
Abstract
Specific channels permit movement of selected ions through cellular membranes, and are of vital importance in a number of physiological processes, particularly in excitable tissues such as nerve and muscle, but also in endocrine organs and in epithelial biology. Disorders of channel proteins are termed channelopathies, and their importance is increasingly recognised within medicine. In the kidney, ion channels have critical roles enabling sodium and potassium reuptake or excretion along the nephron, in magnesium homeostasis, in the control of water reabsorption in the collecting duct, and in determining glomerular permeability. In this review, we assess the channelopathies encountered in each nephron segment, and see how their molecular and genetic characterisation in the past 20–30 years has furthered our understanding of normal kidney physiology and disease processes, aids correct diagnosis and promises future therapeutic opportunities.
Keywords
Introduction
Channelopathies in other organ systems – selected examples.
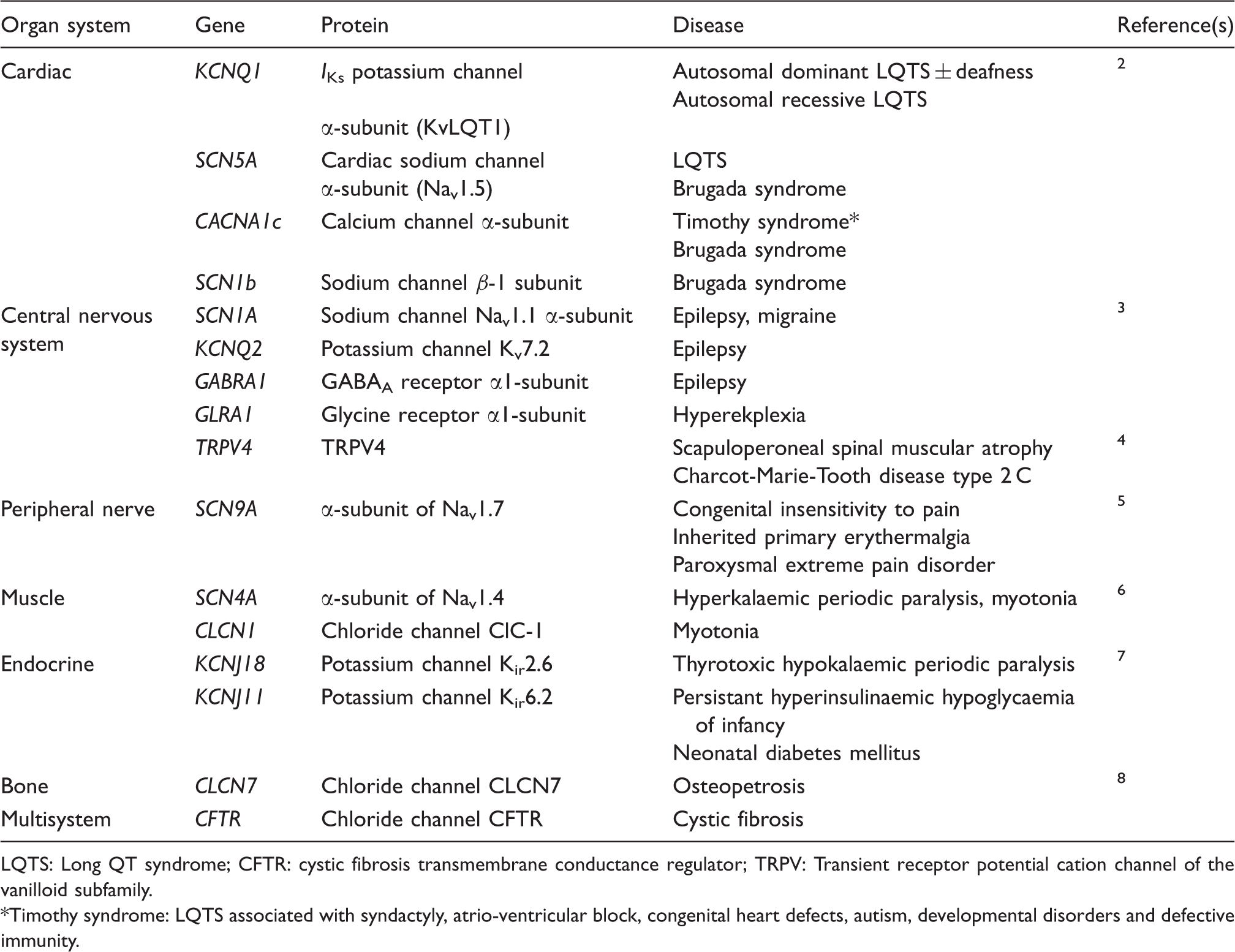
LQTS: Long QT syndrome; CFTR: cystic fibrosis transmembrane conductance regulator; TRPV: Transient receptor potential cation channel of the vanilloid subfamily.
Timothy syndrome: LQTS associated with syndactyly, atrio-ventricular block, congenital heart defects, autism, developmental disorders and defective immunity.
Disease-causing mutations may result in ion channels that exhibit defective function, regulation or expression. 1 Mutations affecting proteins that directly regulate ion channels, and which lead to a defect in channel function, are also grouped under the channelopathy heading. Different mutations within the same channel protein can lead to diametrically opposed clinical syndromes, for example congenital excessive pain – or insensitivity to pain – with different mutations in SCN9A (encoding a voltage-gated sodium channel). 5 In addition, mutations may have very different effects on channel function (and disease phenotype) where the channel is expressed in a variety of tissues. Whilst outside the scope of this review, acquired channel defects may also arise through autoimmune disease (e.g. myasthenia gravis) or as a paraneoplastic phenomenon (e.g. Lambert-Eaton myaesthenic syndrome).
The renal channelopathies
When considering the renal channelopathies, one might first think of tubular disorders where there is abnormal handling of salt or water, such as the salt-wasting syndromes of Bartter and Gitelman, or nephrogenic diabetes insipidus (NDI). Genetic characterisation of these conditions over the last 20 years has led to a far greater understanding of the molecular players involved in ion transport within the loop of Henle and distal nephron, not least because as the condition is ‘solved’ in one pedigree it becomes clear that other families do not possess the same mutant gene, so other candidate genes are sought and sequenced, and thus a more detailed picture gradually develops. The renal channelopathies are not restricted to salt and water, however, and different channelopathies have now been described affecting every nephron segment, from glomerulus to collecting duct (Figure 1). In this review, we travel along the nephron, describing channelopathies by anatomical location, and illustrating the molecular basis of normal physiology plus disease syndromes, with brief outlines of diagnosis and treatment. Other proteins implicated in the same disorders, but which do not function as ion channels – for example NKCC2 in Bartter syndrome – will not be described in detail.
The renal channelopathies – by location within the nephron.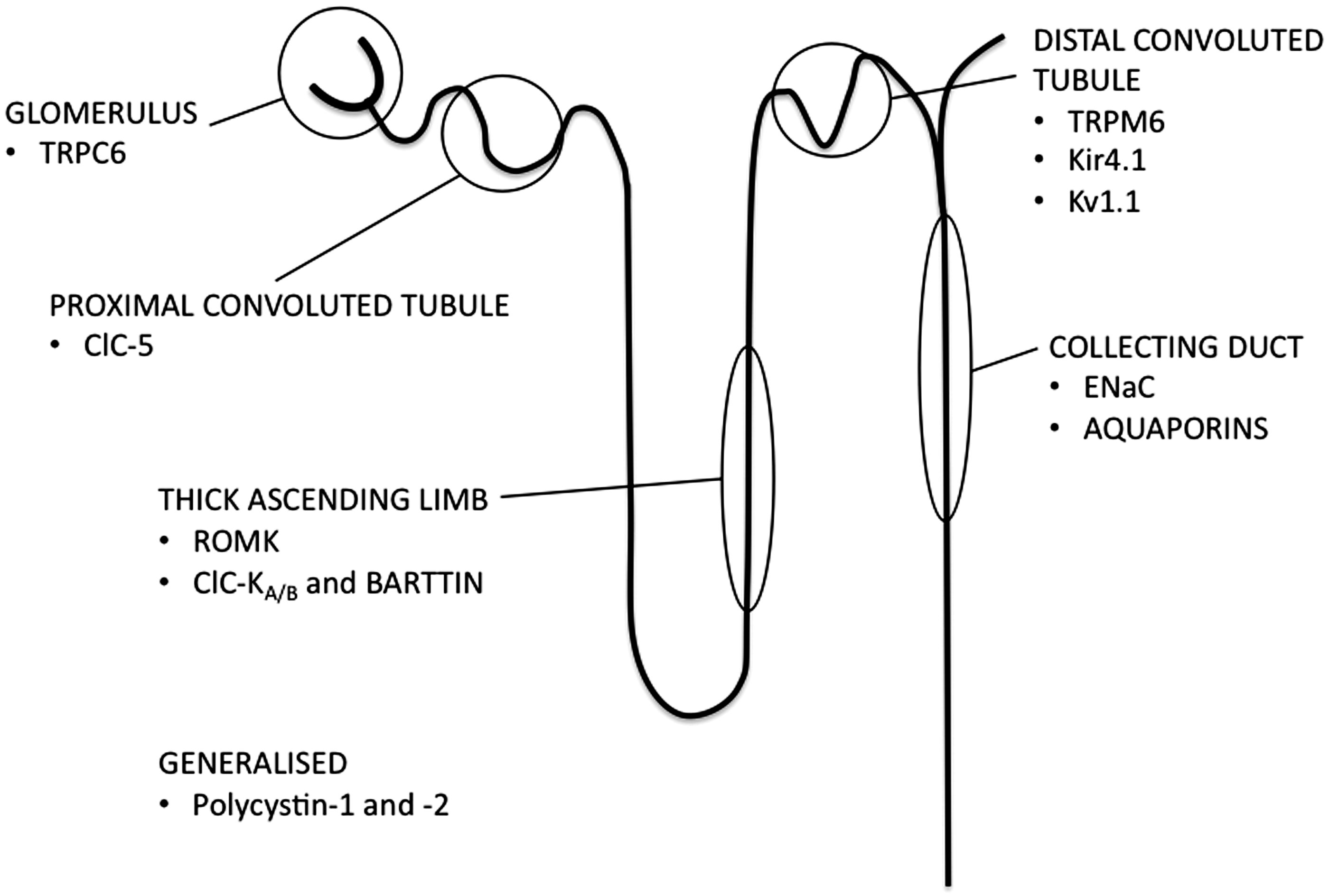
Glomerular disorders
Focal and segmental glomerulosclerosis (FSGS) is a common cause of the nephrotic syndrome in adults, presenting with significant proteinuria, hypoalbuminaemia and peripheral oedema, and frequently resulting in progressive loss of renal function and ultimately end-stage renal failure (ESRF). The glomerular filtration barrier is formed by the fenestrated capillary endothelium, the glomerular basement membrane (maintained by glomerular podocytes) and slit diaphragms between podocyte foot processes. The early hallmark of FSGS is loss of podocyte architecture, with disruption of the slit diaphragm and glomerular basement membrane, and consequent loss of normal glomerular permeability. The prevalence of this condition in the dialysis population may be as high as 20%. 9 A number of familial forms of the condition exist, which together cause approximately 18% of primary FSGS. 10 Disease-causing mutations have thus far been described in several podocyte-expressed genes, including TRPC6, which encodes a member of the transient receptor potential (TRP) superfamily of cation channels.
TRP non-selective, voltage-independent, cation channels are widely expressed in a variety of tissues, including kidney, brain, lung and muscle, and are implicated in ion homeostasis, mechanosensation and signal transduction.
11
TRPC6 contains six transmembrane spans and forms homotetramers or heterotetramers with other class members to create calcium-permeable cation channels, expressed both in the glomerulus and collecting duct (reviewed in Woudenberg-Vrenken et al.
11
). Within the glomerular podocyte, it localises to the slit diaphragm and interacts with podocin and nephrin (mutations in which have also been described in familial FSGS) (Figure 2).
12
Phospholipase C-ɛ 1 (PLCE1, itself linked to childhood nephrotic syndrome
13
) cleaves phosphatidylinositol-3,4-bisphosphate to form diacylglycerol (DAG), which activates TRPC6, leading to an influx of calcium, rearrangement of the podocyte actin cytoskeleton and potential closure of the slit diaphragm.
14
TRPC6 and the podocyte. TRPC6 activation results in calcium influx into the podocyte (foot process is shaded in grey), with downstream effects on the cytoskeleton and the calcineurin-NFAT pathway (among other effects – see text). PLC: phospholipase C; NFAT: nuclear factor of activated T-cells; DAG: diacylglycerol; AT1R: Angiotensin II receptor 1; ANG: angiotensin; ACE-I: angiotensin converting enzyme inhibitor; ARB: angiotensin receptor blocker; CNI: calcineurin inhibitors (cyclosporin and tacrolimus).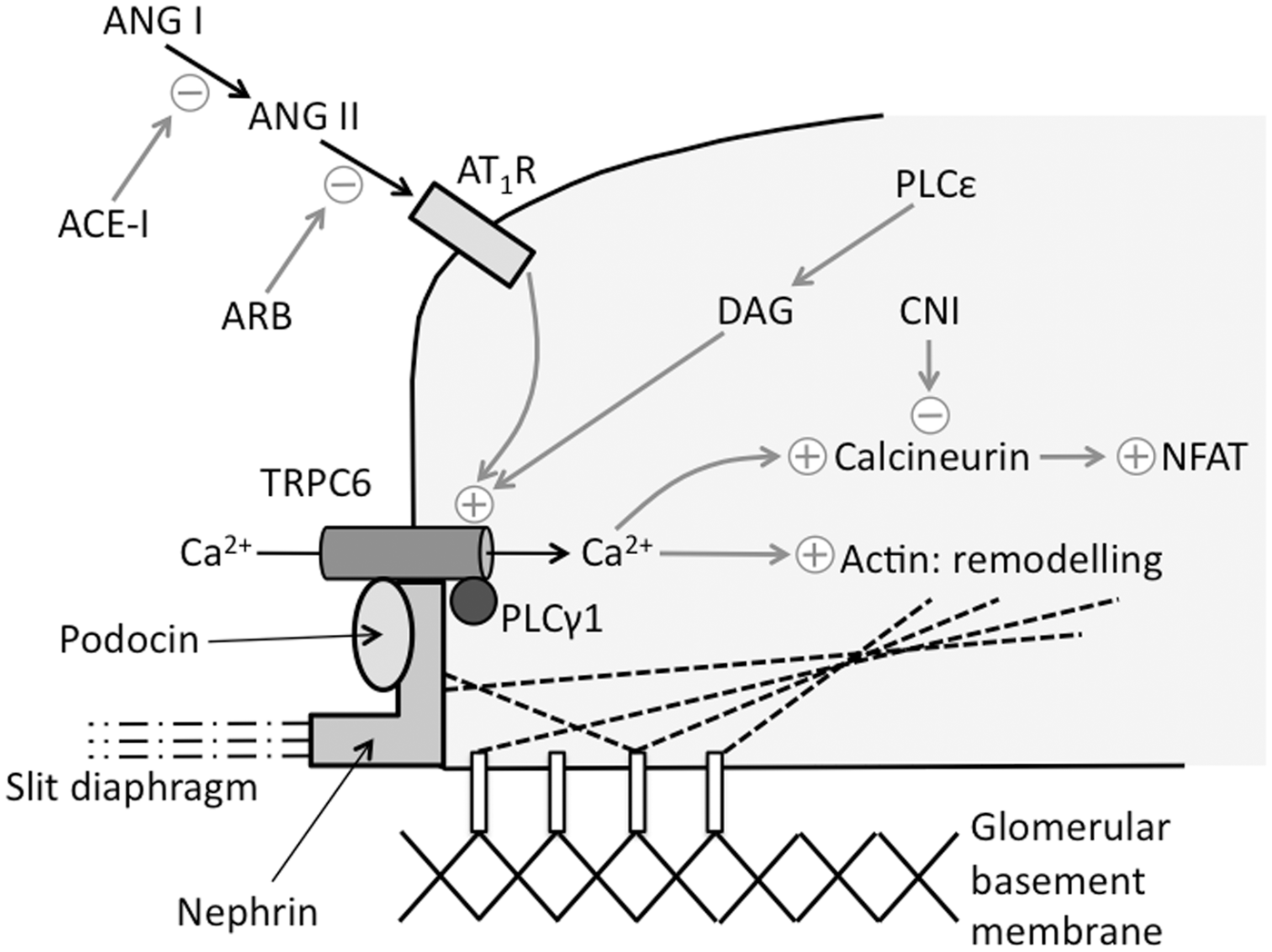
TRPC6 mutations were first described in a large New Zealand cohort with autosomal dominant FSGS, which followed an aggressive clinical course with affected individuals presenting in their third or fourth decade with high-grade proteinuria, and progression to ESRF in approximately 60%. 15 Additional TRPC6 mutations have now been described in over a dozen different families – in both paediatric- and adult-onset disease, and also in sporadic cases of FSGS.12,16–19 When studied in cell culture, the effect of most mutations is to increase calcium influx, potentially through increased cell surface expression of TRPC6; increased glomerular expression of TRPC6 has also been demonstrated in a series of biopsies from patients with FSGS.12,15–17 Increased expression of wild-type TRPC6 has also been found in other acquired proteinuric renal diseases, such as membraneous glomerulonephritis and minimal change disease, and over-expression of wild-type TRPC6 in mice induces proteinuria, adding further credence to the theory that excessive TRPC6-mediated calcium influx is pathogenic.20,21 Recent work has demonstrated that tyrosine-phosphorylation of TRPC6 enables complex formation with phospholipase C (PLC)-γ1, a prerequisite for cell surface expression of TRPC6. Nephrin can interact with phosphorylated TRPC6 to prevent PLC-γ1 binding; FSGS-causing nephrin mutations reduce its interaction with TRPC6, thus leading to increased cell surface expression and channel activity. 22
In terms of therapeutic strategies for FSGS, the involvement of TRPC6 is particularly interesting (Figure 2). TRPC6-mediated calcium influx activates the calcineurin-NFAT pathway; 23 the calcineurin inhibitor ciclosporin effectively treats some patients with FSGS. 24 Non-immunosuppressive treatment of proteinuric renal disease centres on the use of angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin-receptor blockers (ARBs) inhibiting the angiotensin II receptor type 1 (AT1R), which mediates downstream effects through activation of TRPC6 calcium influx.25,26 Angiotensin II (ANG-II) infusion induces albuminuria in wild-type mice – this effect is ameliorated in mice deficient for TRPC6. 25 Blockade of TRPC6 itself may provide a future therapeutic target for proteinuric renal disease.
Disorders of the proximal tubule
Dent disease is a rare, X-linked disorder, typified by proximal tubular dysfunction with low-molecular-weight (LMW) proteinuria, hypercalcuria, nephrolithiasis, nephrocalcinosis and progression to ESRF. There is often additional proximal tubular (PCT) dysfunction (partial Fanconi syndrome) with aminoaciduria, phosphaturia and glycosuria. The skeletal phenotype includes rickets in children and osteomalacia in adults. There is significant variability in phenotype, even within families, with approximately 60% of patients progressing to ESRF, usually by the age of 30–40. 27 Female carriers exhibit a variable degree of LMW proteinuria and mild hypercalciuria. Screening for LMW proteinuria can be performed by assays for urinary beta-2 microglobulin or retinol binding protein – the latter is more sensitive and specific. 28
Dent disease is most commonly (60% of cases – type 1) due to an inactivating mutation of the CLCN5 gene, located on Xp11.22. 29 The vast majority of mutations is either nonsense or missense and severely truncate the protein with complete loss of function. CLCN5 encodes CLC-5, part of the voltage-gated chloride channel (CLC) family (see later section on Bartter syndrome). This forms an ion translocation pore with chloride ion selectivity, also demonstrated in vitro to exhibit chloride/hydrogen ion exchange activity. 30 CLC-5 is predominantly expressed within the proximal convoluted tubule and also to a lesser extent within the thick ascending limb (TAL) and the alpha-intercalated cells of the distal nephron, and co-localises with the H+-ATPase to subapical vesicles where it is thought to contribute to endosomal acidification. 31 Endosomal acidification is necessary for endocytosis from the tubular lumen through the megalin-cubulin receptor pathway, and also for recycling of cell surface receptors – loss of chloride conductance by CLC-5 leads to defective endosomal acidification and accounts for the LMW proteinuria seen in Dent disease. Reduced uptake of parathyroid hormone (PTH) from the glomerular filtrate by CLC-5 dependant endocytosis in the early proximal tubule increases the PTH available to activate the apical PTH receptor in later segments of the proximal tubule, in turn downregulating expression of the phosphate transporter NaPi-2 a, reducing phosphate reabsorption, and causing hyperphosphaturia. Increased concentrations of PTH also activate the renal 1α-hydroxylase, increasing formation of active 1,25(OH)2-vitamin D and enhancing intestinal calcium reabsorption – ultimately leading to the combination of hyperphosphaturia and hypercalciuria which favours renal stone formation and nephrocalcinosis. 32 Two strains of CLC-5 knockout mice have been published, both with a similar phenotype to Dent disease, although only one is hypercalciuric – but this has elevated concentrations of 1,25(OH)2-vitamin D and develops renal failure, indicating the importance of hypercalciuria in the pathogenesis of renal impairment.33,34
Dent disease type 2 (15% of cases) is phenotypically identical but caused by mutations in OCRL at Xq24, encoding OCRL1, a phosphotidylinositol-4,5-bisphosphate phosphatase. 35 Mutations in OCRL also cause Lowe syndrome, with similar renal features as Dent disease, but also congenital cataracts, cognitive impairment and renal tubular acidosis. OCRL1 is widely distributed, but has a role in protein trafficking, probably affecting the subapical endosomes implicated in pathogenesis with dysfunctional CLC-5. 36 A number of families exist in whom the genetic defect remains to be elucidated. 35
Management strategies for Dent disease are supportive, focusing primarily on the prevention of nephrocalcinosis and nephrolithiasis, generally through maintenance of a high fluid intake, but also utilizing thiazide diuretics to reduce hypercalciuria. 37 Promising results have been shown with a high citrate diet in CLC-5 knock-out mice, retarding progression to advanced renal disease compared to controls, but this has yet to be translated to human studies. 38
Disorders of the TAL of the Loop of Henle
The TAL of the Loop of Henle and the distal convoluted tubule (DCT) mediate reabsorption of filtered sodium and other ions, whilst the TAL also generates medullary hypertonicity enabling water reabsorption by the collecting duct. Failure of sodium reabsorption results in the clinically related syndromes of Bartter (in the TAL) and Gitelman (in the DCT). The biochemical picture is dependent on the site of the reabsorptive defect. Salt-wasting by any nephron segment leads to a contraction in extracellular volume, activation of the renin-angiotensin-aldosterone axis and consequent sodium retention at the expense of potassium – the resulting hypokalaemia indicating intact collecting duct function (more precisely, the aldosterone-sensitive distal nephron (ASDN) – the Principal cells of the collecting duct.). Potassium secretion by BK channels in the distal nephron is also upregulated by increased luminal flow (seen with natriuresis), further augmenting hypokalaemia. 39 Chloride reabsorption occurs in the TAL and DCT, so any defect of sodium reabsorption here also impairs chloride reabsorption, which cannot be compensated for as no distal uptake exists. Chloride associates with ammonium and potassium in the collecting duct (further increasing urinary potassium losses), and all are subsequently lost in the urine – TAL and DCT defects produce a metabolic alkalosis with profound hypokalaemia. Magnesium and calcium re-uptake is also affected by the site of the sodium reabsorptive defect – as we shall see later.
Bartter syndrome
Bartter syndrome results from malfunction of one of the transport proteins of the TAL epithelial cells (Figure 3). Within these cells, apical membrane NKCC2 (Na+/K+/2Cl- cotransporter – blocked by loop diuretics, e.g. furosemide) mediates sodium, potassium and chloride reuptake, primarily driven by the low intracellular sodium concentration consequent on the basolateral Na+/K+-ATPase. Recycling of potassium occurs via the apical membrane channel Renal Outer-Medullary K+ channel (ROMK), providing for persistent NKCC2 function along the length of the TAL, and generating an intraluminal positive charge and the electrical driving force for the paracellular reuptake of sodium, calcium and magnesium (50% of sodium reuptake within the TAL is via cation-selective paracellular pathways, and 50% via NKCC2). Chloride exits the cells via basolateral membrane chloride channels ClC-Kb and –Ka, which require the presence of a β-subunit barttin, both for passage to the cell surface and correct assembly once there.40,41 Bartter syndrome is thus characterised by urinary losses of sodium and chloride, causing loss of extracellular volume, which combined with intact ASDN function results in a hypokalaemic alkalosis with low blood pressure. Clinical subdivision into three subtypes is possible, based on clinical features, which correlate with the underlying genetic defect: antenatal Bartter syndrome, antenatal Bartter syndrome with sensorineural deafness and classic Bartter syndrome (usually presenting in infancy) (Table 2). Classification systems have recently been discussed by Seyberth.
42
Genotype-phenotype correlation relates to the known roles of each transport protein within the nephron – the antenatal forms are most severe, as the genetic defect leads to complete loss of TAL function, whereas in the milder classic variant loss of ClC-Kb function can be compensated for by the additional presence of ClC-Ka in the TAL (with the loss of ClC-Kb from the DCT resulting in a phenotypic overlap with Gitelman syndrome).
Transport proteins involved in Bartter syndrome. An epithelial cell of the thick ascending limb of the Loop of Henle – transport proteins labeled 1–4 correspond to the dysfunctional proteins in the four subtypes of Bartter syndrome (see text). NKCC2: Na+/K+/2Cl− co-transporter, ROMK: Renal Outer-Medullary K+ channel; ClC-Ka/b: chloride channel Ka/b; TJ: tight junction. Salt-wasting disorders with hypokalaemia. TAL: thick ascending limb; DCT: distal convoluted tubule.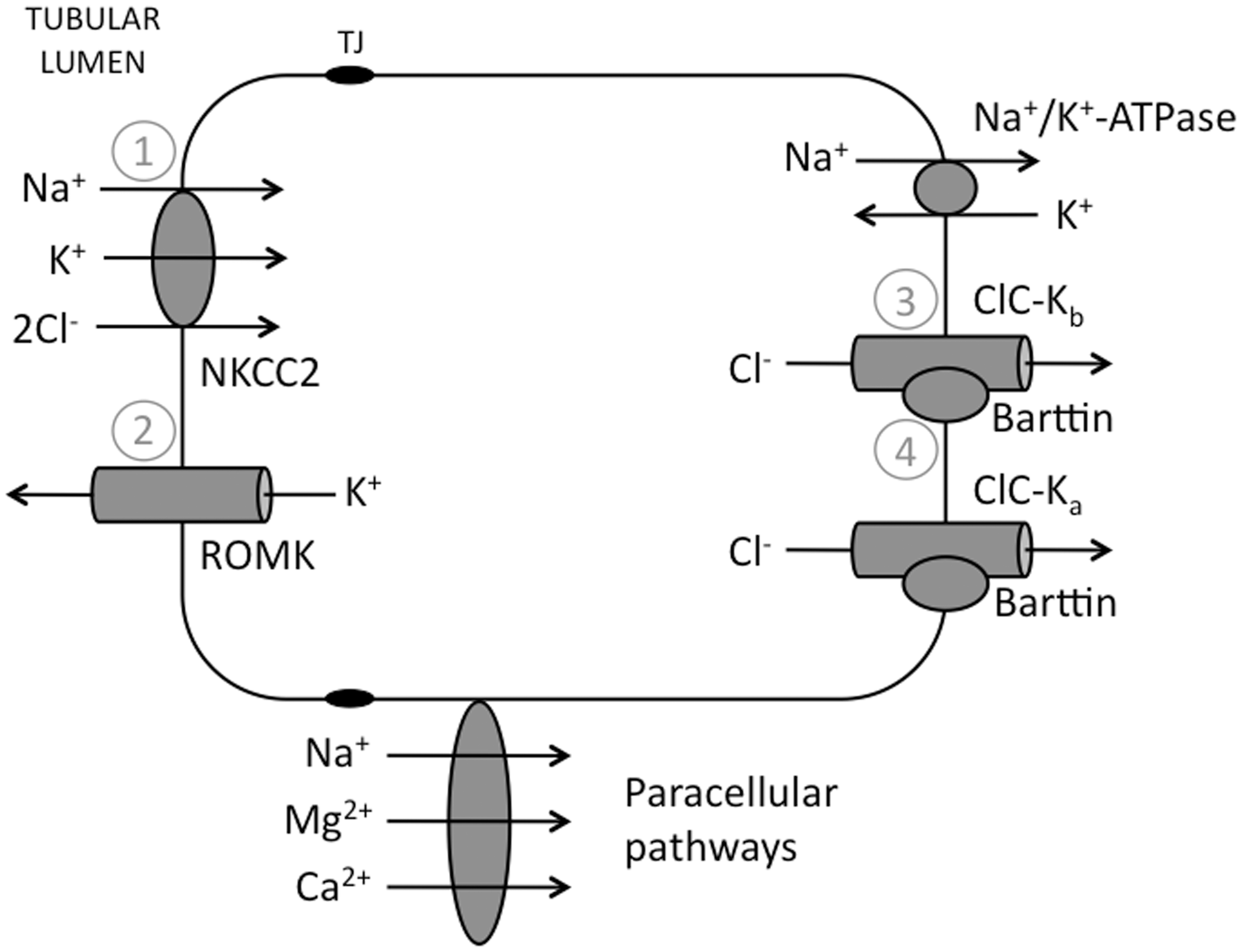
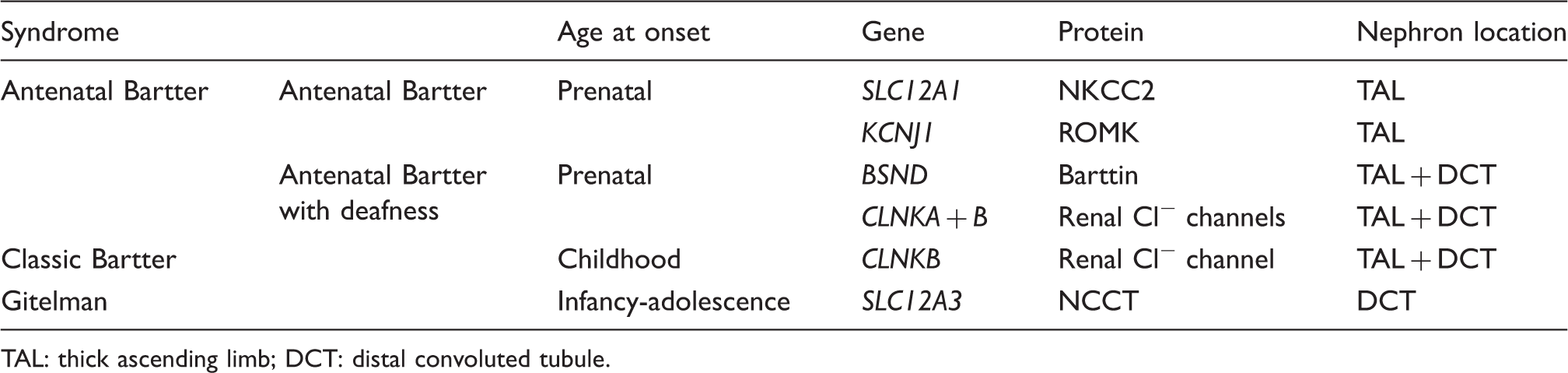
Salt-wasting in Bartter syndrome may also relate to the presence of all the implicated transport proteins in the cells of the macula densa as well as the TAL. The macula densa is a specialised region of epithelial cells at the transition point between the TAL and DCT, which contact extra-glomerular mesangial cells and granular cells of the afferent arteriole supplying that nephron, and act in concert to modify (a) glomerular perfusion pressure in response to the tubular fluid composition at that point – a mechanism termed tubuloglomerular feedback and (b) vascular tone and blood pressure systemically. Normal NKCC2 and ROMK function in the cells of the macula densa generates a net negative intracellular charge, with depolarisation of the basolateral membrane, calcium entry and release of vasoactive mediators (prostaglandins, ATP, nitric oxide – affecting afferent arteriolar tone – and renin – activating the angiotensin/aldosterone axis). Loss of TAL function is thus compounded by a failure of the macula densa to sense the increased distal delivery of sodium, consequent failure to reduce glomerular filtration appropriately, and increased salt wasting.
The specific channelopathies implicated in Bartter syndrome are discussed below:
Renal outer-medullary K+ channel
ROMK is a member of the inward rectifying family of K+ channels (Kir) that provide powerful conduits for K+ flux. 43 They are present in many cell types where they play an important role in regulation or maintenance of the cell membrane potential, such as neurons, myocytes, leukocytes and epithelial cells. Mutations in the KCNJ family of genes encoding Kir channels are linked with numerous conditions, including epilepsy, sensorineural deafness, disorders of pancreatic insulin secretion, long Q-T syndrome and cardiac arrhthymias, as well as renal salt-wasting. Functional Kir channels formed from four subunits provide for robust outward K+ flow at physiological membrane potentials, with a long-lived active conformation attained through phosphorylation or ligand binding.
KCNJ1 encodes ROMK or Kir1.1, the major K+ secretory channel in the kidney (reviewed in Welling and Ho). 44 Alternative splicing generates three different ROMK isoforms (ROMK1-3) with differential expression along the nephron: ROMK2 mediates apical K+ recycling in the TAL, whilst ROMK1 and ROMK3 in the distal nephron mediate K+ secretion. 45 Regulation of ROMK activity occurs by modulating the number of channels present at the cell surface, or the activity of membrane-resident channels. N- and C-terminal phosphorylation by protein kinase A (PKA) determines both cell surface expression and maintains the channel in a high PO state.46,47 Phosphatidylinositol-4,5-bisphosphate (PIP2) directly binds ROMK and acts as an essential cofactor for maintaining the channel in an open state. 48 ATP inhibits ROMK activity in the presence of the cystic fibrosis transmembrane conductance regulator (CFTR), itself highly expressed along the nephron. 49 CFTR and ROMK interact through binding to the scaffolding protein NHERF via PDZ-based interactions. 50 Channel activity is inhibited by a fall in intracellular pH, possibly via an effect on PKA and ROMK phosphorylation.51,52
Loss-of-function mutations in ROMK cause Bartter syndrome (type 2), with recapitulation of the disease phenotype in ROMK knockout mice.53,54 Mutations range from those producing a non-functional truncated protein to those that affect trafficking to the cell surface, and to mutations in key residues involved in channel regulation. The phenotype is very similar to that for Bartter patients with NKCC2 mutations – antenatal presentation with polyhydramnios, prematurity with a median gestational age of 33 weeks and from the neonatal period profound polyuria, hypokalaemia, hyponatraemia and hypercalciuria with secondary nephrocalcinosis later in life. Interestingly, there is often a period of transient hyperkalaemia – frequently severe – in the first few days of life; combined with the hyponatraemia this may be misdiagnosed as mineralocorticoid deficiency. 55 This is thought to reflect the failure of ROMK-mediated K+ secretion in the CCD; alternative secretory pathways (e.g. BK channels in the apical membrane of CCD cells 39 ) are not well-developed in prematurity and require a period of upregulation. With an intact ASDN essentially restored, hypokalaemia ultimately results, although to a lesser degree than with NKCC2 mutations. Not all subjects develop transient hyperkalaemia: in the 2 (out of 10) initially normokalaemic patients from the series of Jeck et al., 55 the mutation site affected the promoter region for ROMK2 and 3, but not 1, leading to the hypothesis that maintained CCD K+ secretion via still functional ROMK1 prevented hyperkalaemia.
Cl− channels (ClC-K) and Barttin
The CLC family of genes are widely expressed in pro- and eukaryotes and encode both chloride channels and hydrogen/chloride exchangers, forming the largest known family of anion transporters.
56
Within mammals, ClC channels are localised to either the plasma or endosomal/lysosomal membranes, and CLC mutations cause a number of human diseases, including myotonia (Becker and Thomsen syndromes), Bartter syndrome and Dent disease (see earlier), together with a potentially causative role in idiopathic generalised epilepsy.
57
ClC-Ka and –Kb are chloride channels specific to the kidney – their discovery shortly followed that of their rat orthologs ClC-K1 and –K2, with which they share approximately 80% homology.58–60 The two channels (-Ka and –Kb) have differential expression within the nephron and thus different roles:
ClC-Ka is predominantly located in the thin ascending limb (possibly at both basolateral and apical membranes) where it allows Cl− reabsorption, thus generating increased medullary hypertonicity and the osmotic gradient for water reabsorption from the collecting duct.56,61,62 Knockout mice lacking ClC-K1 suffer from NDI.
63
ClC-Kb is more widely expressed through the distal nephron, localised at the basolateral membrane of the tubular epithelial cells, with highest concentrations of expression seen in the TAL and DCT where it permits NaCl reabsorption.
56
Both channels are also found in the marginal cells of the stria vascularis of the inner ear where they form part of the mechanism for K+ secretion into the endolymph – disturbance in their function thus also leads to sensorineural deafness. Changes in intracellular volume, inhibition of NKCC2 and local effects of the renin-angiotensin system are thought to regulate ClC-Ka function; a high salt diet and direct effects of furosemide regulate ClC-Kb.
64
Expression of functional channels (ClC-Ka, -Kb, –K2, but not –K1) in Xenopus and epithelial cell culture requires the presence of an additional subunit Barttin, a small protein with 2 transmembrane domains that is necessary for membrane insertion and stability, plus anion conductance.41,65,66 Mutations in BSND, encoding Barttin, result in perinatal Bartter syndrome with sensorineural deafness. 67 A genotype-phenotype correlation exists – whilst all patients are profoundly deaf, different BSND mutations have different effects on channel trafficking or activation, leading to a variable renal phenotype – from absent renal symptoms to early-onset renal failure. 56
No disease-causing mutations have yet been described in CLCNKA (with a single exception – see below), whereas mutations in CLCNKB result in Classic (type 3) Bartter syndrome, with onset in childhood, and salt-wasting with hypokalaemia, hypochloraemia, alkalosis and failure to thrive. CLCNKB mutations reflect the nephron distribution of ClC-Kb, with a later-onset, milder Bartter phenotype and features of Gitelman syndrome also present (including a tendency to develop hypomagnesaemia). 64 A hallmark of the antenatal Bartter syndromes is hypercalciuria (with resultant nephrocalcinosis), but urinary Ca2+ excretion is normal in those with CLCNKB mutations, indicating some preservation of TAL function, likely through basolateral ClC-Ka activity.
A single patient has been described with deficiency of both ClC-Ka and –Kb (subsequent on a CLCNKA loss-of-function mutation and CLCNKB gene deletion), and a severe disease phenotype mimicking that of patients with BSND mutations. 68 The severity of the clinical picture illustrates the degree of compensation that can normally occur when only one of the two channels is affected.
Genetic variants in both CLCNKA and CLCNKB have been associated with salt-sensitive hypertension; the best-characterised example is that of the ClC-Kb(T481 S) polymorphism which demonstrates increased channel activity, and a higher prevalence of the variant allele in an Afro-Caribbean population.69–71 As mentioned, furosemide may exert some of its affect through an inhibitory action on ClC-K channels, and recently a number of novel compounds with direct ClC-Ka/b inhibition have been proposed as a future class of antihypertensive agents, following demonstration that they provoke a diuresis in rats. 72
Diagnosis and treatment
A disorder of renal salt-handling should be considered in those infants presenting with polyuria and dehydration, particularly if accompanied by polyhydramnios in pregnancy and preterm birth, and as a differential in the older child with growth retardation. Plasma Na+ and Cl− concentrations will be reduced, together with a hypokalaemic metabolic alkalosis, and blood pressure is low. Renin concentrations are very high. The finding of an elevated urinary chloride concentration (>20 mmol/L) indicates a renal defect (in the absence of diuretic use) – hypercalciuria indicates TAL dysfunction, and serum Mg2+ should be normal (both not constant findings in the classic variant). Imaging may demonstrate nephrocalcinosis. Genotyping confirms the diagnosis, and helps distinguish Bartter and Gitelman variants in the case of CLCNKB mutations. Fluid and electrolyte replacement will be guided by the immediate clinical picture. Long-term treatment in Bartter syndrome consists of a high-salt diet, potassium and magnesium replacement and potassium-sparing diuretics such as spironolactone/eplerenone and amiloride (potentially augmented with angiotensin-converting enzyme inhibitors [ACE-I]). In addition, volume depletion leads to upregulation of Prostaglandin E2 (PGE2) by both renal and non-renal mechanisms – blockade with cyclooxygenase inhibitors such as indomethacin is highly beneficial.
The distal tubule and disorders of Magnesium handling
Magnesium (Mg2+) is essential for many physiological processes, and hypomagnesaemia results in disturbed function in excitable tissues, with muscle cramps, convulsions, arrhythmias and cardiac arrest. Absorption via the gut, excretion by the kidneys, and release from bone and muscle (<1% of total body Mg2+ is circulating in the plasma) regulate serum Mg2+ concentrations; 2400 mg of Mg2+ is filtered each day by the kidney, of which 90–95% is ultimately reabsorbed. A small proportion (10–25%) is reabsorbed in the proximal tubule, but the bulk (70%) is reabsorbed by a passive paracellular route in the TAL, driven by a positive transepithelial voltage (generated by ROMK – see earlier). 73 Mutations in CLDN16 and -19, encoding the tight junction proteins claudin-16 (paracellin-1) and claudin-19, result in failure of TAL Mg2+ and Ca2+ reabsorption and the syndrome Familial Hypomagnesaemia with Hypercalciuria and Nephrocalcinosis (FHHNC).74,75 Interestingly, ROMK/NKCC2 mutations (affecting the same segment) only result in hypercalciuria; whilst paracellular Mg2+ uptake is also reduced, increased distal sodium delivery and NCCT (Na+/Cl− cotransporter, thiazide-sensitive) upregulation in the DCT increases Mg2+ reabsorption in this segment, allowing compensation.
Fine-tuning of Mg2+ homeostasis occurs through the reabsorption of 5–10% of the Mg2+ load via an active transcellular route in the DCT (Figure 4); no Mg2+ reabsorption occurs distal to this. Within this nephron segment, mutations in NCCT, TRPM6, Kv1.1 and Kir4.1 all result in clinical phenotypes characterised by hypomagnesaemia – Gitelman syndrome, hypomagnesaemia with secondary hypocalcaemia (HSH), autosomal dominant hypomagnesaemia and EAST syndrome, respectively.
The distal tubule and disorders of Magnesium handling. Magnesium enters the cell by the apical TRPM6 channel, along an electrochemical gradient generated by K+ efflux through Kv1.1 and Kir4.1 channels, itself dependent on the activity of the basolateral Na+/K+-ATPase, and exits the cell at the basolateral membrane via an as-yet unidentified route, possibly a Na+/Mg2+ exchanger or ATP-dependant Mg2+ pump. Na+ and Cl− enter the cell at the apical membrane via the Na+/Cl− co-transporter NCCT, mutated in Gitelman syndrome and the target of thiazide diuretics. Mutations in the channel proteins TRPM6, Kv1.1 and Kir4.1 all cause different forms of inherited renal magnesium wasting, as do mutations in EGFR, γ-subunit of the Na+/K+-ATPase (FXYD2) and hepatocyte nuclear factor 1 homeobox B (HNF1B) – a transcription factor regulating expression of FXYD2. EGFR activation by EGF ligand binding results in activation of TRPM6 – this is blocked by the monoclonal antibody cetuximab. EGF: epidermal growth factor; EGFR: EGF receptor.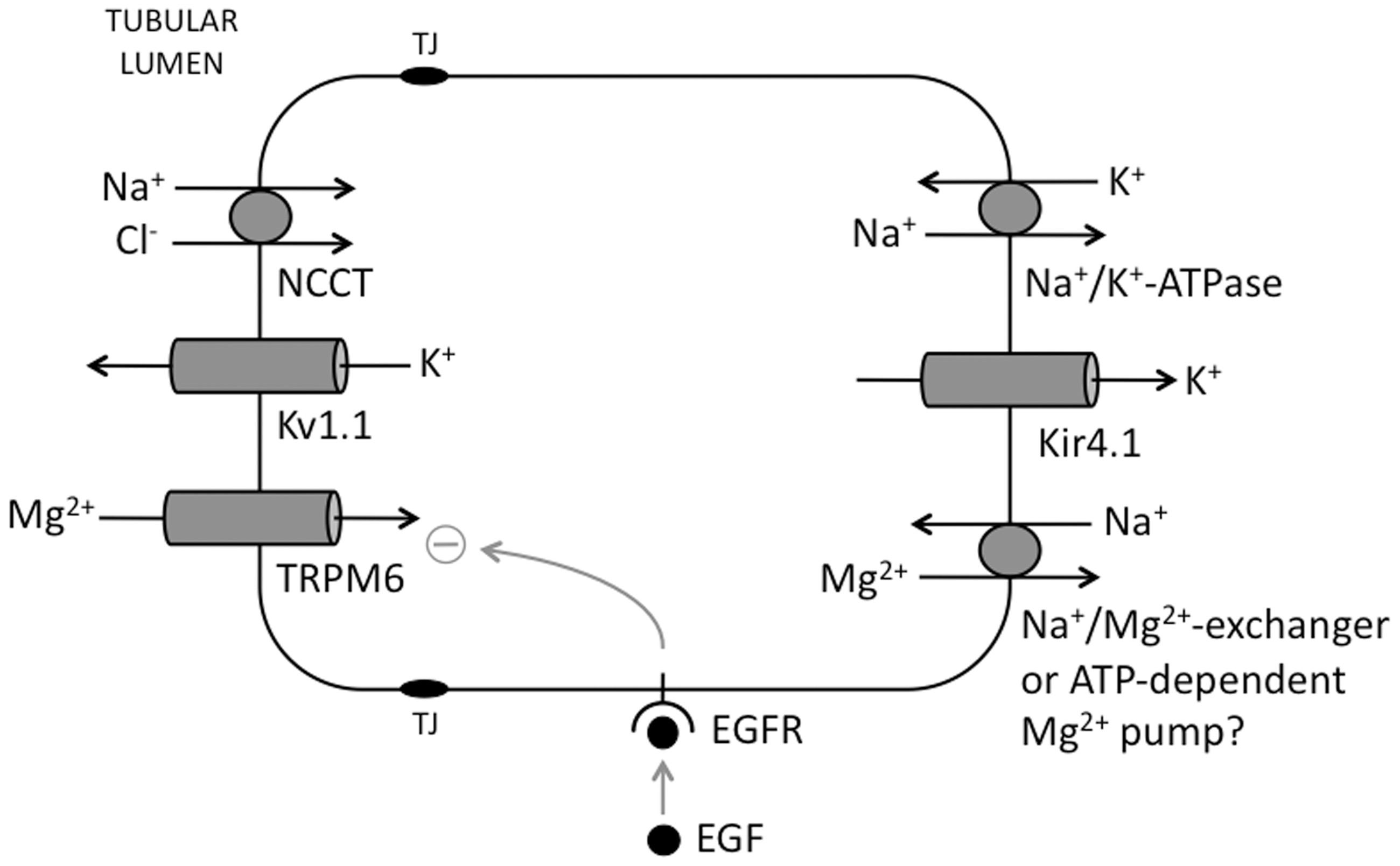
TRPM6 – HSH
Autosomal recessive HSH presents in infancy with muscle spasms and generalised seizures, and results from mutations in TRPM6 (encoding TRPM6 protein) which forms the apical epithelial Mg2+ channel in the gastrointestinal tract and renal DCT.76,77 In affected patients, both serum Mg2+ and Ca2+ are very low – the hypocalcaemia is thought consequent to the suppressive effect of hypomagnesaemia on PTH receptor function or PTH release, although this remains incompletely understood. Prompt treatment with magnesium replacement is necessary to prevent death or long-term neurological damage.
TRPM6 is a member of the TRP family of cation channels and is a large protein consisting of six transmembrane-spanning domains; four TRP protein subunits assemble to constitute a functional ion channel capable of conducting both Mg2+ and Ca2+, although with five-fold higher affinity for Mg2+.78,79 Within the DCT TRPM7 is also present, and hetero-oligomerisation between TRPM6 and –7 is thought by some researchers essential to enable Mg2+ conductance – TRPM7 alone cannot substitute. 14 Uniquely amongst ion channels, the TRPM channels have been found to possess a C-terminal domain with enzymatic activity – a protein kinase in the case of TRPM6 and –7, and an ADP-ribose pyrophosphatase in TRPM2. 80 Termed ‘chanzymes,’ the precise role of the enzyme domain remains unclear. TRPM6 is tightly regulated by the intracellular Mg2+ concentration – rising Mg2+ concentrations reducing channel activity. 79
Loss of function mutations in TRPM6 cause HSH, the majority of which are likely to result in a complete lack of protein expression. Of the five point mutations described, four are located in the cytoplasmic N-terminus and are considered to affect protein trafficking, or possibly association with TRPM7. 14 A single mutant – P1017 R – has been reported with an amino acid substitution in the putative pore-forming region of the protein, with consequent reduced channel activity of TRPM6P1017R/TRPM7WT complexes. 81
Hormonal regulation of renal Mg2+ excretion through the action of epidermal growth factor (EGF) on TRPM6 activity was first postulated when patients treated with cetuximab, a monoclonal antibody directed against the EGF receptor (EGFR) – used in the treatment of colorectal cancer, developed renal Mg2+ wasting and symptomatic hypomagnesaemia. 82 Subsequently, mutations in pro-EGF were identified in a family with Isolated recessive Renal Hypomagnesaemia (IRH). 83 Pro-EGF is sorted to both apical and basolateral membranes of polarised kidney epithelial cells, and cleaved at the surface to release active EGF. In the DCT, EGFR is present only at the basolateral surface – EGF acts here in an autocrine/paracrine fashion with activation of the EGFR leading to increased Mg2+ flux through TRPM6 (due to increased expression concentrations at the plasma membrane) via an intracellular signalling cascade. 84 EGF may also act on other anatomically juxtaposed nephron segments (e.g. the PCT) in a paracrine fashion. The point mutation identified disrupts pro-EGF targeting to the basolateral membrane, preventing EGF-mediated stimulation of TRPM6 activity – and cetuximab was also demonstrated to abolish EGF-augmented Mg2+ flux though TRPM6 in cell culture. 83
Kv1.1 – autosomal dominant hypomagnesaemia
The presence of the potassium channel Kv1.1 (voltage-gated potassium channel subtype 1.1) at the luminal membrane in DCT cells establishes the apical membrane potential essential for Mg2+ reabsorption through TRPM6. A mutation in KCNA1, encoding Kv1.1, results in a non-functional channel, and has been identified in a family with autosomal dominant hypomagnesaemia.85,86 Interestingly, other mutations in the same protein cause the neurological syndrome of Episodic Ataxia type 1 – these patients are not reported to be hypomagnesaemic. 87
Kir4.1 and EAST/SeSAME syndrome
The potassium channel Kir4.1 recycles K+ at the basolateral membrane, enabling Na+/K+-ATPase activity and regulating intracellular voltage and Mg2+ transport. 88 Kir4.1 is expressed in the DCT, glial cells in the brain and spinal cord, and in the inner ear. Mutations in KCNJ10, its encoding gene, result in hypomagnesaemia associated with a severe neurological phenotype, alternatively termed EAST (epilepsy, ataxia, sensorineural deafness, and tubulopathy) or SeSAME (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance) syndrome.89,90 The tubular disorder resembles Gitelman syndrome with renal salt-wasting, a hypokalaemic metabolic alkalosis, hypomagnesaemia and hypocalciuria. KCNJ10 mutations are highly varied in their effects on the protein, and individually have been demonstrated to abolish channel function, impair protein folding, reduce surface expression and alter pH sensitivity and pore gating.91,92
Disorders of the collecting duct
ENaC and sodium transport
Approximately 5% of filtered sodium is reabsorbed in the collecting duct, where there are three cell populations – the principal cell, responsible for salt and water reabsorption, the α-intercalated cell, responsible for acid secretion and bicarbonate reabsorption, and its mirror image, the β-intercalated cell with a role in bicarbonate secretion. Na+ uptake from the tubular lumen occurs through apical epithelial sodium channels (ENaC), the gradient for Na+ reabsorption driven by basolateral Na+/K+-ATPase activity. Apical K+ excretion occurs through ROMK1 and Maxi-K K+ channels, the latter activated by increased tubular flow (Figure 5). ENaC is a heteromultimeric channel, formed from α, β and γ subunits (encoded by SCNN1A, SCNN1B, and SCNN1G) – the exact stoichiometry remains unclear – which consist of cytoplasmic N− and C−termini, and a large extracellular domain.
93
The C−terminus of each subunit contains a PPPXY (PY) motif recognised by the WW domain of E3 ligases (e.g. Nedd 4-2) – these ubiquitinate ENaC and target it for internalisation and proteasomal degradation, thus regulating surface concentrations and hence activity.
94
Hyperkalaemia and hypovolaemia increase aldosterone concentrations – this acts via the cytosolic mineralocorticoid receptor in principal cells to promote transcriptional activation of ENaC, and also inhibition of Nedd 4-2 activity (Figure 5).
95
The principal cell and ENaC. Apical Na+ uptake occurs through ENaC, driven by basolateral exit through the Na+/K+-ATPase. The accumulated K+ is secreted via apical ROMK and Maxi-K channels. Hypovolaemia and hyperkalaemia increase circulating aldosterone concentrations, which acts on the intracellular mineralocorticoid receptor to both enhance transcription of ENaC and to promote phosphorylation and inhibition of Nedd 4-2 (which would otherwise reduce ENaC activity) – the net result is increased surface expression and activity of ENaC. ENaC: Epithelial Na+ Channel; MCR: Mineralocorticoid receptor.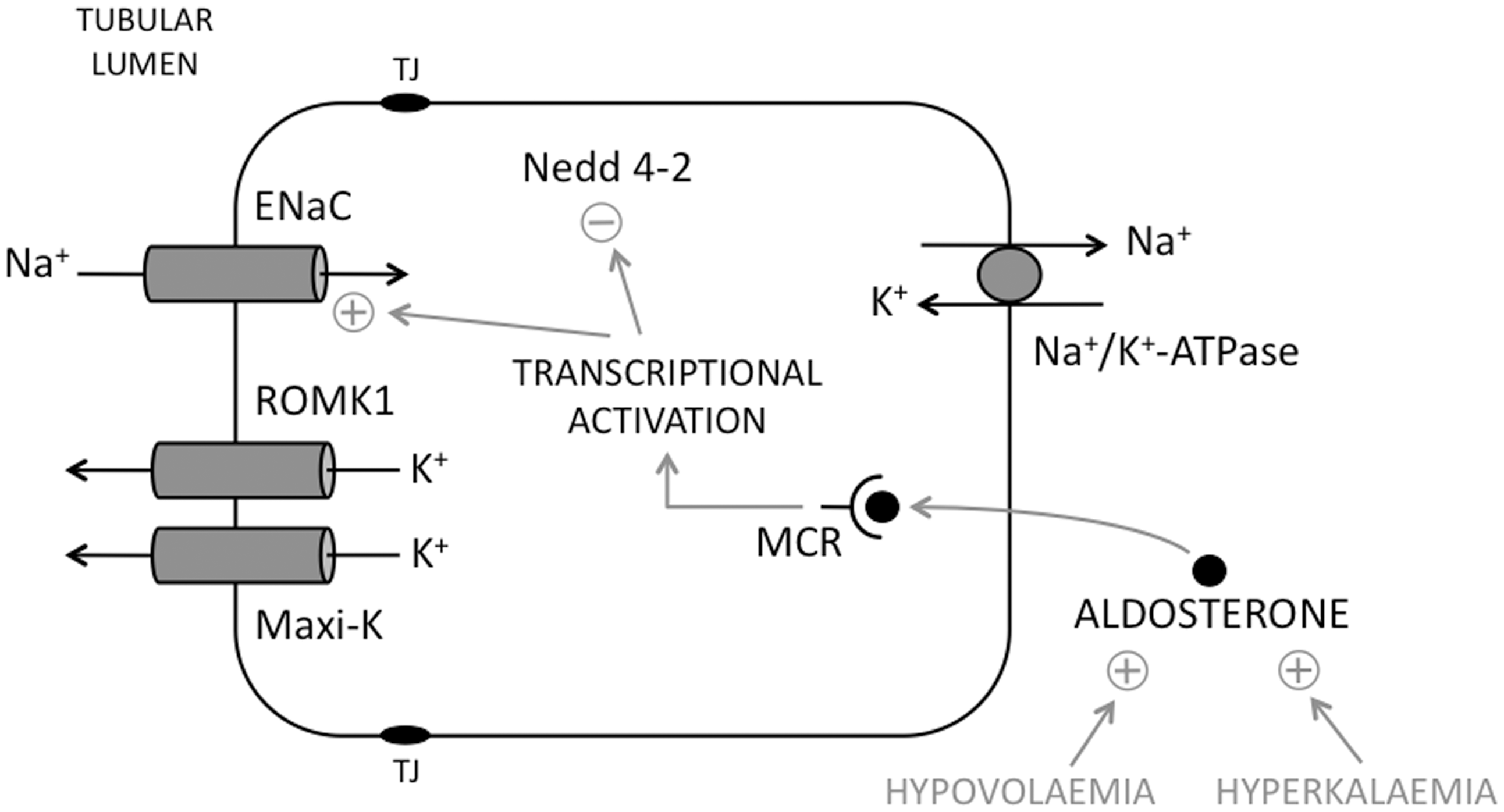
Disorders of ENaC function
(A) Overactivity – Liddle syndrome
Liddle syndrome arises as a consequence of an activating mutation in ENaC resulting in an increase in distal sodium reabsorption. First described by Liddle in 1963, the proband presented at the age of 16 with early-onset severe hypertension, associated with hypokalaemia and a metabolic acidosis and suppressed aldosterone concentrations. 96 Hypertension and hypokalaemia responded to triamterene, a Na+ channel blocker, but not to the mineralocorticoid receptor antagonist spironolactone. Despite treatment, persistent hypertension led to the development of renal impairment and ultimately ESRF, treated by renal transplantation in 1989. This led to resolution of her metabolic abnormalities and confirmed the initial hypothesis of an intrinsic defect in distal nephron sodium uptake. 97
Analysis of the original kindred demonstrated linkage to the β-subunit gene (β-ENaC), containing a truncation mutation with loss of the cytoplasmic C-terminus. Mutations in other families are heterogeneous but all result in loss of the C-terminal PY domain in β- or γ-ENaC; mutant channels are not targeted for internalisation and remain at the cell surface resulting in enhanced activity.98–102 A gain of function mutation has also been described in the extracellular domain of γ-ENaC, but no mutations have yet been described in α-ENaC. 103 Recombinant mice expressing ENaC with the same truncating mutation as the initial proband recapitulate the features of the human condition when fed a high salt diet – hypertension, hypokalaemic metabolic alkalosis and evidence of end-organ damage (cardiac hypertrophy). There is also increased Na+ reabsorption in the distal colon suggesting an additional contribution to salt-loading and human hypertension. 104 Further studies in this mouse model confirmed that renal Na+ retention is a result of abnormal ENaC over-activity in the apical membrane of collecting duct epithelial cells. 105
Clinically, Liddle syndrome may require differentiation from other causes of early-onset hypertension with hypokalaemic alkalosis – namely Conn syndrome (primary hyperaldosteronism), glucocorticoid-remediable hypertension (GRH), apparent mineralocorticoid excess and 11β-hydroxylase deficiency. Therapy consists of sodium restriction, potassium supplementation (where needed – not all patients are hypokalaemic) and treatment with the diuretics amiloride or triamterene, which directly block ENaC.
(B) Underactivity – autosomal recessive pseudohypoaldosteronism type 1 (PHA1)
Pseudohypoaldosteronism defines a renal (and more widespread) lack of responsiveness to circulating aldosterone. Two forms of PHA1 exist – autosomal dominant, renal-limited and a consequence of mutations in the mineralocorticoid receptor 106 – and autosomal recessive, a result of inactivating mutations in any of the three (α, β or γ) subunits of ENaC, and which has ‘systemic” features, affecting the kidneys, lungs, colon and sweat glands. 107 Autosomal recessive PHA1 presents in early infancy with lethargy, failure to thrive and volume depletion, secondary to excessive salt-wasting. Laboratory investigation demonstrates hyponatraemia, hyperkalaemia and a metabolic acidosis, together with elevated renin and aldosterone concentrations. The systemic effects of loss of ENaC function include high sweat sodium concentrations (exacerbating renal salt-wasting and creating a tendency to skin infections) and respiratory symptoms (recurrent infections, chronic cough, increased airway secretions), such that it mimics cystic fibrosis. 108
Loss of ENaC function in the collecting duct results in a failure to reabsorb <5% of filtered Na+, and lack of Na+ influx into principal cells results in a failure to excrete K+ (causing hyperkalaemia, worsened by the lack of responsiveness to aldosterone, the main K+ regulatory hormone), and a failure to excrete H+ by α-intercalated cells (causing metabolic acidosis). Treatment requires lifelong sodium chloride supplementation and strict restriction of dietary potassium intake, together with prophylactic antibiotics for the lung or skin sequelae.
Nephrogenic diabetes insipidus
NDI results from a failure of the kidney to concentrate the urine in response to antidiuretic hormone (ADH, a.k.a. vasopressin) secreted by the posterior pituitary, and in the inherited form is a consequence of mutations in key proteins involved in water reabsorption in the collecting duct (Figure 6). Clinically, acquired disease predominates, caused by drugs (lithium [Lithium enters collecting duct principal cells via ENaC and causes downregulation of AQP2; amiloride can be used to block this if lithium cannot be discontinued. Lithium also causes a chronic tubulointerstitial nephritis, which will also contribute to a loss of urinary concentrating ability], amphoteracin, tetracyclines), chronic kidney disease – particularly chronic tubulointerstitial nephritis, hypokalaemia and hypercalcaemia, ureteric obstruction and sickle cell disease.
Aquaporin expression and regulation in collecting duct principal cells (left image) and mistargeting of mutant forms of AQP2 (right image). In normal circumstances, ADH acts on the V2R to increase the formation of cAMP, leading to the activation of PKA, phosphorylation of the water channel AQP2 and its insertion at the apical membrane to increase water permeability. Autosomal recessive mutants are retained in the ER, whilst dominant mutants (black) form heterotetramers with wild-type AQP2 (white; only one of each is shown for clarity) and are retained in various organelles or expressed at the basolateral membrane, depending on the individual mutation. ADH: anti-diuretic hormone; V2R: V2 vasopressin receptor; PKA: protein kinase A; AQP: aquaporin.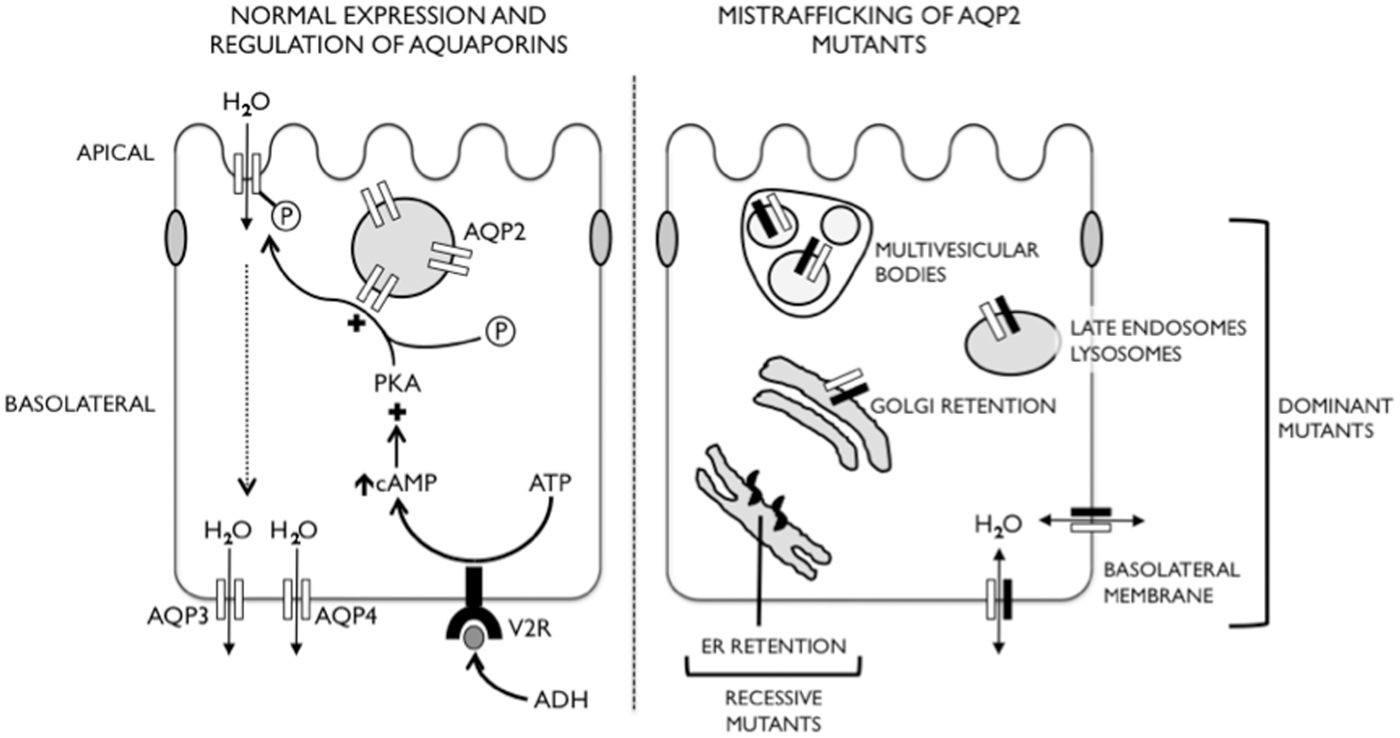
X-linked recessive NDI is the most common inherited form (90–95%), resulting from inactivating mutations in vasopressin type-2 receptor (V2R). Autosomal recessive disease (5–10% of cases) results from endoplasmic reticulum retention (and then degradation) of misfolded aquaporin-2 (AQP2) (with mutations in the segments forming the water pore), but autosomal dominant disease (<1% of cases) is the consequence of incorrect targeting of functional protein. Autosomal dominant mutations variously retain the mutant protein in the Golgi, or target it to late endosomes, multivesicular bodies, lysosomes or the basolateral plasma membrane. A dominant negative effect is exerted as the mutant forms heterotetramers with wild-type protein, also preventing it from reaching its correct apical membrane destination. 109 AQP2 mutations in dominant NDI are all located in the C-terminal cytoplasmic tail which contains the phosphorylation site for PKA and an apical targeting motif. The disease-causing mutations which direct AQP2 to the basolateral membrane represent gain of basolateral sorting motifs; they are frameshift mutations which result in C-terminal extension of AQP2 to include dileucine and/or tyrosine-based motifs, the presence of which results in mistargeting of both mutant protein alone and wild-type/mutant heterotetramers to the basolateral membrane.110–112 One of these dominant NDI mutations was used to create a knock-in mouse model, in which heterozygotes had a severe impairment of urinary concentration, and provided immunohistochemical confirmation that both wild-type and mutant AQP2 were mistargeted to the basolateral membrane of collecting duct cells. Treatment with the phosphodiesterase-4 inhibitor Rolipram increased renal cAMP and phosphorylated AQP2 concentrations, increased wild-type AQP2 expression at the apical membrane and resulted in an increase in urine osmolality in heterozygous mice. 113
Patients with congenital NDI present in infancy with polyuria, obvious excessive thirst and – if not recognised early – recurrent episodes of hypernatraemic dehydration that can lead to irreversible brain and kidney damage. During episodes of dehydration, plasma biochemistry demonstrates hypernatraemia, potentially raised urea and creatinine, and high plasma (>300 mOsm/kg) and low urine (<200 mOsm/kg) osmolality (the latter combination highly suggestive of nephrogenic or cranial DI). The vasopressin test distinguishes between cranial and nephrogenic DI – NDI patients fail to increase urinary osmolality in response to intranasal administration of DDAVP (a synthetic ADH analogue). X-linked and autosomal forms of the disease are generally clinically indistinguishable, although autosomal dominant forms may present later. 114 Responses of the clotting system to DDAVP may help – factor VIII activity and von Willebrand Factor concentrations increase after administration in AD and AR forms, whereas these responses are absent in X-linked disease as a result of mutant extrarenal V2R.
Treatment of affected patients includes the maintenance of an adequate water intake, together with the use of thiazide diuretics and a low-salt diet to reduce polyuria. Thiazides inhibit sodium reuptake in the DCT, leading to stimulation of proximal tubule salt and water reabsorption. Amiloride and NSAIDs are also used – the former reducing potassium losses and the latter inhibiting synthesis of prostaglandins which antagonise the effect of ADH. 115
Autosomal dominant polycystic kidney disease
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common monogenic disorders – with a prevalence of 1:400–1:1000 – and the commonest inherited renal disease, accounting for 5–10% of ESRF. Characterised by progressive renal cyst formation, clinical presentation may occur as a result of flank pain through bleeding into or infection of a cyst, haematuria, urinary tract infections, hypertension or renal insufficiency. Extrarenal manifestations are also common, including intracranial aneurysms, hepatic cyst formation and cardiac valvular abnormalities. Patients are increasingly identified through screening of affected families – ultrasonography provides a simple, non-invasive diagnostic tool.
ADPKD arises from mutations in two genes, PKD1 (chromosome 16p13) and PKD2 (chromosome 4q21), accounting for 85% and 15% of cases, respectively. PKD2 mutations give rise to a milder phenotype – the mean age at which ESRF is reached is 54 years for PKD1 and 74 years for PKD2. PKD1 and −2 encode polycystin−1 (PC1) and polycystin-2 (PC2) respectively – PC1 is a very large (462 kDa) protein, with a substantial extracellular N-terminal domain, 11 transmembrane repeats and a cytosolic C-terminus that interacts with a number of proteins, including PC2. PC2 is a smaller (110 kDa) integral membrane protein with a number of transmembrane domains that have structural homology to TRP channels.116,117 Indeed, PC2 (a.k.a. TRPP2) acts as a non-selective cation channel permeable to calcium. PC1 is found predominantly at the cell plasma membrane, whilst PC2 is also resident in intracellular organelles. Both are also localised to the primary cilium of renal epithelial cells where it is thought that they have a role in transducing flow sensed by the primary cilium, either by channel activity and an increase in intracellular calcium, through mediating other signalling pathways (such as the mTOR, JAK/STAT, wnt and NFAT pathways), or a combination of both. 116 PC2 also indirectly regulates intracellular calcium concentrations through interaction with the ryanodine receptor and inositol 1,4,5-triphosphate receptor. 118
The mechanisms by which PC1/2 dysfunction cause cyst formation remain unclear. A ‘two-hit’ model has been proposed for cystogenesis, whereby somatic mutation of the normal PKD allele – in addition to germline mutation of the other – is necessary for cyst formation. The rate of cystogenesis is subject to considerable variation, dependant on an individual’s genetic background and on environmental factors. Cysts form initially as dilatations in the walls of intact tubules, but as they grow they lose their connection with the parent nephron. At this stage they enlarge both through accumulation of more fluid within the cyst cavity and also through proliferation of the lining epithelium. Once cysts have formed, Cl- flux into the cyst lumen via apical membrane CFTR is thought to drive Na+ and H2O flow (through paracellular pathways) into the cyst, and hence facilitate cyst expansion. Interestingly, the small numbers of patients with both cystic fibrosis and PKD demonstrate retarded progression of renal cystic disease, 119 expression of mutant CFTR reduces cyst growth in an epithelial PKD model, 120 and small molecule inhibitors of CFTR function reduce cyst growth and renal enlargement in an animal model. 121 CFTR activity is stimulated by cAMP, the concentrations of which are elevated in renal cyst epithelial cells, and which is also a potent mitogen.122,123 cAMP thus provides an intriguing link to both fluid accumulation and cyst expansion.
Current treatment modalities in ADPKD are based on preserving renal function and addressing cardiovascular complications. Ultimately, renal transplant is the treatment of choice in those approaching/reaching ESRF. Novel therapies are emerging based on targeting the various normal functions of polycystins. The V2-receptor antagonist tolvaptan reduces cAMP production in the collecting tubule: recent phase-III clinical trials have been promising, showing significant reduction in cyst development (kidney volume) and lower rates of renal decline. 124 The mTOR inhibitor sirolimus is also under investigation, with promising initial results in animal models, although no significant results to date in human trials. 125
Conclusion
In essence, the study of channelopathies reminds us of the utility of human monogenic disorders to provide unique insights into both normal physiology and mechanisms of disease. It enables a precise diagnosis to be made in affected families, and may lead to future advances in therapeutics.
Footnotes
Acknowledgement
We would like to thank Professor Fiona Karet Frankl for her critical review of the manuscript.
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not required.
Guarantor
ACF.
Contributorship
Both authors contributed to writing the review.