
Abstract
Background
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is rapidly becoming the technology of choice for measuring steroid hormones. We have developed a rapid LC-MS/MS assay for the routine analysis of serum oestradiol and oestrone. The assay uses a relatively small volume and has a rapid run time.
Methods
Supported liquid extraction was performed on 250 µL of sample using methyl tertiary butyl ether. The extract was dried and reconstituted with 100 µL of 40% methanol. Online automated solid phase extraction was performed on 75 µL of extract using C18 cartridges on a Waters OSM coupled to a Waters TQS mass spectrometer. Serum samples (n = 197) were analysed by LC-MS/MS and a commercial immunoassay.
Results
The lower limit of quantitation for oestradiol and oestrone was 10 and 6 pmol/L, respectively. The coefficient of variation (CV) of the assay for oestradiol and oestrone concentrations of 125 pmol/L was <7%. The assay had a CV of 10% at 22 pmol/L for oestradiol and 5% at 16 pmol/L for oestrone. The average recovery for oestradiol was 102% and oestrone was 106%. The comparison with a commercial immunoassay gave the following equation: Immunoassay = 0.94 × LC-MS/MS + 21 pmol/L. The run time was 4.5 min per sample.
Discussion
We have developed a rapid assay for the LC-MS/MS measurement of oestradiol and oestrone which does not require derivatization in the sample preparation. The assay is suitable for routine clinical use or for clinical trials. The assay demonstrated superior performance compared to immunoassays at lower concentrations making it more suitable for use in males and patients on aromatase inhibitors.
Introduction
High-sensitivity measurement of oestradiol is advantageous in certain patient populations such as males, amenorrhoeic premenopausal females, postmenopausal females and patients receiving aromatase inhibitors. Many patients older than 40 years undergoing chemotherapy for breast cancer will develop amenorrhoea; however, this can spontaneously reverse in some women. 1 Third-generation aromatase inhibitors have also been shown to increase the rate of reversal of amenorrhoea.1,2 Therefore, establishing the ovarian function in patients undergoing therapy for breast cancer is important due to reduction in anticancer efficacy of the therapy and risks of pregnancy associated with resumed menstruation. Suggested guidelines also state that oestradiol concentrations should be below 10 pmol/L in patients taking aromatase inhibitors. 2 Unfortunately, the performance of routine immunoassays is not sufficient to measure oestradiol at these concentrations or the concentrations required for the accurate assessment of the degree of ovarian failure.
A previous study has demonstrated this poor performance of many immunoassays using comparisons with a gas chromatography-mass spectrometry (GC-MS) assay. 3 This is also supported by observations of external quality assurance data which show that a wide range of concentrations can be obtained by immunoassays when measuring samples for oestradiol over the full concentration range but particularly at lower concentrations.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is increasingly becoming the method of choice for steroid hormone measurements due to the poor specificity of immunoassays. LC-MS/MS assays also allow the measurement of other relevant hormones, e.g. oestrone, and this would assist in the calculation of total oestrogen status. Some assays have been reported in the literature for the measurement of oestradiol and oestrone using dansyl chloride derivatization4,5; however, the use of dansyl chloride has limitations such as non-specific fragmentation of derivatives and an increase in isobaric interferences. 5 Assays which do not require derivitization have been reported recently6,7; however, these require lengthy sample preparation and are difficult to introduce into the routine clinical laboratory. Chromatographic separation of oestradiol and oestrone is desirable to prevent natural isotopes of oestrone interfering with the measurement of oestradiol; however, extended chromatography to achieve this can result in lengthy run times.
We developed a direct, sensitive and rapid LC-MS/MS assay for serum oestradiol and oestrone using a novel combination of extraction techniques for a fast method that is suitable for use in the routine clinical setting.
Materials and methods
Sample preparation
Stock solutions were prepared by dissolving oestradiol or oestrone (Sigma, Poole, UK) in ultra pure methanol (Fluka, Poole, UK) and were stored at −20℃ for up to six months. Separate stock solutions were prepared for standards and quality controls (QCs). Stocks of 5 and 5.2 mg/mL were prepared in methanol for oestradiol and stocks of 5.7 and 5.5 mg/mL were prepared in DMSO (Sigma, Poole, Dorset) for oestrone. Working standards and weighed-in QCs were prepared by diluting the stock solution with phosphate-buffered saline (PBS), pH 7.4 (Sigma, Poole, UK), containing 0.1% (weight/volume; w/v) bovine serum albumin (BSA, Sigma, Poole, UK) to give concentrations of 0–2000 pmol/L and 125, 400 and 1500 pmol/L, respectively. Aliquots (300 µL) of these were stored at −30℃ for up to six months. In addition, serum QCs (360, 630 and 1220 pmol/L for oestradiol and 430, 720 and 1300 pmol/L for oestrone) were used to assess imprecision in a matrixed samples. Deuterated 17β-oestradiol-2, 4, 16, 16, 17 (D5 E2) and deuterated oestrone-2, 4, 16, 16 (D4 E1) were used as internal standards (CDN Isotopes, Quebec, Canada) at a working concentration of 10 µg/L each (37.0 nmol/L oestrone and 36.7 nmol/L oestradiol) in ultra pure methanol.
Standard, QC or sample (250 µL) was manually pipetted into a 1.5-mL microcentrifuge tube (Clinicon, Petworth, UK). To this, 10 µL of working internal standard and 150 µL of de-ionized water were added. After a 10-s vortex, all diluted sample was transferred to the wells of a supported liquid extraction plate (SLE + Biotage, Uppsala, Sweden), while it was positioned on a vacuum manifold. A vacuum was applied to the plate to draw the entire sample into the diatomaceous earth layer for approximately 10 s. The plate was then allowed to rest for 5 min during which a 2-mL square well 96-well collection plate (Porvair, Leatherhead, UK) was positioned under the SLE plate. Following this, 900 µL of methyl tertiary butyl ether (MTBE) was loaded into each well and allowed to pass through the plate into the collection plate. When no MTBE was visible in the wells of the SLE plate, the vacuum was reapplied to draw the remains of the MTBE into the collection plate. The MTBE was evaporated to dryness using a Vacmaster 96 (Biotage, Uppsala, Sweden). The extract was reconstituted with 100 µL of 40% ultra pure methanol. The block was heat-sealed (Thermo, Hemel Hempstead, UK), vortexed for 1 min and then centrifuged at 8000 g for 5 min. Following centrifugation, the plate was transferred directly to the autosampler for analysis; 75 µL of sample was injected into the Acquity ultra high pressure liquid chromatography (UPLC) and on-line solid phase extraction (SPE) system (OSM; Waters, Manchester, UK) using partial loop mode.
Chromatography and solid phase extraction
The mobile phases utilized were (A) distilled water with 0.3 mmol/L ammonium fluoride (Sigma, Poole, UK) and (B) methanol (ultra pure grade, Fluka, Poole, UK). MassTrak C18 10 µm cartridges were used for on-line sample extraction. The cartridges were conditioned with 0.5 mL of ultra pure methanol and then equilibrated with 0.5 mL of distilled water. The sample (75 µL) was loaded onto the cartridge using 0.5 mL of distilled water. The cartridge was washed with 0.25 mL of 30% ultra pure methanol. Mobile phase was then passed through the cartridge for 0.75 min to facilitate elution on to the analytical column. The analytical column used was a 2.1 × 30 mm 1.8 µm Waters Acquity UPLC HSS C18 SB column coupled to a Waters Vanguard filter. Oestradiol and oestrone were eluted from the cartridge and analytical column isocratically using 57% mobile phase B at a flow rate of 0.45 mL/min. After 1.5 min, the composition was stepped up to 100% B for 1 min before returning to starting conditions for 0.75 min. Both guard and analytical columns were maintained at 50℃.
Mass spectrometry
The eluate was injected from the LC directly into a XEVO™ TQS tandem mass spectrometer (Waters, Manchester, UK). MassLynx NT 4.1 software was used for system control and the MassLynx TargetLynx programme allowed data processing. This software used the area of the detected peaks, 1/x weighting and linear least squares regression to produce a standard curve. The 1/x weighting gives greater accuracy at lower concentrations. The mass spectrometer was operated in electrospray negative mode, the capillary was maintained at 2.5 kV and the source temperature was 150℃. The desolvation temperature and gas flow were 600℃ and 1000 L/h, respectively. The source offset was maintained at 50 V. The quantifier and qualifier transitions identified were m/z 271.1 > 145.1 and 271.1 > 183.1 for oestradiol, respectively, and 269.1 > 159.1 and 269.1 > 183.1 for oestrone, respectively. The internal standards had transitions of m/z 276.1 > 147.1 for D5 oestradiol and 273.1 > 147.1 for D4 oestrone. Transitions were monitored in multiple reaction monitoring mode, with a dwell time of 0.025 s.
Validation
The assay was validated against published acceptance criteria for linearity, precision, recovery and sample stability. 8
Ion suppression
Ion suppression is a matrix effect which occurs when compounds in a sample compete with the analyte for ionization in the source. To investigate this, we infused a 30-nmol/L solution of internal standards in 50% (v/v) ultra pure methanol/water directly into the mass spectrometer to give a constant background signal. This concentration was chosen as it allowed visualization of the increase in background signal, while concentrations in the physiological range did not. Extracted serum samples (n = 6) were injected simultaneously via the autosampler. A reduction in the background signal is observed when ion suppression is occurring and ion suppression is deemed significant if a reduction in signal of >10% is observed where the compound of interest elutes.
Accuracy
The accuracy of the assay was determined by the analysis of certified reference materials BCR 576, 577 and 578 for oestradiol. Accurate measurement of this matrix-matched reference material would also confirm the lack of matrix effects given the use of PBS-based calibrators. To further confirm this, the method of standard additions was used with serum and PBS-based spiked samples with a target of within 5% of each other. Unfortunately, no suitable reference materials were available for oestrone.
Recovery
Four different concentrations of oestradiol and oestrone (200, 400, 800 and 1500 pmol/L) were spiked into six different serum samples with concentrations ranging from 11 to 57 pmol/L for oestradiol and 68 to 164 pmol/L for oestrone. The recovery was calculated from measured compared to expected concentrations and absolute ion counts were compared for any variances between matrices.
Imprecision
Imprecision of the method was assessed against a range of concentrations using PBS-based and serum-based QC samples. These samples were analysed daily for 10 days to calculate inter-assay imprecision. To determine intra-assay imprecision, the same samples were analysed 10 times within one batch. Further serum samples with lower concentrations were also analysed for intra-assay imprecision. Percentage deviation was calculated from the difference between mean observed and nominal concentrations to assess bias for the PBS-based calibrators.
Linearity
To evaluate linearity of the calibration curves, three curves were prepared and analysed in separate batches. The ratios of analyte peak area to internal standard peak area were plotted against oestradiol or oestrone concentration in pmol/L. Calibration curves were judged linear if the correlation coefficient (R2) was better than 0.9900 as calculated by weighted linear regression.
Limit of quantitation and detection
The lower limit of quantitation (LLOQ) was defined as the concentration for which 10 replicates of serum-based samples prepared with low concentrations of oestradiol or oestrone gave a CV of less than 20%. The lower limit of detection was defined as the concentration which gave a peak with a 3 to 1 signal to noise ratio.
Specificity
Solutions of various related steroids, both natural and synthetic, were prepared in 50% (w/v) methanol/water and injected directly into the mass spectrometer via the autosampler, without internal standard. Steroids tested to a final concentration of 1000 nmol/L were testosterone, dehydroepiandrosterone sulphate, dehydroepiandrosterone, cortisol, progesterone, aldosterone, cortisone, corticosterone, 17 hydroxyprogesterone, 11 deoxycortisol, 21 deoxycortisol, pregnenolone, epitestosterone, dihydrotestosterone and androstenedione. Others tested to 1 mg/L were fludrocortisone, dexamethasone, methylprednisolone, budesonide, prednisone, beclometasone, fluocinolone, cyproterone, ethinyl oestradiol, norethisterone, triamcinolone and prednisolone.This excess concentration was chosen to allow easy identification of any potentially interfering peaks. The retention times of any compounds found to give a signal in the specific channels for oestradiol, oestrone or their internal standards were subsequently assessed to determine if they would cause interference.
Stability
We investigated the stability of oestradiol and oestrone after supported liquid extraction had taken place. Serum samples (n = 35) were prepared in duplicate. One plate was analysed immediately, while the second plate was stored at 4℃ for 24 h before analysis. The results of the duplicates were compared using a paired sample t-test to determine if a significant difference was apparent.
Comparative assays
Serum samples (n = 197) were analysed by the LC-MS/MS method and compared to a commercially available immunoassay (Abbott Architect, Abbott, Maidenhead, UK).
Ten serum samples from females taking aromatase inhibitor therapy were analysed.
Serum samples (n = 91) from adult males analysed by this method were also previously analysed by GC-MS. 9 Samples were collected and analysed according to the European Male Ageing Study (EMAS) protocol. 10
Statistical analysis
All statistical analyses were performed using Analyse-it software (Analyse-it Software Ltd, Leeds, UK).
Results
Both oestradiol and D5 oestradiol had retention times of 1.04 min and oestrone and D4 oestrone had retention times of 0.92 min (Figure 1). The cycle time, injection to injection, was 4.5 min. There was no significant suppression of ionization in the region of the chromatogram where oestradiol or oestrone elutes. The mean recoveries following the addition of oestradiol and oestrone to six serum samples was 102% (range 96–107%) and 106% (range 100–112%), respectively.
Chromatogram of a serum sample containing 12 pmol/L and 49 pmol/L of oestradiol and oestrone, respectively.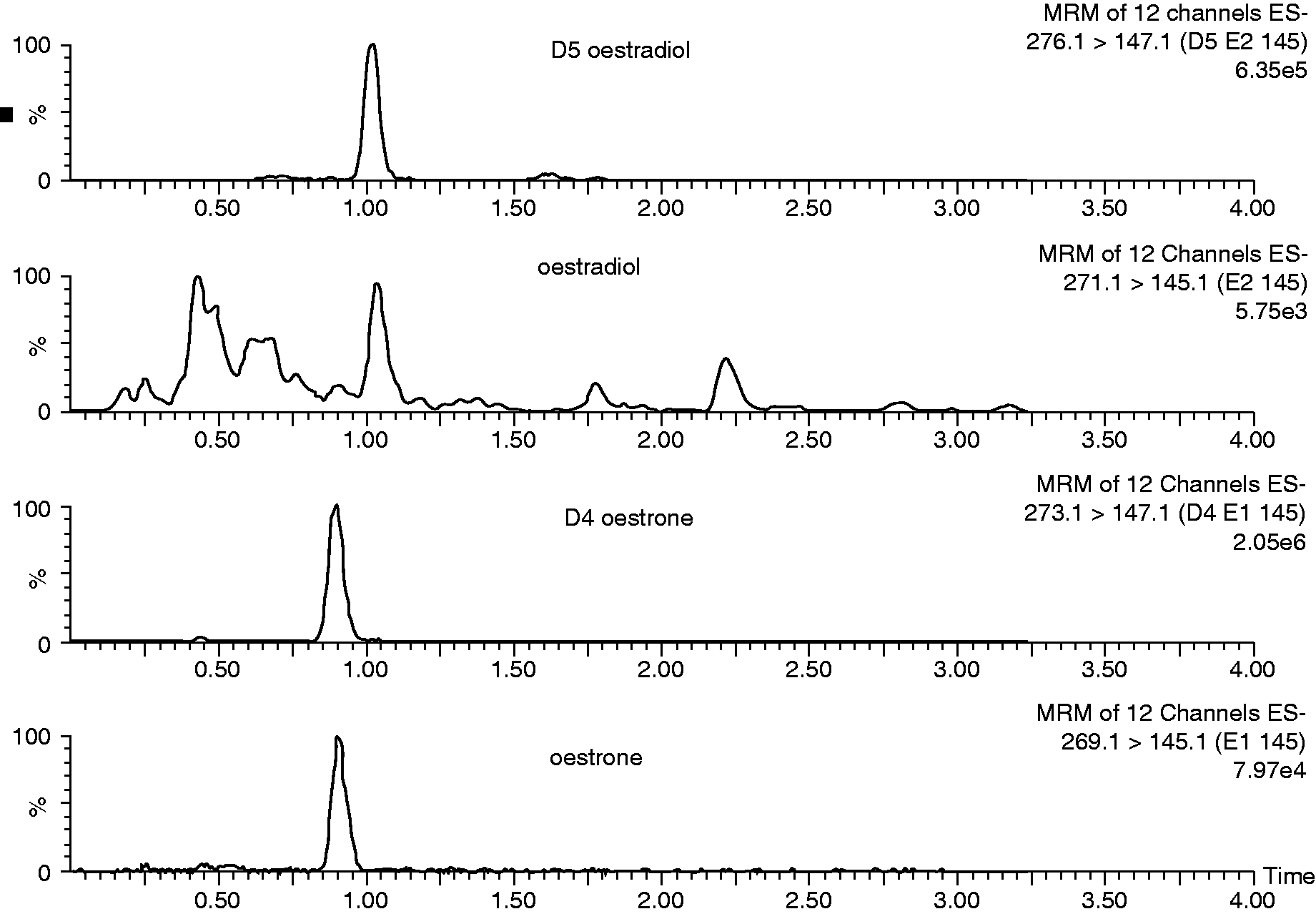
Analysis of matrix-matched reference materials gave concentrations within the stated measurement uncertainty for each of the target concentrations. The method of standard additions showed that the PBS containing 0.1% (w/v) BSA and serum spiked with 0–2000 pmol/L of oestradiol and oestrone were within the 5% target.
The inter-assay imprecision for the PBS-based QCs was 5.4, 3.7 and 4.9% for oestradiol and 5.3, 3.8 and 5.1% for oestrone at concentrations of 125, 400 and 1500 pmol/L. The inter-assay imprecision for the serum-based QCs was 3.4, 5.0 and 4.2% for oestradiol at concentrations of 352, 615 and 1184 pmol/L and 2.5, 3.4 and 2.1% for oestrone at concentrations of 425, 716 and 1300 pmol/L. The intra-assay imprecision for the PBS-based QCs was 3.1, 3.5 and 4.0 for oestradiol and 4.0, 3.4 and 5.0% for oestrone. The intra-assay imprecision for the serum QCs were 3.6, 4.1 and 2.4 pmol/L and 2.4, 3.1 and 1.9% for oestrone. Further serum samples were analysed for intra-assay imprecision at lower concentrations. A serum sample of 19 pmol/L gave a CV of 6.7% for oestradiol and a sample of 15 pmol/L gave a CV of 4.7% for oestrone. All means were within 8% of the PBS-based QC targets.
Standard curves were made by plotting oestradiol or oestrone concentrations on the x-axis and oestradiol or oestrone/D5 oestradiol or D4 oestrone peak area ratios on the y-axis. The curve was linear over the standard range and was reproducible between batches. The curves showed good correlation with the assigned standard values with an R2 value of 0.99.
The lower limits of detection were 8.0 and 3.9 pmol/L for oestradiol and oestrone, respectively. The lower limits of quantitation were 10 pmol/L for oestradiol and 6 pmol/L for oestrone. Norethisterone and ethinyl oestradiol gave peaks at a retention time similar to oestradiol and oestrone in the transitions for both compounds. The peaks obtained by ethinyl oestradiol are likely to be below the LLOQ at pharmacological concentrations; however, a more significant interference was seen with norethisterone. The retention time for this was slightly earlier than oestradiol and could be discriminated from oestradiol. None of the other steroids, endogenous or exogenous, gave a response above the LLOQ at the retention time for either compound or their internal standards.
The extract was stable for 24 h at 4℃ for oestradiol (P = 0.07); however, a statistically significant difference was observed for oestrone (P = 0.02) but the differences were not clinically significant.
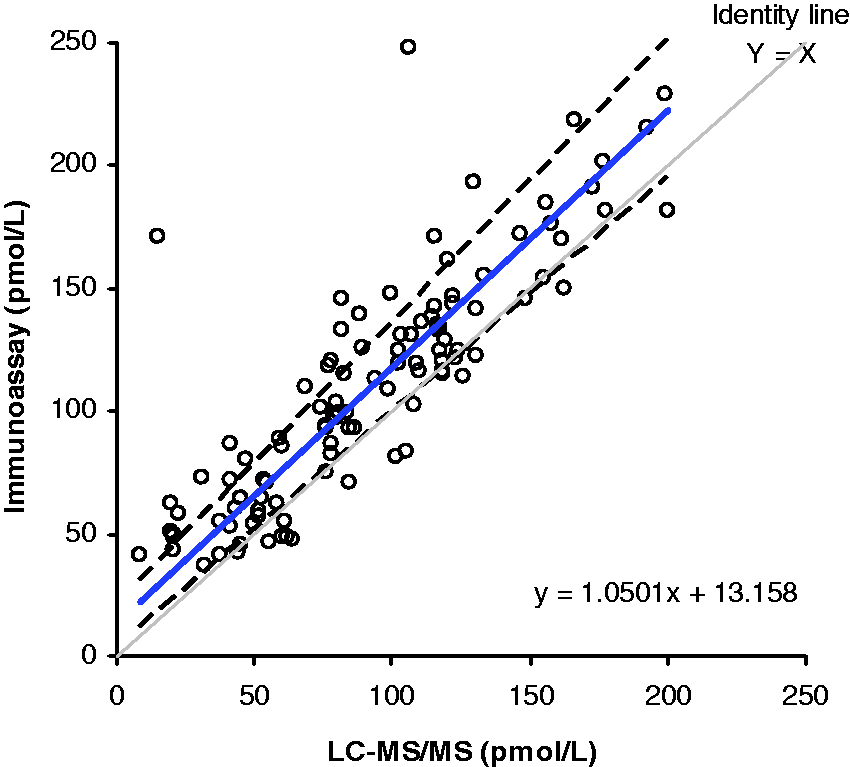
A method comparison of serum samples analysed by this assay (n = 197) and an oestradiol immunoassay (Abbott Diagnostics, Maidenhead, UK) gave a Bland–Altman bias plot shown in Figure 2 with a mean bias of 3.2 pmol/L (95% CI = 3.8–10.2 pmol/L). Passing–Bablok analysis gave an equation of Abbott = 0.94 × LC-MS/MS + 21.07 pmol/L. The correlation coefficient (R2) was 0.98. Of these samples, 105 had oestradiol concentrations <200 pmol/L. The Passing–Bablok method comparison for these lower concentration samples was Abbott = 1.05 × LC-MS/MS + 13.16 pmol/L. This group displayed a mean bias of +19.2 pmol/L (95% CI = 14.1–24.2 pmol/L) and a correlation coefficient (R2) of 0.84 (Figure 3). Analysis of the reference materials was also performed on this assay. The low level gave a value that was 17% higher than the target concentration, the medium level was 18% lower than the target concentration and the high level was estimated at 4% lower than the target concentration.
Comparison of oestradiol concentrations (n = 197) when measured using the Abbott immunoassay and the LC-MS/MS assay. The correlation coefficient (R2) was 0.98. Comparison of low oestradiol concentrations (<200 pmol/L, n = 105) when measured using the Abbott immunoassay and LC-MS/MS assay. The correlation coefficient (R2) was 0.84.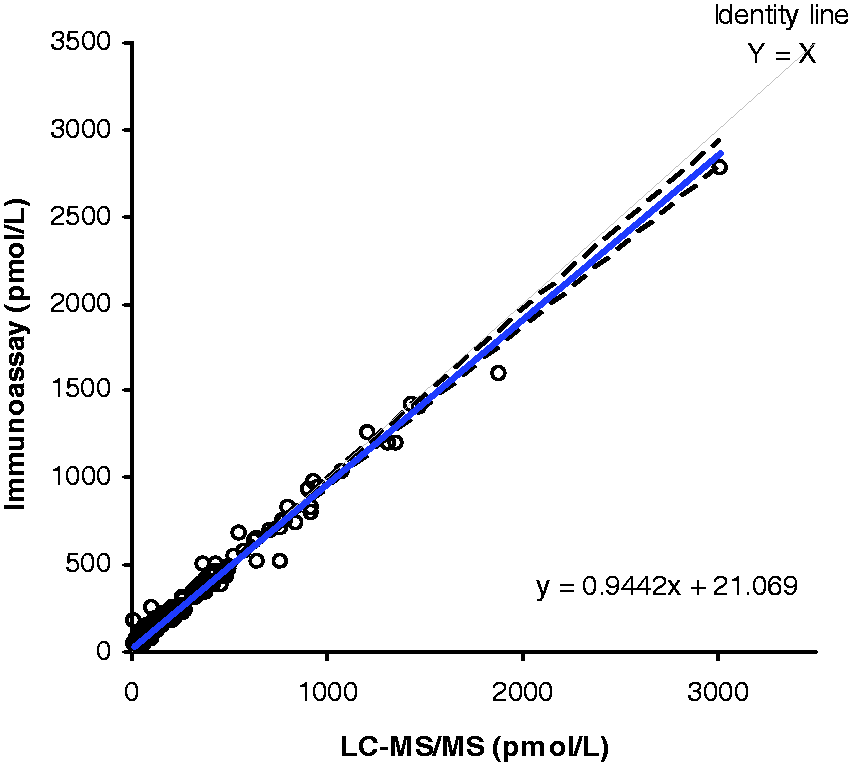
Preliminary measurement of 10 serum samples taken from female patients undergoing aromatase inhibitor therapy for breast cancer showed all oestradiol and oestrone concentrations to be below the LLOQ for the assay.
The method comparison against GC-MS gave the equation GC/MS = 1.01 × LC-MS/MS – 0.56 pmol/L. The Pearson correlation coefficient (R2) was 0.92. The mean bias (Figure 4) was 1.2 pmol/L (95% CI = 1.6–4 pmol/L). The correlation between LC-MS/MS and GC/MS showed good agreement whereas the correlation between the LC-MS/MS and immunoassay was poor particularly at low concentrations. This confirms previous data using this GC/MS assay showing the poor performance of immunoassays for measuring oestradiol.
9
These results also confirm the superiority of LC-MS/MS measurements over immunoassay.
Comparison of oestradiol concentrations (n = 91, males) when measured using a GC-MS assay and the LC-MS/MS assay. The correlation coefficient (R2) was 0.92.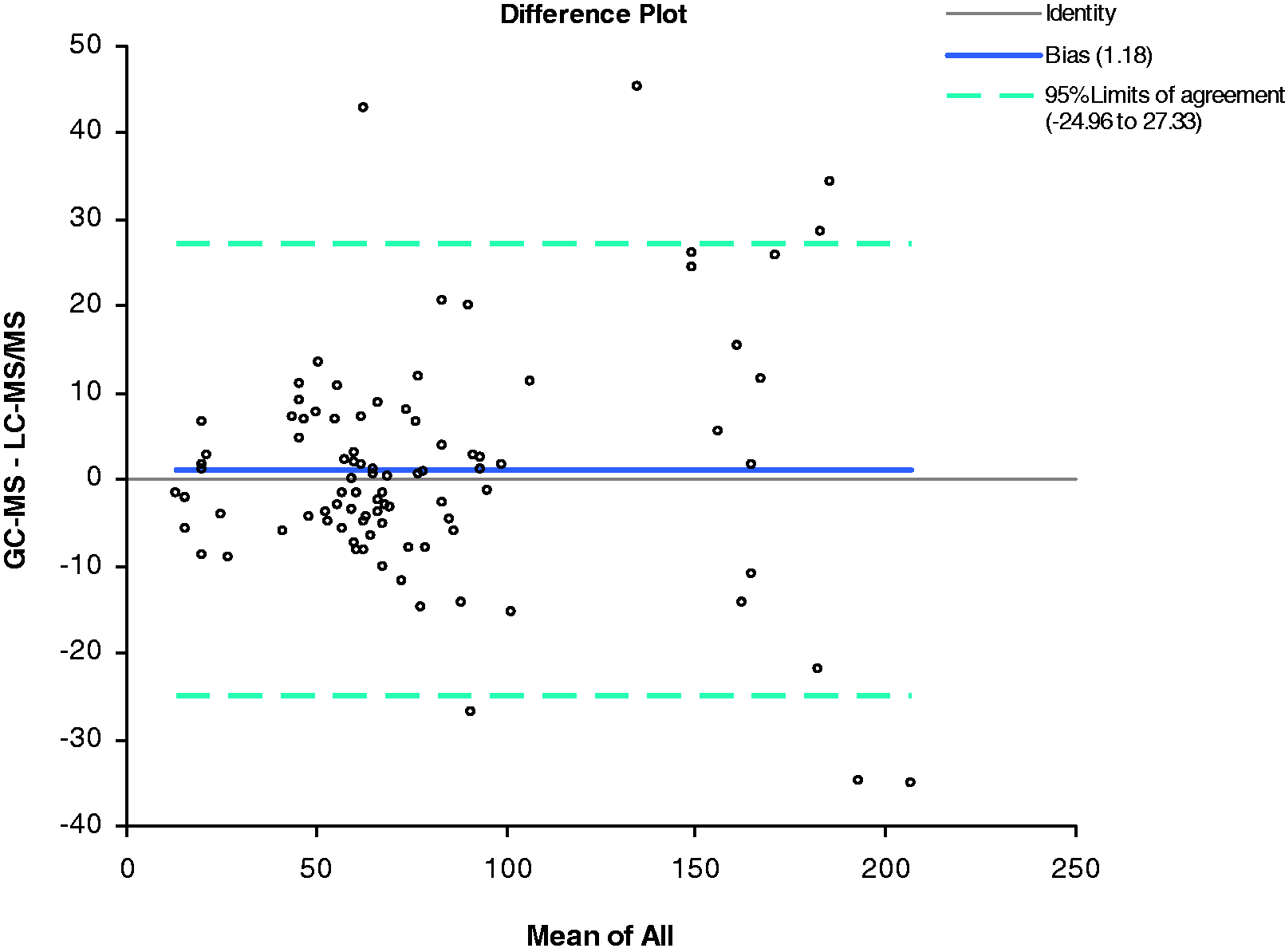
Discussion
We have developed a novel direct assay for the measurement of oestradiol and oestrone suitable for routine clinical use. The selectivity of the chosen column allowed a combination of adequate resolution of the two compounds with a fast run time. Adequate resolution is required to prevent natural isotopes of oestrone interfering in oestradiol measurement in contrast to a previously reported direct assay where this was not ensured. 6 The sample preparation in this assay uses a unique combination of supported liquid extraction and on-line SPE using a Waters OSM. This approach, coupled with the short chromatography column allows for rapid sample preparation and analysis; it takes approximately 90 min to prepare 96 samples and is hence a high-throughput assay. The introduction of the OSM for extraction allows a large volume of extract to be loaded onto a short column without deterioration of peak shape which is observed without the OSM. The large volume of extract is required to optimize the LLOQ. In addition, the use of fluoride in the mobile phase increases sensitivity by facilitating more efficient ionization. 11
The lack of ion suppression, good alignment of reference materials and the use of the method of standard additions support the use of PBS-based calibration material. The assay also demonstrates excellent recovery, precision and linearity.
The peaks observed in the chromatograms from ethinyl oestradiol are not likely to be of any significance at pharmacological concentrations; however, norethisterone can be observed as a shoulder to the oestradiol peak in the chromatogram. There are no recent guidelines recommending the measurement of oestradiol in patients receiving this drug; therefore, this interference may not be clinically relevant, but it is worthwhile to be aware of this as a comment may be appended to the reports of routine samples if this is observed.
The immunoassay that we evaluated against our LC-MS/MS method showed a negative bias at higher oestradiol concentrations and a positive bias at lower oestradiol concentrations. These results were similar to a previous study using the same immunoassay but with GC-MS as the comparator. The method comparison and analysis of reference materials using the immunoassay supports previous findings of the inaccuracy of this type of analysis, 12 particularly at the low levels of oestrogens.
As GC-MS is considered the gold standard for oestradiol measurement, this assay was compared against an established GC-MS assay for low concentrations expected in males. An excellent agreement was observed, suggesting the suitability of this assay at these lower concentration ranges.
The assay was very sensitive with lower limits of quantitation of 10 and 6 pmol/L for oestradiol and oestrone, respectively. This makes the assay applicable to measuring samples in patient groups where immunoassays have previously been insufficiently sensitive and specific.
Initial investigation into the oestrogen status of samples taken from patients on aromatase inhibitors show results to be below the LLOQ for both oestradiol and oestrone indicating adequate suppression. 1
Footnotes
Acknowledgements
The authors would like to acknowledge the help received from Martin Eastwood of Waters particularly regarding column selection.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable.
Guarantor
LJO.
Contributorship
The development and validation of the assay described was performed by LJO and BK. FCW contributed to the GC-MS comparison. All authors have contributed to the manuscript.