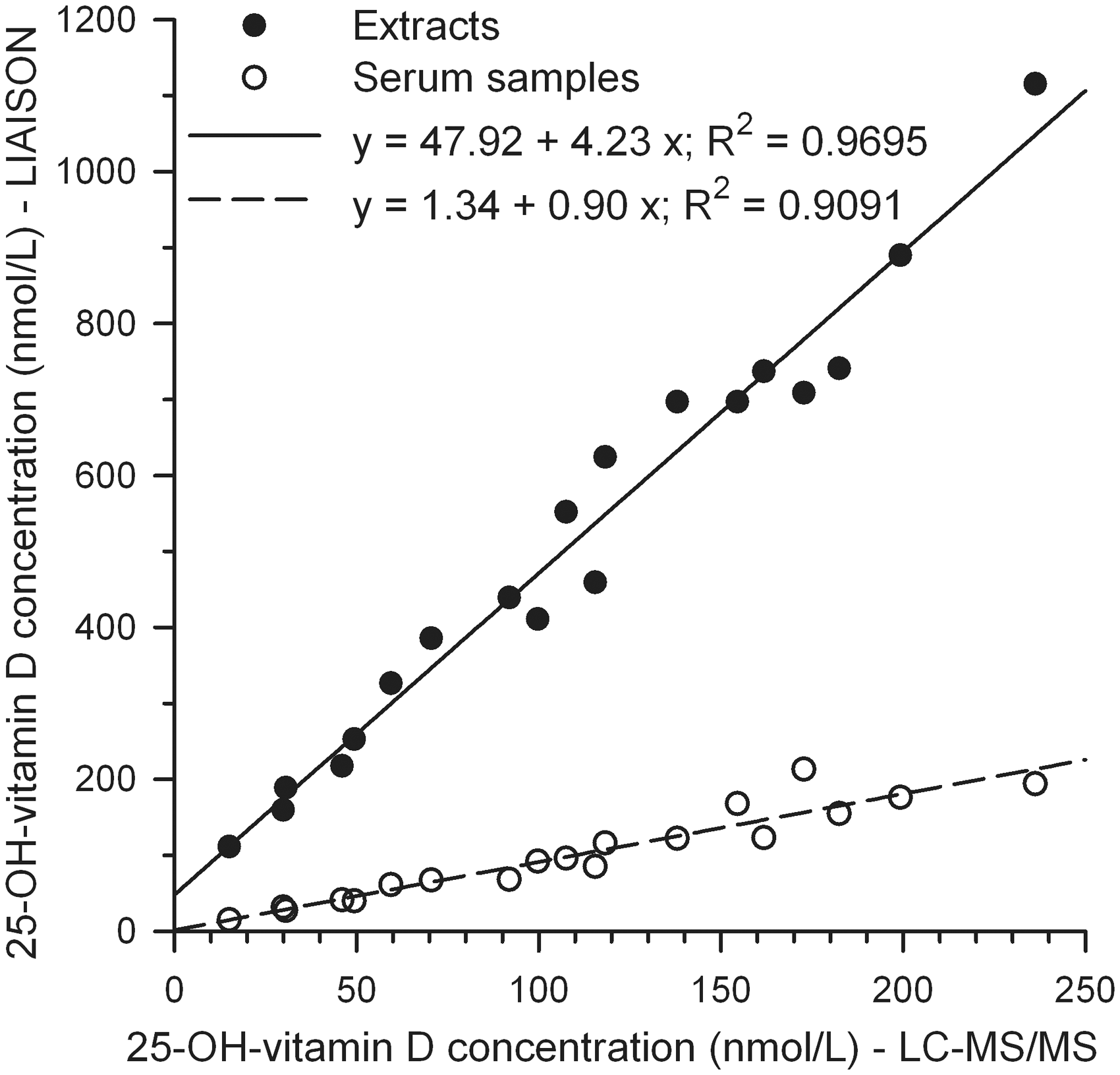
The regression analysis of the serum samples reveals a constant bias of 1.34 nmol/L and a proportional bias of 0.90 at a coefficient of determination of 0.9091. Linear regression data of singlicate measurements are sufficiently consistent with triplicate data shown in Figure 4(a). The proportional bias of the regression analysis for the extracts is 4.23, i.e. a factor of 4.7 higher than for the serum samples. The constant bias of 47.92 nmol/L is extremely high and caused by matrix changes from ‘serum’ to ‘buffer’. The coefficient of determination (R2 = 0.9695) is better for the extracts compared to the serum samples.
Discussion
The mathematical analysis of the extraction series led to an optimization of our prior ‘in-house’ LC-MS/MS assay. 17 An increase of the 70% methanol-to-sample ratio from 0.75 to 2.5 and a decrease of the sample-to-hexane ratio from 0.2 to 0.04 significantly increased the relative ‘extracted’ 25-OH-vitamin D yield to 85%. As a consequence we were able to use a smaller sample volume of 60 µL and achieve a reduced imprecision (%CV = 7.3).
We found, as opposed to Tai et al., 13 that equilibration of the exogenous IS is not a prerequisite for quantification of endogenous 25-OH-vitamin D, which saves 1 h of sample preparation time according to Ding et al. 18 Instead, the prerequisite for quantification is a 70% methanol-to-sample ratio of 2.5, where the systematic difference in relative extracted 25-OH-vitamin D yield between non-equilibrated IS, spiked equilibrated 25-OH-vitamin D2 and endogenous 25-OH-vitamin D3 is minimal. At a ratio of 2.5, the 25-OH-vitamin D-to-IS signal ratio is barely affected by a parallel shift of the relative extracted 25-OH-vitamin D yield curves between non-equilibrated and equilibrated components.
The higher slope in extraction profiles of +0.50 ± 0.08 (SD) for endogenous, natural 25-OH-vitamin D3 compared to exogenous, spiked 25-OH-vitamin D and IS raises concern. Firstly, a different slope of exogenous 25-OH-vitamin D in calibrators compared to endogenous 25-OH-vitamin D in samples causes calibration issues if the methanol-to-sample ratio is chosen incorrectly. Secondly, we presume that the different slope is caused by incomplete release of endogenous 25-OH-vitamin D from its DBP. Given the above differences in slope, this would mean that the total 25-OH-vitamin D concentration is about one-third higher than the measured extractable 25-OH-vitamin D concentration. We conclude that this portion is precipitated with the DBP and undetected by our LC-MS/MS method because the IS does not account for this portion.
The mathematical data analysis confirms that we are able to determine the extractable 25-OH-vitamin D concentration accurately. The data for exogenous 25-OH-vitamin D show that there is virtually no adsorptive loss during the extraction procedure. The extractable concentrations of endogenous 25-OH-vitamin D3 obtained from the curve fit analysis agree with the concentrations determined with our optimized ‘in-house’ LC-MS/MS assay. Although different sample matrix (highly turbid and grossly haemolysed samples) alters the extraction characteristics, the ‘in-house’ extraction procedure of the LC-MS/MS assay is sufficiently robust to reveal accurate concentrations of the extractable 25-OH-vitamin D for different matrix conditions.
In general, the results of the automated analysers showed a moderately good correlation to the LC-MS/MS results. Previous reports of a negative bias of the LIAISON assay against LC-MS/MS assays could not be confirmed. 7 Carter suggested that a negative bias may be due to the presence of interfering 3-epi-25-OH-vitamin D3 and other isomers. 19 Farrell et al. 20 reported an average epimer proportion of 8% for about 40% of their samples. Albeit the 3-epi-25-OH-vitamin D3 signal interferes with our 25-OH-vitamin D3 signal, we find a sample-specific bias of +0.5% for our samples.
The absence of the sample-specific bias is accompanied by a large inaccuracy of the LIAISON assay for individual samples. A similar variability and imprecision of the LIAISON method was reported previously.21,22 Both haemolysed samples showed major deviations on the LIAISON analyser. The concentrations of spiked 25-OH-vitamin D2 are 56% lower at the LIAISON analyser compared to the LC-MS/MS assay. A similar tendency has been shown by De Koning et al. 23
Our upstream extraction procedure on the LIAISON analyser revealed a slope of the regression line which is a factor of 4.7 higher than the slope of the regression line between the unaltered LIAISON assay and the LC-MS/MS assay. The extraction procedure made the analyte much more accessible for detection on the LIAISON analyser. Vice versa, when analysing serum samples, only a minor portion (comprising 21%) of the 25-OH-vitamin D3 is detected by the LIAISON analyser compared to our ‘in-house’ LC-MS/MS assay.
The alteration of the matrix by extracting the analyte from a serum matrix into a buffer matrix causes a huge constant bias of 47.9 nmol/L. From those results we conclude that the poor detection yield and sample matrix effects are incorporated into a correction function within the LIAISON method, so that the LIAISON instrument displays concentrations that actually agree on average with LC-MS/MS concentrations. However, minor matrix differences may cause small variable detection yields, which are responsible for amplified high inaccuracies of 25-OH-vitamin D concentrations of individual samples. Becker et al. 24 showed that discordant higher concentrations found on the LIAISON analyser could be reduced by simple measure of twofold diluted samples. Our data support the finding that reducing matrix differences between samples within the LIAISON method via extraction or dilution reduces discordance of LIAISON results.
The ‘all laboratory’ LIAISON method revealed a much lower sample-specific bias for the DEQAS samples compared to the ‘in-house’ LC-MS/MS assay. This inconsistency in the results of the method comparisons between our samples (no bias) and the DEQAS samples (moderate negative bias) may be due to sample processing procedures like sera pooling and subsequent filtration which alters the matrix of DEQAS samples compared to freshly frozen serum samples. As discussed earlier, the LIAISON assay is likely to be affected easily by slight changes of the matrix.
The concentrations of our LC-MS/MS assay showed a similar positive sample-specific bias to the ‘local’ ARCHITECT assay for 59 samples and to the ‘all laboratory’ ARCHITECT method for the DEQAS samples. The ARCHITECT assay might be less prone to matrix effects which could also result in a significantly better precision than the LIAISON assay.
The concentrations of the ‘in-house’ LC-MS/MS assay also showed a positive sample-specific bias compared to the mean concentrations of the ‘all laboratory’ LC-MS/MS assays for the DEQAS samples. A similarly raised level was found for our ‘in-house’ samples after 11 days of storage at ambient temperature, while previous studies did not find any measurable effect on 25-OH-vitamin D concentrations at extended sample storage in the fridge for 7 days 25 or at ambient temperature for 8 days. 26 This is in accordance with the hypothesis of non-detected 25-OH-vitamin D bound to DBP in freshly frozen samples. The dispatch of DEQAS samples at ambient temperature may be an issue due to oxidation and degradation of proteins, which may affect the dissociation of 25-OH-vitamin D from its DBP and non-specific adsorption characteristics to other plasma proteins. This could lead to higher concentrations at remote laboratory locations due to the longer travel time of the samples.
In general, we conclude that it is necessary to optimize the extraction procedure in LC-MS/MS assays to a similar relative extraction yield of the different analytes and IS. It is desirable to reduce the difference in extraction profiles between exogenous and endogenous protein bound hormones. In addition for 25-OH-vitamin D, automated, non-extracted chemiluminescence immunoassays are unsuitable for quantitative evaluation as they detect only a minor portion of the 25-OH-vitamin D present in samples and are therefore prone to matrix effects. Finally, basic comparisons of current 25-OH-vitamin D assays do not improve the accuracy of a particular method. Rigorous investigation is required to examine the dissociation of 25-OH-vitamin D from DBP in an effort to approach the accurate determination of the 25-OH-vitamin D concentration in blood.
Footnotes
Acknowledgements
We would like to thank John Wallace for technical assistance with the ARCHITECT measurements.
Declaration of conflicting interests
The manufacturers (DiaSorin and Abbott) provided reagents for the automated immunoassay analyses. DiaSorin also temporarily provided the LIAISON analyser. Neither of these companies played any role in the interpretation of the data nor in the preparation and review of the manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable.
Guarantor
PG.
Contributorship
UL drafted the study, performed the experimental work and data analysis, and wrote the manuscript. PAE, JGL and PG contributed to the design of the study and to the interpretation of the data. They reviewed and edited the manuscript.