
Abstract
17-Hydroxyprogesterone (17-OHP) is an intermediate steroid in the adrenal biosynthetic pathway from cholesterol to cortisol and is the substrate for steroid 21-hydroxylase. An inherited deficiency of 21-hydroxylase leads to greatly increased serum concentrations of 17-OHP, while the absence of cortisol synthesis causes an increase in adrenocorticotrophic hormone. The classical congenital adrenal hyperplasia (CAH) presents usually with virilisation of a girl at birth. Affected boys and girls can have renal salt loss within a few days if aldosterone production is also compromised. Diagnosis can be delayed in boys. A non-classical form of congenital adrenal hyperplasia (NC-CAH) presents later in life usually with androgen excess. Moderately raised or normal 17-OHP concentrations can be seen basally but, if normal and clinical suspicion is high, an ACTH stimulation test will show 17-OHP concentrations (typically >30 nmol/L) above the normal response. NC-CAH is more likely to be detected clinically in females and may be asymptomatic particularly in males until families are investigated. The prevalence of NC-CAH in women with androgen excess can be up to 9% according to ethnic background and genotype. Mutations in the 21-hydroxylase genes in NC-CAH can be found that have less deleterious effects on enzyme activity. Other less-common defects in enzymes of cortisol synthesis can be associated with moderately elevated 17-OHP. Precocious puberty, acne, hirsutism and subfertility are the commonest features of hyperandrogenism. 17-OHP is a diagnostic marker for CAH but opinions differ on the role of 17OHP or androstenedione in monitoring treatment with renin in the salt losing form. This review considers the utility of 17-OHP measurements in children, adolescents and adults.
Introduction
17-Hydroxyprogesterone (17-OHP) in serum is a critical diagnostic test for congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency which is amongst the commonest genetic disorders in man. The clinical features of CAH have been known for some time, even as far back as 1865, in an autopsy account of a patient described by Luigi de Crecchio. The basis for the problems could not be ascribed at that time. Bongiovanni published a partial translation of the original account. 1 ‘The cadaver of a “man” having a large penis with first degree hypospadias and without externally visable or palpable gonads. On dissection a vagina, uterus, fallopian tubes and ovaries were found within the abdomen’. Unusual steroids in the urine of such patients with adrenal virilism and ambiguous genitalia were first chemically characterised in 1937. 2 The nature of the steroid hormones and their link with cholesterol, sex steroids, cortisol and aldosterone became apparent over the next 20 years. 3 A biosynthetic pathway was defined and generally accepted, although deviations to the route are possible. 4 Prior to 1968, excess androgen production in CAH was defined by analysis of 17-ketosteroids (androgens) in urine, an obsolete colorimetric test since specific steroids can now be measured in blood. Pregnanetriol was known to be elevated in urine of patients with virilism before this was accepted as the main metabolite of 17-OHP.
CAH due to 21-hydroxylase deficiency was first accepted in 1955 5 and now constitutes a spectrum of disease with varying severity and biochemical features consistent with expression of mutations in the CYP21A2 gene. The laboratory often gets requests for 17-OHP during investigations of patients with androgen excess. 6 This review addresses the utility of measuring 17-OHP concentrations when investigating children, adolescent and adult patients with androgen excess. The information herein will help a clinical biochemist answer questions raised by clinicians. The scientist can help with interpreting steroid/biochemical data and suggest protocols for further investigations of patients according to the clinical presentation.
Clinical presentation of hyperandrogenism
In a newborn female infant, clitoromegaly is a sign of androgen excess in utero. In some cases, virilisation is so profound that without careful inspection, the child can get male gender assignment. Ambiguous genitalia can also reflect a failing of 46XY male development, so karyotyping is important. Androgen excess in a child and adult can cause changes in body hair, growth patterns and apocrine sweat gland function. Bone age may be advanced by excess androgen exposure in children although before the end of puberty the normal growth spurt is aborted by epiphyseal fusion and the adult patient will be short. Adrenarche needs to be considered. This is a period of development characterised by the appearance of pubic and axillary hair and increase in body odour in girls around 8 years of age and boys around 9 years of age. 7 Early adrenarche (premature adrenarche/pubarche) is a condition reflecting early development of the zona reticularis in the adrenal gland between the cortex and medulla secreting androgen. 8 CAH and adrenal tumours need to be excluded. Imaging will be needed in some patients. Premature adrenarche, once characterised, is usually a benign diagnosis. The hair growth pattern in adrenarche in the pubic and axillary areas is different to the general hair growth (hypertrichosis) with use of certain drugs such as cyclosporin, diazoxide, minoxidil and phenytoin. The hair pattern is also different to the androgenic pattern seen in adrenal tumours, polycystic ovary syndrome (PCOS) and other states where hair is on the arms, legs, back and chin.
Puberty results when pulsatile secretion of gonadotrophins activates the gonads. In precocious puberty, the secondary sex characteristics develop prematurely. This can include breast development in girls (oestrogen exposure) and growth of penis, facial hair and musculature in boys from androgen excess without the normal pubertal changes in ovaries seen on ultrasound and size of the testes. Hirsutism is defined as excess body or coarse facial hair in females in a male-like pattern. Hair growth should be assessed on chin, face, chest, abdomen, back, thighs and upper arms. A scoring system by Ferriman and Gallwey in 1961 9 is a visual assessment and a recent version with patient photographs is a useful reference point. 10 A polycystic ovarian appearance is inevitable in any of these clinical situations.11,12 PCOS is also often associated with insulin resistance and hyperinsulinemia. Insulin resistance may also develop through the metabolic effects of androgens.7,13,14 The precise mechanism for cardiovascular risk is still unclear.
In the adolescent and adult, androgen excess may be due to peripheral or central defects and tend to be rarer in origin. Anovulation, menstrual irregularity and infertility in CAH are also linked with the androgen and progesterone excess. 15 The hypothalamic-pituitary ovarian axis is disrupted and gonadotrophin releasing hormone (GnRH) pulse frequency is rapid, favouring LH secretion. Disorders of dihydrotestosterone (DHT) production may also manifest for the first time at 12–15 years of age through symptoms of androgen excess.
In rare cases, there may be additional dysmorphic features. Exogenous steroid use needs to be questioned. A dialogue between clinician and the laboratory is important in the diagnosis of an ever broadening field. A multidisciplinary team meeting should assemble clinical and laboratory experts to debate issues around difficult cases. 16
Steroid synthesis
Adrenal steroid synthesis of cortisol and aldosterone from cholesterol (Figure 1) starts with the uptake of cholesterol into mitochondria through the action of steroidogenic acute regulatory protein (StAR) followed by side chain cleavage (a cytochrome P-450 enzyme, now called CYP11A1), to liberate pregnenolone.
17
Cholesterol and pregnenolone have β-hydroxyl groups at carbon 3 (C3) and double bonds at C5 to C6 (5-ene). In the smooth endoplasmic reticulum of the adrenal zona fasciculata, pregnenolone can be hydroxylated at C17 by 17α-hydroxylase (CYP17A1) to produce 17-hydroxypregnenolone which is converted to the steroid hormone 17-OHP (17-hydroxy 4-pregnene-3,20-dione) with carbonyl at C-3 and C4 to C5 double bond (4-ene) by the action of 3β-hydroxysteroid dehydrogenase and 5,4-isomerase (HSD3B2). 17-OHP is converted to 11 deoxycortisol by C-21 hydroxylase (CYP21A2). 11-deoxycortisol has to migrate to the mitochondria for 11-hydroxylation by 11 beta hydroxylase (CYP11B1) to produce cortisol in the fasciculata zone. 17-OHP is thus an intermediate steroid in the adrenal pathway from cholesterol to cortisol but not significantly for androgens because 17-OHP is a poor substrate for CYP17A1. Cortisol and cortisone are interconverted by oxidase and reductase activities of two 11-hydroxysteroid dehydrogenases (HSD11B2 and B1 respectively). The electron transfer of the reductase is coupled with hexose-6-phosphate dehydrogenase.
18
In the outer zona glomerulosa, pregnenolone is converted to progesterone, deoxycorticosterone, corticosterone and aldosterone through the actions of HSD3B2, CYP21A2, and then CYP11B2 that has 18-hydroxylase and aldosterone synthetase activity.
Biosynthesis of steroids, including alternative path to DHT from 17-OHP via androsterone and 5α-androstane-3α,17β-diol.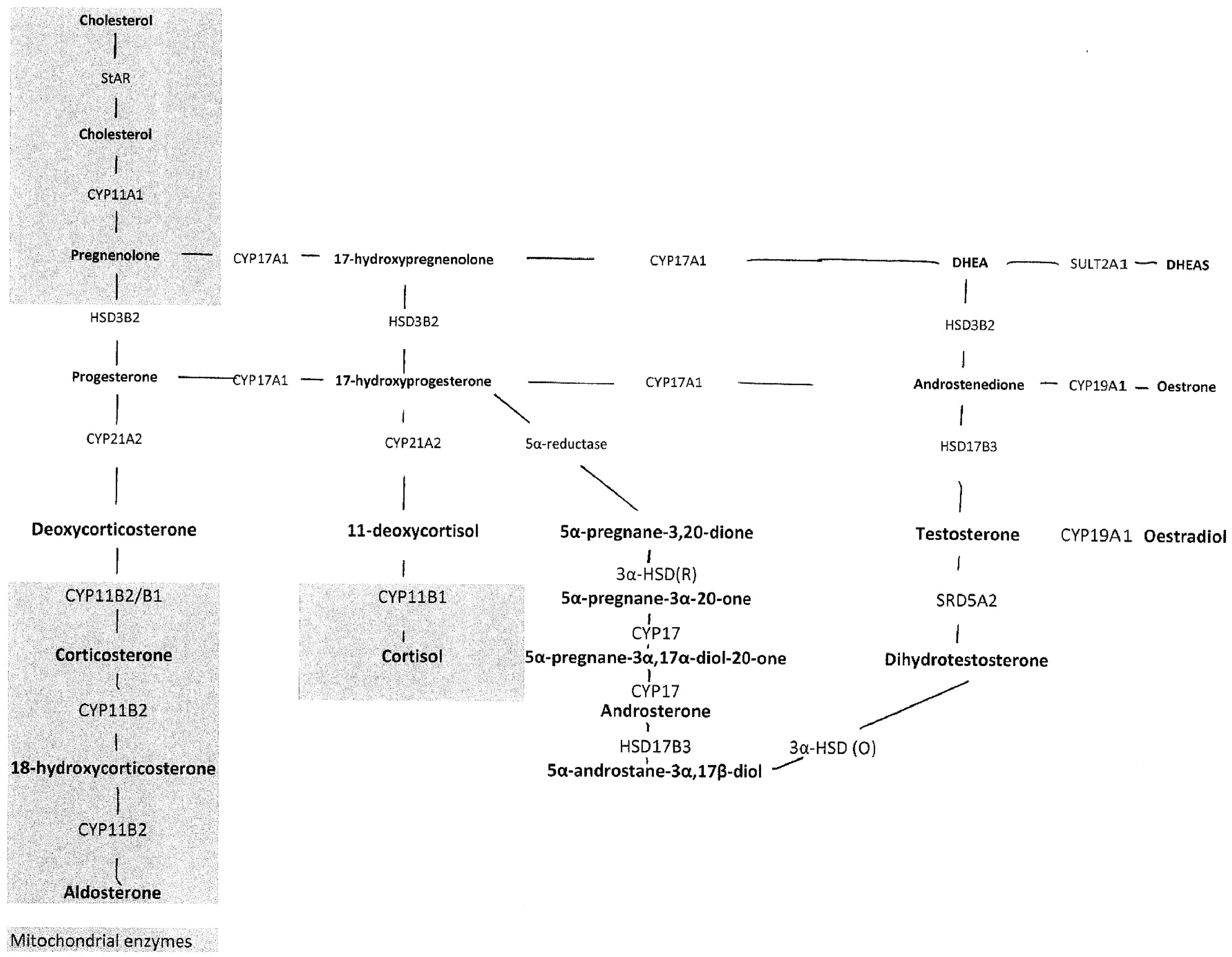
In the human adrenal cortex, CYP17A1 can efficiently convert pregnenolone to dehydroepiandrosterone (DHA) but cannot convert progesterone to androstenedione. DHA is conjugated with sulphuric acid through the action of sulphotransferase to the sulphate (DHAS). In adults, DHAS is the main steroid secreted by the adrenal cortex from the inner zona reticularis leading to circulating concentrations of 2–10 micromoles per litre. The adrenals secrete small amounts of testosterone but greater amounts of androstenedione through the action of CYP11A1, HSD3B2 and 17-hydroxysteroid dehydrogenase (an aldo-keto reductase, AKR1C3). 19 The gonads also produce 17-OHP and high concentrations of 17-OHP can be found for example in ovarian follicles. 20 17-OHP may also be converted to DHT through a ‘backdoor path’.21,22 This may provide a more efficient route to DHT under conditions of high 17-OHP accumulation as in CAH due to 21-hydroxylase deficiency. 23 In the past decade, other aspects of steroid synthesis have attracted clinical interest. A cytochrome P450-oxidoreductase (POR) is part of the electron transport system for CYP21A2 and CYP17 (and other enzymes). 24
Congenital adrenal hyperplasia
Increased concentrations of the 17-OHP in the peripheral circulation indicate a deficiency of 21-hydroxylase activity. 25 Failure to synthesise cortisol leads to an increase in ACTH through activation of the cortisol negative feedback and is the basis of CAH. Aldosterone synthesis can also be affected, so progesterone may also be elevated failing its conversion to deoxycorticosterone. There are variable consequences to glucose and electrolyte problems due to steroid deficiencies that need to be characterised. Electrolyte disturbance is the key feature of the male neonate with CAH. The classical form of 21-hydroxylase deficiency presents usually in childhood. Virilisation is the result of hyperandrogenaemia, and in many cases salt loss is the consequence of reduced aldosterone synthesis. The androgen excess in CAH must derive from 17-hydroxypregnenolone to produce DHA through the 17,20-lyase activity of CYP17A1.
The non-classical CAH form (NC-CAH) was first called late-onset (sometimes cryptic) because of the time of presentation notably in adolescents and adults. The effects of cortisol and aldosterone deficiency are not usually apparent and the presentation is due to signs of androgen excess. Precocious puberty, acne, hirsutism and subfertility are the commonest presenting features of NC-CAH requiring investigations. The prevalence of NC-CAH in women with androgen excess can be from 0.6 to 9% according to ethnic background and genotype (see reference 26 for survey). The disorder is more common in Ashkenazi Jews, Mediterranean, Middle-Eastern and Indian populations. NC-CAH is more likely to be suspected clinically in females.
Genetic basis of 21-hydroxylase deficiency
The gene for steroid 21-hydroxylase was sequenced in 1985,27–29 some 6 years before genetic mutations specific to mild forms of 21-hydroxylase deficiency were described in family studies that included asymptomatic members.30–31 The clinical presentation are usually related to the magnitude of 21-hydroxylase deficiency, being severe in neonates and milder during childhood and adults. Two CYP21 genes are located in chromosome 6 within the coding region for the major histocompatability complex and fourth component of complement, C4. The active 21-hydroxylase gene (CYP21A2) is about 30 kb downstream of a pseudogene (CYP21A1P) that does not encode protein. The genes have 98% nucleotide homology in 10 exons and 9% homology in introns. 27 Human leucocyte antigen (HLA) typing has been used to track gene mutations of CYP21A2. HLA-B14 is found in about 40% of NC-CAH haplotypes. The common mutations of CYP21A2 are genetic recombinations with CYP21A1P as well as deletions and point mutations. The mutations cause varying degrees of enzyme deficit as shown by in vitro expression studies 32 as well as genotype–phenotype correlations.33–34 In the classic defect, more than 95% of enzyme activity is lost through mutations in the CYP21A2 gene coding the protein. 27 Where the enzyme activity is lost, the phenotype is of salt-wasting disease with virilisation. When 21-hydroxylase activity is as little as 2% of normal, the phenotype is simple virilisation. If the 21-hydroxylase activity is 10–75% of normal, patients have the mild NC-CAH picture. This was recently reviewed. 35
Work with molecular models of the enzyme substituted for the absence of crystalline structure of the human 21-hydroxylase protein.36–39 NC-CAH is associated most commonly with homozygous Valine to Leucine conversion at position 281 that alters haem binding. This mutation is found in NC-CAH often as a compound heterozygote with a severe mutation on the other allele. Another mutation commonly found in NC-CAH is Proline to Leucine at position 30 that is at the point for membrane insertion and enzyme stability. The NC-CAH mutations have been found to affect 21-hydroxylase activity at oxidoreductase interactions, salt-bridge and hydrogen bonding networks and non-conserved hydrophobic interactions. 40 Multiple copies of the genes are found due to unequal crossover in meiosis at various points in the sequence. 41
The approach for genetic analysis of gene deletions, gene conversions, point mutations and small gene alterations has been reviewed recently. 42 A reference laboratory skilled in this analysis should be consulted. Different concentrations of service are offered from linkage analysis to complete sequencing of the gene. Analyses are available for detection of deletions, duplications and targetted mutations. The impact of changes in the gene on protein expression may be required in a few cases. CAH is now recognised as a spectrum of clinical presentations from mild androgen excess (NC-CAH) to prominent androgen excess with adrenal failure of cortisol (simple virilising form) and often of aldosterone (salt-wasting form) production.
Related enzyme defects
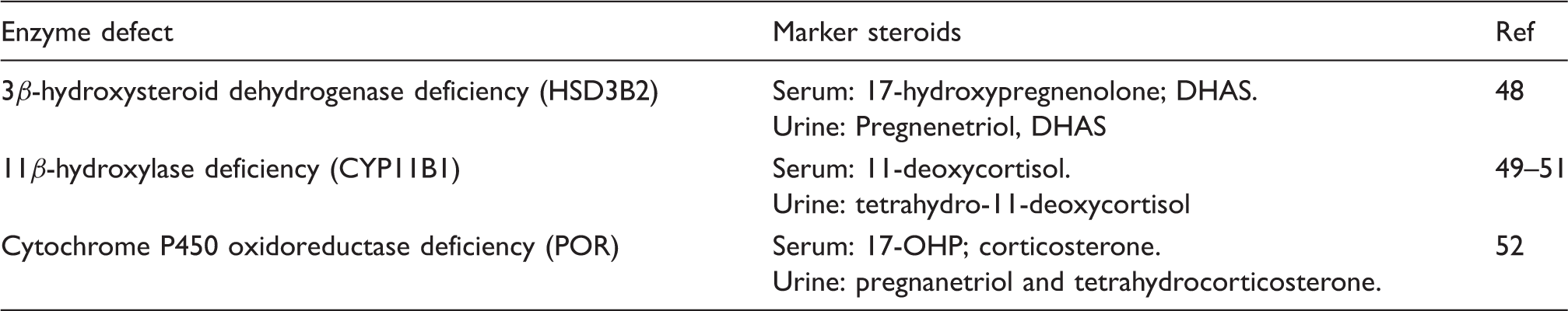
Androgen excess can also be the consequence of defects of HSD3B, CYP11B1 and POR. Cases are usually detected after birth because of typical presentations. The markers for these defects are DHAS, 11-deoxycortisol and in the case of POR both 17-OHP and corticosterone. In CAH due to 3β-hydroxysteroid dehydrogenase (HSD3B2) deficiency, the aldosterone production may also be blocked and, in the absence of this mineralocorticoid, salt loss leads to stimulation of renin production. 43 DHA is the substrate for HSD3B2 and a circulating marker for the defect along with 17-hydroxypregnenolone. In a newborn infant with CAH due to HSD3B2, the 17-OHP can be apparently elevated either through cross reaction in an immunoassay with 17-hydroxypregnenolone (and more so it’s sulphate form) or because 17-OHP is genuinely raised through the action of other 3β-hydroxysteroid dehydrogenase isoenzymes on 17-hydroxypregnenolone. 44 In CAH due to 11-hydroxylase deficiency, the production of deoxycorticosterone is increased and this potent mineralocorticoid causes renal sodium retention with suppression of renin release form the renal juxtaglomerular apparatus. 45
‘Functional’ deficiency of 21-hydroxylase and 17-hydroxylase has been described with POR defects.46,47 17-OHP and corticosterone concentrations in serum are raised although assays for the latter are not generally available. Although POR deficiency is usually detected in neonates with ambiguous genitalia and skeletal abnormalities (Antley-Bixler syndrome), mild forms can present with adrenal insufficiency and later polycystic ovaries. A urinary steroid profile (USP) shows characteristic high excretion of both 17-OHP and corticosterone metabolites. 47 Non-classic forms of CAH may also be attributable to mutations leading to mild defects in HSD3B2 48 and CYP11B149–51, POR 52 and StAR. 53
The interconversion of cortisol to cortisone can be defective in the reverse reaction by cortisone reductase (HSD11B1) and leads to a need for an increase in ACTH to raise cortisol and androgen production. This condition presents like adrenarche but is best characterised in a USP from the high ratio of cortisone to cortisol metabolites. 54 This can be distinguished from apparent cortisone reductase deficiency (ACRD) due to inactivating mutations in hexose-6-phosphate dehydrogenase gene by even lower cortisol metabolites and higher cortisone metabolites. High ACTH also leads to some increased 17-OHP production.
17-OHP analysis
The first assay for 17-OHP per se was based on competitive protein binding with radioactive 17-OHP using a crude preparation of cortisol binding globulin as ligand.55,56 Radioimmunoassays were available from 1971 57 and have remained the prominent test although are now being superceded by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) methods.58,59 Two main analytical methods are thus in use for plasma 17-OHP; immunoassay and chromatographic.
Immunoassay (IA) is the most frequently used technique for steroid measurement. 17-OHP is quantitatively displaced from its binding proteins and measured immunometrically using antibodies supposedly specific to 17-OHP. In practice, some cross-reactivity, e.g. with 11-deoxycortisol, is inevitable. Commercial assays that use antibody-coated tubes are designed for 17-OHP concentrations up to 100 nmol/L; reflecting the more common requirement, in clinical practice, for 17-OHP assays able to detect NC-CAH. If these assays are used for samples from patients with 21-hydroxylase deficiency, sample dilution is needed. There are interference problems in the neonate with steroids from the fetal adrenal zone (e.g. 17-hydroxypregnenolone sulphate) and extraction assays should be used. 60 Steroids made during fetal development are produced in lesser amounts over the first six months of life after delivery. The serum matrix in this period has many unusual steroids with the potential to produce unreliable results when using immunoassays. The free steroids are extracted into a solvent that is dried and reconstituted in a buffer for IA. Radioimmunoassay, enzyme-linked immunosorbent and time-resolved fluorescence immunoassays are in general use,61–63 though there is an expected move away from this technology.
Chromatographic methods to measure 17-OHP include LC with ultra-violet (UV) detection, 64 gas chromatography coupled with mass spectrometry (GC-MS), 58 GC with tandem mass spectrometry 65 and LC-MS/MS. 58,59 These methods have the advantage of specificity in that they separate and distinguish 17-OHP from other steroids and metabolites with visual display of results in contrast to immunoassay methods that produce results as numbers by comparison of signal with calibration signals. Some chromatographic methods require derivative formation before analysis. 65 Mass spectrometric methods are based on isotope dilution analysis against stable isotope-labelled internal standards. GC-MS approaches the standards of a reference method 66 because of the power of the GC to achieve higher separation of steroids in the sample. Method validation is important particularly with LC-MS/MS methods where ion suppression can affect results despite the improved specificity gained by the chromatographic step. 67 There are examples of methods where 17-OHP is part of a steroid panel or profile.68–74 Hyperandrogenism is an important diagnostic determinant and steroid panels using LC-MS/MS and importantly new reference ranges are now in use.75–90
17-OHP assay performance
17-OHP results with RIA and GC-MS (Supraregional Assay Service data) basally and after ACTH stimulation.

ACTH stimulated 17-OHP concentrations up to 2000 nmol/L need to be measured for classical CAH. Specialist laboratories should be used to get rapid results for newborn infants. Commercial kits will suffice in most other cases provided that the laboratory make clinicians aware of any differences in reference ranges from published data. If GC-MS or LC-MS methods are available, blood samples can be taken in the first day of life but if IA methods are used (especially direct IA without free steroid extraction) then sampling should be delayed until the third day by which time placental steroids that can interfere in the assay will have been cleared. Blood spots are used in neonatal screening programmes and saliva concentrations can be used for monitoring treatment but are not relevant to this review. Distinction between normal, heterozygotes, non-classic and classic CAH forms by measuring 17-OHP after ACTH stimulation was first described by Maria New and then others93,94 This has not been repeated with modern MS assays though basal reference ranges according to age and gender have been reported.82,83,90 An equivalent chart for GC-MS analysis is shown in Figure 2 with results closer to LC-MS/MS methods than IA methods. As a guide, 17-OHP after ACTH stimulation is >30 nmol/L (10 ng/mL) in NC-CAH.
95
Approximate ranges of 17-hydroxyprogesterone concentrations by RIA basally and 60 min after ACTH stimulation and GC-MS of heptafluorobutyrate derivatives basally and 30 min after 250 µg synacthen. Results are shown for normal subjects (N) and heterozygote carriers of CAH (Hz).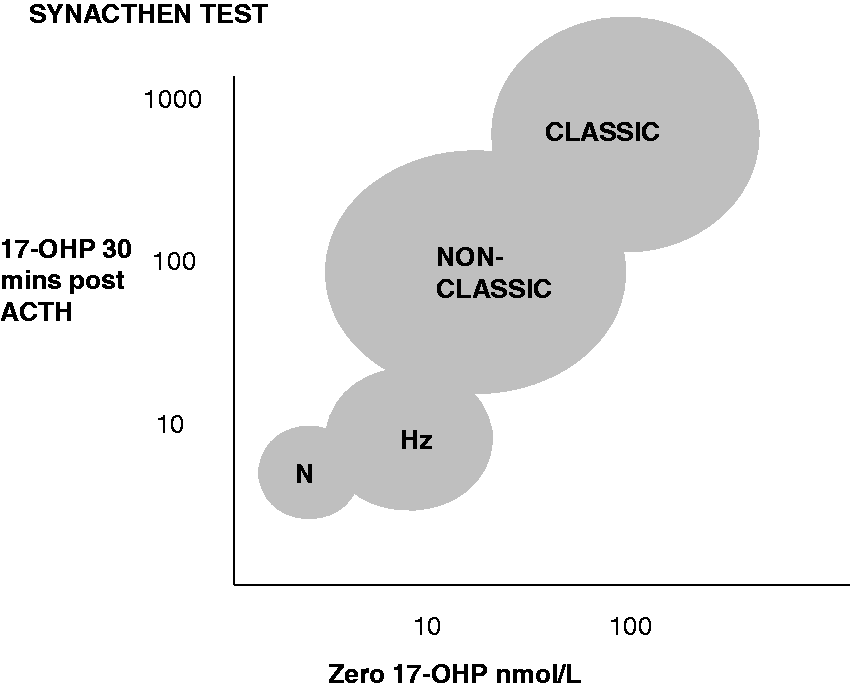
Sample requirements for 17-OHP
The laboratory must clearly communicate to clinicians/nurses/phlebotomists whether plasma or serum is suitable for the tests to diagnose CAH. When monitoring treatment, saliva and blood spot samples are used in some centres in some cases over a 24-h period with defined frequent sampling. Blood should be transferred into a plain tube or EDTA/heparin and in the case of a newborn infant with suspected CAH, before any emergency administration of corticosteroids and preferably in the early morning. Timing of the blood sample is important with any test and depends on age of the patient and the method for 17-OHP analysis. 17-OHP concentrations are lower in the afternoon than the morning. 94 The 17-OHP concentrations are higher after ovulation in the menstrual cycle, so an early phase or latest mid-follicular sample is preferred (if menstrual cycles are normal, which is often not the case in the context of a patient with high androgen production). The laboratory needs to be informed of corticosteroid treatment. In order to identify NC-CAH and diagnose heterozygotes in a female patient with menstrual cycles, the blood sample for 17-OHP and cortisol should be carried out between 08:00 h and 10:00 h in the first five days following the start of menstruation. In some cases, a basal panel of 17-OHP, testosterone DHAS, LH, FSH and thyroxine may reveal abnormal results. A normal 17-OHP does not exclude NC-CAH. Realistically, an ACTH stimulation test is required to detect NC-CAH because the basal result for 17-OHP may still be within the reference range.
Procedures for adrenal stimulation will vary but adrenocorticotrophin (ACTH 1-24, Tetracosactrin/Synacthen, 250 µg) is usually injected i/m of i/v with blood samples taken 30 min, and/or in some protocols l h, after synacthen injection. 96 Care must be taken with low-dose ACTH stimulation tests (<1 µg) 97 because peak concentrations of 17-OHP occur around 10 min and return to basal concentrations by 30 min after the ACTH injection. Many publications use inappropriate protocols with samples at 0 then at 30 and/or 60 min. If adrenal failure is suspected, a single dose of 1000 µg tetracosactrin is given and cortisol and 17-OHP concentrations are measured after 6 h and 24 h. A gradual rise in cortisol to exceed 500 nmol/L occurs in normal subjects, whereas 17OHP concentrations will not exceed 30 nmol/L.
A gonadotrophin releasing hormone agonist (GnRHa) has been used. The test is performed after dexamethasone suppression of adrenal steroids with 0.5 mg at 6 h intervals for four days. 98 On admision to hospital at 08:00 h, an iv catheter is positioned and baseline blood samples taken at 20-min intervals for 1 h. Nafarelin is then given as a single 100 µg subcutaneous injection. Blood is taken at 0.5, 1, 2, 3, 4, 8, 12, 16 and 24 h for assay of gonadotrophins. Additionally, 17-OHPreg, DHA, oestradiol, progesterone, 17-OHP, androstenedione and testosterone are determined in the samples taken at 16, 20 and 24 h after the GnRHa. A supranormal response of 17-OHP in PCOS has been recognised in response to GnRHa.
Related analytes
In order to explore other defective enzymes in cortisol synthesis (CYP11B1, CYP17A2 and HSD3B2, respectively), steroid measurements are used as the markers of substrates raised by the disorder. Androstenedione is also raised in CAH due to defects of CYP11B1 and HSD3B2 but low with defect of CYP17A2. Androstenedione is a better androgenic marker for adrenal disease than testosterone. The elevation of androstenedione can be marginal in NC-CAH due to CYP21A2 deficiency. 99 DHAS concentrations will be raised in defects of HSD3B2. 21-deoxycortisol has been proposed as an additional marker for 21-hydroxylase deficiency, 100 but the assay is not widely available although metabolites are detected in steroid profiles. Other tests useful in the investigation of adrenal function include measurement of cortisol, ACTH, aldosterone and renin principally to determine whether adrenal failure is primary or secondary. Anti-mullerian hormone (AMH) at birth is around a mean concentration of 6 ng/mL rising to mean of 120 ng/mL at six months then decreasing to around 40 ng/mL and normal results would be seen in male neonates with CAH, 101 but this test is unfortunately not widely available with acceptable turn-around times. In females, AMH is less than 2 ng/mL. Low sodium and glucose concentrations with high potassium may occur in adrenal hypofunction. High glucose and low potassium concentrations are found in adrenal hyperfunction, particularly when due to ectopic secretion of ACTH (Cushing’s syndrome). To detect a defect of cortisone reductase, an LC-MS/MS method for cortisol and cortisone in saliva could be usefully applied in some cases. 102 Some of the serum profile assays include cortisol and cortisone.77,89
A USP by high-resolution gas chromatography (GC) with GC and mass spectrometry (GC-MS) affords separation and detection of metabolites and provides a composite picture of the quantitatively major steroid biosynthetic and catabolic pathways. 103 Metabolites of cortisol, progesterone, corticosterone and DHA sulphate are readily detected, whereas those of testosterone, oestradiol and aldosterone are not. Selected ion monitoring can be used for steroids at low level to extend the range of metabolites detected. A profile is more informative on adrenocortical than gonadal steroidogenesis and especially useful for investigation of adrenal disorders in newborn infants, taking account of steroid metabolism being markedly different from that of children and adults. A profile overcomes the problems of interference in serum ligand assays from unusual steroids that are present in the neonatal period.
Clinical uses and limitations of 17-OHP measurements
Classical CAH
17-OHP analysis is important in newborns for the diagnosis of CAH due to 21-hydroxylase deficiency because this is the commonest cause of virilisation at birth. Many countries include 17-OHP in newborn screening programmes, and gestation age-related reference ranges are essential. 104 Mass spectrometric methods have been introduced as second tier tests.105–108 Transient high 17-OHP has been seen in premature infants. 109 Since the clinical condition will require life-long treatment, a sound confirmation of the defect is required. A classical CAH patient is at risk of an adrenal crisis. Merely relying on an IA result is not sufficient evidence. A genetic test or a USP is recommended to confirm classical 21-hydroxylase deficiency depending on need within the care path. Classical CAH is diagnosed in childen and adults on the basis of high 17-OHP concentrations (above 100 nmol/L) or other precursor of cortisol such as 21-deoxycortisol, accompanied in many cases with low cortisol concentrations (<100 nmol/L) and activation of the hypothalamo-pituitary-adrenal axis. The steroid metabolome is very complicated in the newborn period and changes over six months after. 110 Urine samples should be sent to specialist laboratories and reference ranges are important for interpretation.110,111 A unique polar metabolite marker for 21-hydroxylase has been partially charaterised. 112 A high ratio of pregnanetriolone (as a metablite of 21-deoxycortisol) to tetrahydrocortisone (cortisol metabolite) can also be used to confirm 21-hydroxylase deficiency and distinguish the pattern from POR deficiency. 113
Non-classical CAH
A basal plasma 17-OHP concentration >6 nmol/L (2 ng/mL) 95 but <100 nmol/L is accepted to indicate a NC-CAH, but this range may not be an appropriate cut-off for all assays and needs to be locally verified. 17-OHP after ACTH stimulation is crucial to the full diagnosis of NC-CAH. The presentation of NC-CAH is subtle and diagnosis usually requires the use of several different tests to exclude other problems. Investigation is justified because the prevalence of NC-CAH can be 1 in 1000 subjects. ACTH and cortisol concentrations may be normal. False positive and false negative results can occur with all diagnostic laboratory tests for adrenal hyperfunction. This test can confuse results if the patient has 11β-hydroxylase deficiency, where 11-deoxycortisol is the analyte of choice. However, due to cross reactions in steroid immunoassays, 17-OHP can appear raised in 11-hydroxylase deficiency and 11-deoxycortisol can be high in 21-hydroxylase deficiency. 17-Hydroxylase deficiency is suspected in a female with primary amenorrhoea and hypertension with persistently elevated progesterone. Although not reported, the progesterone could cross react in a 17-OHP immunoassay. Even cortisol can be raised in CAH due to CYP11B2 deficiency when an assay with 11% cross reaction with 11-deoxycortisol was used. 114 In a few cases, diagnosis can only be established on the basis of genetic analysis. In some cases of adrenal insufficiency, the steroid results may indicate CAH with raised 11-deoxycortisol raising the possibility of 11-hydroxylase defect. However, this would not explain salt loss. A defect in DAX-1 gene was found on further investigation. 115
Precocious pubarche
Care is needed in integrating biochemical data with other clinical investigations and possibly diagnostic tools such as imaging. Elevated DHAS for age reflects precocious pubarche/adrenarche where the patient presents with early appearance of pubic and axillary hair and increase in body odour in girls younger than 8 years and in boys younger than 9 years of age. 8 Plasma DHAS should be interpreted against age-related reference ranges because concentrations increase from <1 µmol/L at 5 years of age to between 2 and 10 µmol/L by 11 years of age. Early adrenarche is very common especially in children of Indian and African background and is usually benign. 7
Adrenarche can be distinguished in a USP from subtle defects in cortisol synthesis, but the USP is not sufficently sensitive to detect moderate elevation of 17-OHP metabolites in NC-CAH. In adrenarche, urinary androgen metabolites are raised for age so interpretation requires age-related reference ranges; cortisol metabolite excretion is also raised. These findings reflect growth of the adrenal gland and early appearance of the zona reticularis secreting androgens. 8 Dexamethasone will suppress the excess steroids.
Adrenal tumours can produce different patterns of steroids. The tumours can sometimes be palpated, and smaller masses can be seen on imaging. Plasma DHAS is a useful measurement in some cases. For some tumours, serum 17-OHP concentrations can be raised directly or circumstantially, so is not reliable. A USP is often the definitive diagnostic test and this should certainly be performed to exclude a virilising adrenal tumour because two types have been described in children according to high excretion rates of DHAS metabolites or 11-hydroxyandrostenedione metabolites.116,117 Plasma assays for 11-hydroxyandrostenedione are rarely available. Dexamethasone will not suppress the excess steroids. Androgens may also be raised in some cases of Cushing syndrome.
Precocious puberty
Outside the newborn period, 17-OHP measurements should be considered in any presentation of hyperandrogenism in a child or adolescent. The penis may be enlarged without growth of the testes in these boys, clitoromegaly may be seen in girls. Precocious puberty is an indication for 17-OHP measurement. Basal 17-OHP concentration >100 nmol/L suggests a defect of classic 21-hydroxylase and late presentation in boys and is usually the non-salt-losing, classic form (simple virilising form). In the UK, where CAH is not included in the national newborn screening programme, around 30 cases of CAH per year have been diagnosed late which may impact on quality of life. 118 High plasma renin and low aldosterone measurements would indicate further abnormalities in the electrolyte status requiring mineralocorticoid replacement treatment. If both renin and aldosterone are low, then a defect of CYP11B1 should be considered. Other causes of hyperandrogenism need to be excluded and tests should be performed for testosterone, DHAS, oestradiol, chorionic gonadotrophin (hCG), α-fetoprotein (AFP), luteinising hormone (LH), follicle stimulating hormone (FSH), thyroxine (T4) to exclude the conditions testotoxicosis (familial, male-limited precocious puberty due to activated LH receptor), tumours, central precocious puberty and primary hypothyroidism, respectively. In some cases, imaging may be necessary. Obese patients should have measurements of cortisol in 24 h urine collection, or saliva or blood taken at midnight to exclude Cushing’s syndrome. Interpretation of all results should consider pubertal stage.81,119 The timing of blood tests in early puberty is important because the activity of the hypothalamic-gonadal axis is mainly through gonadotrophin pulses during the night, so early morning blood sampling is essential. A luteinising hormone releasing hormone analogue (GnRHa) test with measurements of LH and FSH are sometimes needed in an early pubertal child. Elevated thyroid releasing hormone (TRH) in thyrotoxicosis stimulates both FSH and thyroid stimulating hormone (TSH).
Hyperandrogenism in adults
Recognition of rare NC-CAH.
The gonads also secrete 17-OHP and can contribute to slightly raised 17-OHP. Thecal cysts have been the origin of high secretion of 17-OHP in trophoblastic disease. 120 A hyperresponsiveness of 17-OHP to GnRHa testing is typical of ovarian hyperandrogenism and PCOS 121 and seems to be a result of ovarian thecal cell overactivity with down-regulation of 17,20-lyase activity. Functional ovarian androgenism did not respond to dexamethasone suppression. The overproduction of androgen in PCOS predisposes to oversecretion of oestrogen, but this is limited by a decrease in aromatase activity. Some boys present with gynaecomastia from aromatisation of androgens. 122 In functional ovarian hyperandrogenism, the LH to oestradiol ratio is kept normal in spite of excessive 17-OHP. The ovarian excess of 17-OHP can be seen in a USP where metabolites of 17-OHP (17-hydroxypregnanolone and pregnanetriol) are seen without the metabolites of 21-deoxycortisol (11-oxo-pregnanetriol) characteristically detected in a patient with CAH due to 21-hydroxylase deficiency. The ovarian source of androgens in PCOS can be confirmed by adrenal and ovarian catheterisation during dexamethasone suppression, 123 but this is not recommended for general diagnostic purposes. Anovulation, menstrual irregularity and infertility in CAH are also linked with the androgen and progesterone excess. 15 The LH to FSH ratio is often high. Several hormones play a part in the pathogenesis of acne 124 and defects in 21-hydroxylase can be detected in some cases of acne by 17-OHP measurements. Raised 17-OHP can be the consequence of activation of the HPA axis to increase cortisol production following raised SHBG from oral contraceptive use. 125
Other situations
Exceedingly rare are the disorders of sexual development with male karyotype and defects of DHT production (5α-reductase or 17-ketosteroid reductase) or action (androgen receptor). Presentation is of masculinisation around the time of puberty with infertility but not hirsutism.123–128 The measurements of testosterone, androstenedione and DHT are most useful, although 17-OHP will inevitably be requested. A high ratio of DHT to testosterone supports defect of 5-alpha reductase whilst a high ratio of androstenedione to testosterone is found with defects of 17-ketosteroid reductase. In urine, a high ratio of 5β to 5α reduced metabolites supports 5α-reducatse deficiency.
Aside from diagnosis of adrenal and gonadal disorders, the response of 17-OHP to GnRHa was found to be predictive of the ovarian response to ovarian stimulation in an in vitro fertilisation programme. 129 Serum 17-OHP concentrations correlated with antral follicle count better than did testosterone. A defect in the StAR gene affects all steroid synthesis but can sometimes present only after onset of puberty which then leads to a state of hypergonadotrophic hypogonadism.53,130 Adrenal tumours in adults producing androgens may also secrete other steroids in excess such as 11-deoxycortisol metabolites and a USP is useful to define the metabolome. 131
Monitoring treatment
This review has focussed on the role of 17-OHP measurements and other tests for diagnosis around hyperandrogenism (summarised in Figure 3). Some recent papers have addressed treatment of classical
132
and NC-CAH.133,134 In general, the approach is to treat symptoms (i.e. hyperandrogenism) unless there is evidence of adrenal failure. There is some dispute as to whether 17-OHP or androstenedione should be used for monitoring treatment of CAH. The assays for 17-OHP perform differently and this may affect interpretation of results.
135
Serum androstenedione concentration and plasma renin activity/concentration are preferred for monitoring generally because this is the basis of androgen and salt-losing side-effects of the condition. A rapid lateral flow device for 17-OHP has been developed which could be used by patients to monitor their status.
136
Oral steroid treatment does not follow the natural pulsatile pattern of cortisol release. A modified release preparation of hydrocortisone,
137
and hydrocortisone delivered subcutaneously with an insulin pump
138
is under development to improve treatment of CAH. Growth rate should be monitored in children and near normal timing of puberty is desirable. Oral contraceptive therapy can increase sex hormone binding globulin (SHBG) concentrations and decrease free testosterone in adult females, 17-OHP can also be elevated.
125
Anti-androgens can also be considered in adult patients. The Androgen Excess Society and the PCOS Society defined criteria for PCOS139,140 and a European consortium meeting in Rotterdam produced guidelines
141
that differed largely through incorporating ovarian ultrasound that is unpopular in the USA. A proposal for debate is to separate PCOS into two phenotypes,
142
and patients with hyperandrogenism may be more clearly separated as investigations move to better assays using lC-MS/MS. If pregnancy is sought, suppression of 17-OHP is required before conception due to its progestagenic activity.
15
The place of 17-OHP measurements in the investigations of hyperandrogenism in children, adolescents and adults. Stim: stimulated; A4: androstenedione; T: testosterone; LH: luteinising hormone; FSH: follicle stimulating hormone; S: 11-deoxycortisol; B: corticosterone; DHAS: dehydroepiandrosterone sulphate; 21-DF: 21-deoxycortisol; T4: thyroxine; FAI: free androgen index; Oe2: oestradiol; DHT: dihydrotestosterone; TSH: thyroid stimulating hormone; Prl: prolactin; A/c tumour: adrenal cortical tumour; PCOS: polycystic ovary syndrome; DSD: disorder of sexual development.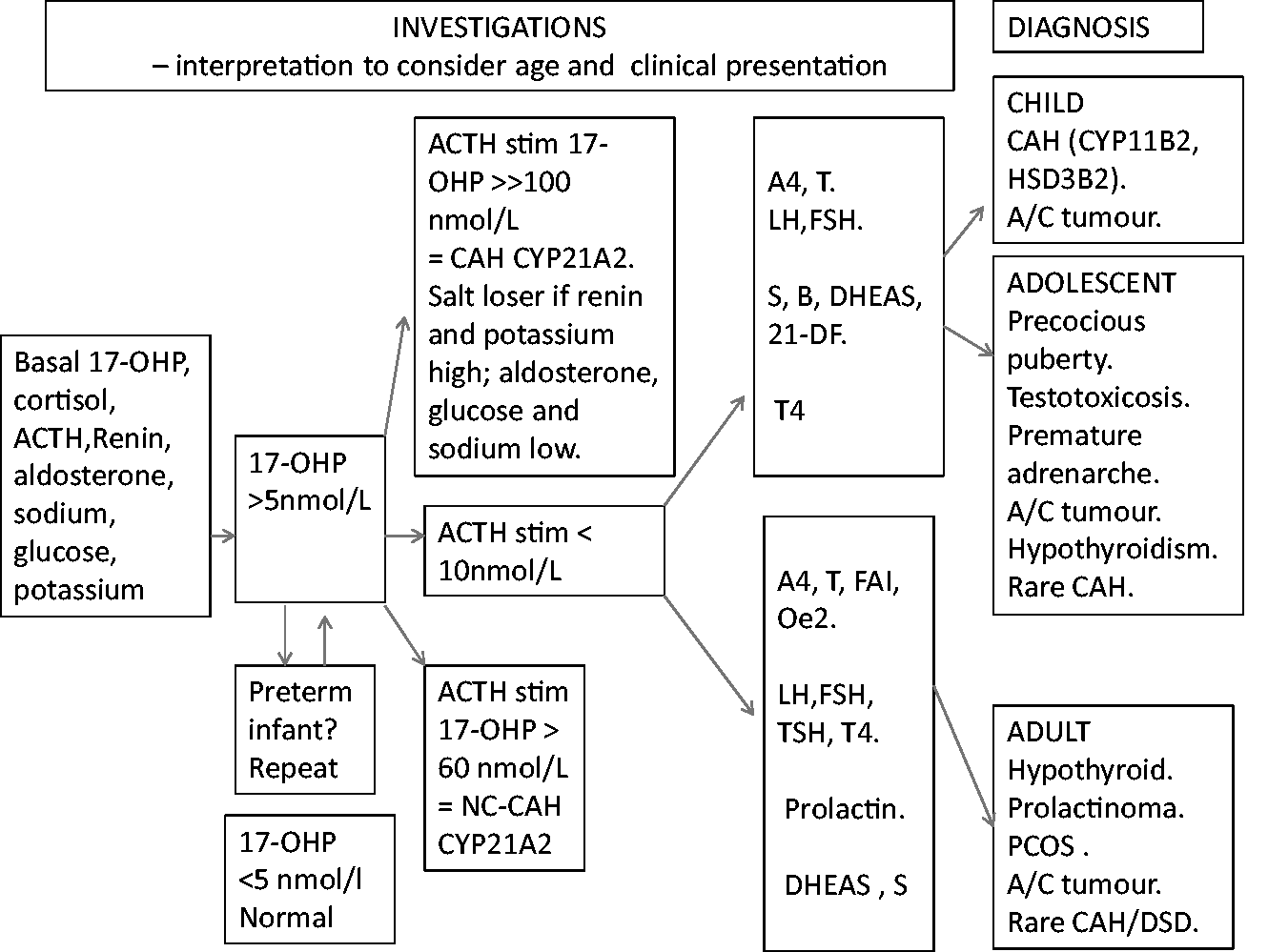
Summary
17-OHP should be measured in any state of hyperandrogenism. Elevation of plasma 17-OHP concentration above 100 nmol/L can be due to CAH, an adrenal tumour or ovarian source. A suspected case of CAH should be confirmed with a definitive test (USP or genetics) because the defect requires life-long treatment. Autonomous 17-OHP can be confirmed when dexamethasone fails to suppress the steroid. The ovarian source can be confirmed from urine steroid data and a GnRHa test. Basal 17-OHP is not reliable in other conditions. To confirm the NC-CAH form in a child or adult with clinical and/or biochemical signs of androgen excess, an ACTH stimulation test (250 microgram i.m.or i.v. tetracosactrin) with measurement of 17-OHP and cortisol at 30 or 60 minutes should be performed. An increase in 17OHP concentration >30 nmol/L indicates non-classic CAH (depending on the assay and reference data). A number of other steroid defects in steroid synthesis and metabolism can be characterised by a USP with, in some cases, further genetic tests for absolute confirmation. All laboratory results should be interpreted using reference ranges for the appropriate assay technology. This is particularly important when steroid assays of greater specificity (for example, LC-MS/MS) are introduced to the laboratory repertoire.
Footnotes
Acknowledgements
The author is grateful to members of the Clinical Science Review Committee, Dr Gill Rumsby and Dr Adel A Ismail for advice and constructive criticism.
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable.
Guarantor
JWH.
Contributorship
JWH researched the literature and is responsible for the manuscript in full.