
Abstract
Inactivation of p53 is one of the most frequent molecular events in neoplastic transformation. Approximately 60% of all human tumors have mutations in both p53 alleles. Wild-type p53 activity is regulated in large part by the proteosome-dependent degradation of p53, resulting in a short p53 half-life in unstressed and untransformed cells. Activation of p53 by a variety of stimuli, including DNA damage induced by genotoxic drugs or radiation, is accomplished by stabilization of wild-type p53. The stabilized and active p53 can result in either cell-cycle arrest or apoptosis. Surprisingly, the majority of tumor-associated, inactivating p53 mutations also result in p53 accumulation. Thus, constitutive elevation of p53 levels in cells is a reliable measure of p53 inactivation, whereas transiently increased p53 levels reflect a recent genotoxic stress. In order to facilitate noninvasive imaging of p53 accumulation, we here describe the construction of a p53-luciferase fusion protein. Induction of DNA damage in cells expressing the fusion protein resulted in a time-dependent accumulation of the fusion that was noninvasively detected using bioluminescence imaging and validated by Western blot analysis. The p53-Luc protein retains p53 function because its expression in HCT116 cells lacking functional p53 resulted in activation of p21 expression as well as induction of apoptosis in response to a DNA damaging event. Employed in a transgenic animal model, the proposed p53-reporter fusion protein will be useful for studying p53 activation in response to exposure to DNA-damaging carcinogenic agents. It could also be used to study p53 stabilization as a result of inactivating p53 mutations. Such studies will further our understanding of p53's role as the “guardian of the genome” and its function in tumorigenesis.
Introduction
The p53 gene product plays an important role in tumor suppression as evidenced by the fact that 60% of all human cancers carry a mutation in the p53 gene. In addition, patients having the Li-Fraumeni syndrome is due to inheritance of a defective p53 gene and this population is much more prone to developing cancer. Similarly, genetically engineered mice lacking the p53 gene are also more prone to developing cancer. Recent reports have also shown that mutations in p53 not only predispose one to cancer, but that the efficacy of chemotherapy is dependent of the presence of functional p53 [1].
Studies on the role of p53 as a tumor suppressor have been complicated because of the fact that p53 has two seemingly opposite functions. First, in response to DNA-damaging events, p53 has the ability to inhibit cell cycle progression at the G1/S border. This effect has been shown to be the result of the ability of p53 to transcriptionaly activate the p21 gene, which is a potent inhibitor of G1 cyclin-dependent kinases. This role of p53 is logical in that if a cell has suffered a DNA-damaging event, replicating the damaged DNA (which occurs in S-phase) could result in propagation of an altered DNA sequence. Therefore, inducing cell cycle arrest before S-phase entry is important to maintain genomic fidelity. In its role as a transcription factor, p53 can also induce genes that are involved in nucleotide excision repair, which are required to repair the DNA damage [2]. In contrast to these protective functions of p53, a second function of p53 is to induce apoptosis following DNA damage in certain cell types. This seemingly contradictory function of p53 has been suggested to be important in cases where the incurred DNA damage is beyond repair and thus rather than propagate a cell that has undergone mutagenesis p53 by transcriptionaly activating genes such as Bax (positive regulator of apoptosis) and death receptors such as killer and Fas [3] which activate the cellular suicide program.
Because p53 negatively regulates cell growth by promoting G1 arrest and by inducing apoptosis, there must be a mechanism in place that suppresses p53 function in dividing tissues. This is accomplished by a proteosome-dependent mechanism which ensures that in actively dividing cells the level of p53 protein is very low. This proteosome-dependent degradation of proteins plays an important role in regulating proteins involved in activities such as cell cycle progression, cell differentiation, stress response, and apoptosis. Degradation of p53 first requires posttranslational ubiquitination of p53 in a series of reactions. MDM2, which has been shown to have ubiquitine ligase activity, plays a major role in the degradation of p53. MDM2 binds to the aminoterminus of p53 which leads to ubiquitination of p53 [4]. The C-terminus of p53 has been proposed to be required for proteosome-mediated degradation.
In order for p53 to transactivate target genes in response to a DNA-damaging event, its degradation must first be inhibited which results in accumulation of the p53 protein (Figure 1). Accumulation of p53 is followed by activation of DNA binding activity through additional posttranslational modification (e.g., phosphorylation), providing p53 with the ability to transcriptionally turn on specific genes. In addition, the inhibitory activity of MDM2 must be overcome. This has been proposed to occur by modification of p53 as well as MDM2 such that they fail to interact thus preventing p53 ubiquitination and degradation.
As stated earlier, mutations in the tumor suppressor gene p53 are common in human cancer, accounting for 60% of all cancers. However, mutations in p53 are quite different from those found in most tumor suppressors. The tumor suppressor genes Rb (retinoblastoma) and APC (adenomatous polyposis coli) are commonly inactivated by nonsense mutations that cause truncation (and therefore loss of function) or instability of the protein. But in p53, more than 90% of the mutations are missense mutations that change the identity of a particular amino acid. Changing the amino acid sequence results in a change in the conformation such that the ubiquitination machinery fails to recognize the protein and thus mutant p53 does not get rapidly degraded and accumulates within the cell.
In general, genotoxic stress to cells results in a transient increase in p53 levels, while long-term stabilization and accumulation of p53 often results from mutation of the p53 coding sequence. Based on this observation, the goal of the present study is to develop a strategy which enables noninvasive imaging of p53 activation in response to genotoxic stress (carcinogenesis) as well as the presence of mutant p53. We hypothesized that fusion of the p53 gene with a coding sequence for a reporter gene such as luciferase would result in degradation and/or accumulation of the reporter in a manner similar to p53. Assuming that reporter activity can be used as a surrogate for p53 levels, this strategy would enable noninvasive detection of increased p53 levels in response to genetoxic stress as well as mutational events within p53 using bioluminescence imaging.
Materials and Methods
Construction of the p53-Luc Fusion
Four synthetic oligonucleotides were used: The first (5′ p53) containing the sequence: ggaattc aagctt cactgcc
To construct the fusion, the p53 sequence was amplified using primers 5′ p53 and 3′ p53 while luciferase was amplified using primers 5′ luciferase and 3′ luciferase. The resulting fragments were purified, and 20 ng of each was used in a second PCR reaction using primers 5′ p53 and 3′ luciferase. Because the 3′ sequence of the p53 fragment and the 5′ sequence of the luciferase have complementarity over 45 bp, the two PCR fragments “join” to generate a single PCR product which codes for the fusion protein. This sequence was then cloned using the EcoRI sites at the 5′ and 3′ends into an expression vector (pCDNA I, Invitrogen, CA).

A strategy for imaging of p53 accumulation in response to genotoxic stress and p53 mutation. The strategy to image p53 is based on the principle that in normal cells, p53 is expressed but rapidly degraded. But in case of a DNA-damaging event, degradation of p53 is prevented resulting in its stabilization (accumulation) and activation (see insert which is a Western blot showing accumulation of p53 in response to ionizing radiation). We therefore hypothesized that physical linkage (by gene fusion) of p53 to luciferase should also result in degradation of the fusion, but in case of DNA damage, the fusion would not be degraded and would thus accumulate enabling imaging of p53 activation by monitoring the level of luciferase activity using bioluminescence imaging. This is based on the assumption that ubiquitination and degradation of the p53-Luc fusion protein will mirror that of p53. This strategy would also be able to image cells wherein p53 itself had mutated and thus accumulate.
Cell Lines and Culture Conditions
The MCF7 and HCT116 were maintained in RPMI 1640 containing 10% heat-inactivated fetal bovine serum, 1%
Irradiations
Freshly seeded Jurkat cells were irradiated at room temperature using a 60Co source at 1–2 Gy/min. Dosimetry was carried out using an ionization chamber connected to an electrometer system that was directly traceable to an NIST standard. Following irradiation, the cells were incubated for the indicated time. Mock irradiations were conducted wherein cells were physically taken to the source but were not irradiated.
Morphological Analysis
Apoptotic morphology was assessed by using propidium iodide (PI, Sigma) staining, cells were plated at a density of 5 × 104 cells onto chamber slides (Nunc). Following irradiation, both treated and control slides were rinsed twice with PBS, fixed in 4% paraformaldehyde at room temperature for 30 min, rinsed three times with PBS, and stained at room temperature for 10 min in a 100 μg/mL solution of PI in PBS. After staining, the slides were rinsed twice with PBS, blotted dry, and mounted using Vectashield mounting medium (Vector Laboratories). Slides were examined immediately after staining using a Leitz Laborlux S microscope.
Western Blot Analysis
Cells were either left untreated or treated with 10 Gy of gamma radiation and at the indicated time points harvested, washed with PBS, and lyzed in sample buffer containing a protease inhibitor mixture (Boehringer Mannheim). The lysates were assayed for protein concentrations and equal amounts of protein were resolved using 10% SDS-PAGE. The resolved samples were transferred onto a nitrocellulose membrane (Gelman Sciences) for Western blot analysis using a luciferase specific antibody as described previously [5].
Bioluminescence Imaging
Bioluminescence imaging was conducted on a cryogenically cooled IVIS system (Xenogen, CA) coupled to a data acquisition PC running LivingImage software (Xenogen) as an overlay on IGOR (Wavemetrics, Seattle, WA) under Windows 98. Luciferin was added to cell cultures to be imaged at (1 μg/mL). An integration time of 1 min was used for luminescent image acquisition. Signal intensity was from the images quantified using LivingImage and IGOR.
Results and Discussion
The p53 coding sequence was fused to the firefly luciferase coding sequence using PCR as described in Materials and Methods. The hybrid cDNA was inserted into a mammalian expression vector (pZ, kindly provided by Genetics Institute, Cambridge, MA). To confirm that the appropriate polypeptide was translated from the resulting expression vector (pZ p53-Luc), a transient transfection into COS cells (African green monkey kidney) was done. Western blot analysis of the transfected cells revealed that pZ p53-Luc transfected but not mock transfected cells expressed a 110 kDa polypeptide that cross reacted with a luciferase specific antisera as well as a p53 specific antibody (data not shown). This polypeptide was of the expected size since 53 kDa of p53 and 60 kDa of luciferase should have resulted in a 113-kDa polypeptide.
pZ p53-Luc was stably transfected into MCF-7 cells and the resulting neomycin resistant clones were analyzed for expression of the fusion polypeptide using a luciferase specific antibody. Although a significant level of the fusion protein was detected in the absence of a DNA-damaging event (Figure 2), when two independent stable cell lines (clones 216 and 217) were gamma-irradiated (10 Gy), there was a reproducible and significant increase in the levels of the p53-Luc fusion protein (216 compared to 216 + IR and 217 compared to 217 + IR). This suggested that like p53, the p53-Luc fusion was stabilized in response to DNA damage induced by ionizing radiation. To investigate if this accumulation of p53-Luc in response to DNA damage could be imaged as an increase bioluminescence activity, clone 216 was imaged at various times after a single 10 Gy dose of radiation. Mock-irradiated clone 216 cells were used as control. As shown in Figure 3, even at the first time point, clone 216 cells had a higher level (two fold) of bioluminescence activity after irradiation. Bioluminescence activity increased in irradiated cells fivefold from the initial levels and reached a plateau at approximately 180 min after irradiation. In this experimental model system, the p53-Luc fusion protein was expressed using a strong promoter (adenoviral major-late promoter) and hence the levels of the fusion under steady state were significantly high (Figure 2). We speculate that this may be due to saturation of the ubiquitination/degradation machinery. This high level of p53-Luc under resting conditions may also explain why only a modest increase in bioluminescence was observed after irradiation. In contrast, mock-irradiated cells had a twofold increase in bioluminescence activity over the entire period after irradiation.

Accumulation of the p53-Luc protein in response to DNA damage. The above described p53-Luc fusion gene was stably transfected into MCF-7 cells and the resulting clones were analyzed for expression of the fusion polypeptide [53 kDa (p53) and 60 kDa (luciferase) = 113 kDa] using a luciferase specific antibody. Although a significant level of the fusion protein was detected in the absence of DNA-damaging events, when two independent stable cell lines (clones 216 and 217) were irradiated, there was a reproducible and significant increase in the levels of the p53-Luc fusion protein (216 compared to 216 + 1R and 217 compared to 217 + 1R).
To demonstrate that the p53-Luc fusion retains p53 function, we utilized HCT116 p53–/– cells (kindly provided by Dr B. Vogelstein). The pZ p53-Luc expression plasmid was stably transfected into HCT116 p53–/– cells and the resulting clones were used for further analysis. To investigate if the fusion protein retains transcription factor activity, clone J was irradiated with 10 Gy of ionizing radiation and the accumulation of the fusion was monitored over time. As shown in Figure 4A, the 110-kDa p53-Luc fusion accumulated over time as observed previously in MCF-7 cells. Analysis of p21, a cell cycle regulatory gene known to be transcriptionally induced by p53, demonstrated that stabilization and accumulation of the p53-Luc protein resulted in a concurrent induction of p21 expression (Figure 4B). The fact that in the parental HCT116 p53–/– cells which lack p53, treatment of these cells with ionizing radiation did not result in p21 expression (our data, not shown and Ref. [6]) provides strong evidence that the p53-Luc fusion protein retains p53's transcription factor function. Next, we investigated whether the p53-Luc fusion retains the ability to induce apoptosis in cells. The above stable cell line expressing the p53-Luc fusion in a p53-deficient background was irradiated and the percentage of cells undergoing apoptosis was determined at 24 and 48 hr. As shown in Table 1, control HCT116 P53 –/– cultures only had 1% apoptotic cells in the absence of radiation which increased to 5% and 8% at 24 and 48 hr, respectively, after irradiation with 10 Gy. In contrast, expression of the p53-Luc fusion in this background resulted in a culture that had 5% apoptotic cells in the absence of irradiation which increased to 11% and 14% at 24 and 48 hr, respectively, after irradiation. This increase in the basal levels of apoptosis in the p53-Luc expressing culture compared to the p53-deficient HCT116 cells is consistent with the fact that the p53-Luc fusion protein was being constitutively overexpressed through a constitutive viral promoter. In response to DNA damage, concurrent with accumulation of the p53-Luc fusion, there was an increase in apoptosis indicating that the p53-Luc fusion retains the ability to induce apoptosis.

Time-dependent accumulation of bioluminescence activity in response to DNA damage. Clone 216 was imaged at various times in the absence (216) and presence (216-IR) of ionizing radiation pretreatment. The absolute number of photons (over background) emitted by the cells per minute was determined and used to plot the data. The data presented are from a single experiment but multiple experiments were conducted that yielded a similar pattern of increase in photon counts over time although the absolute numbers varied significantly.
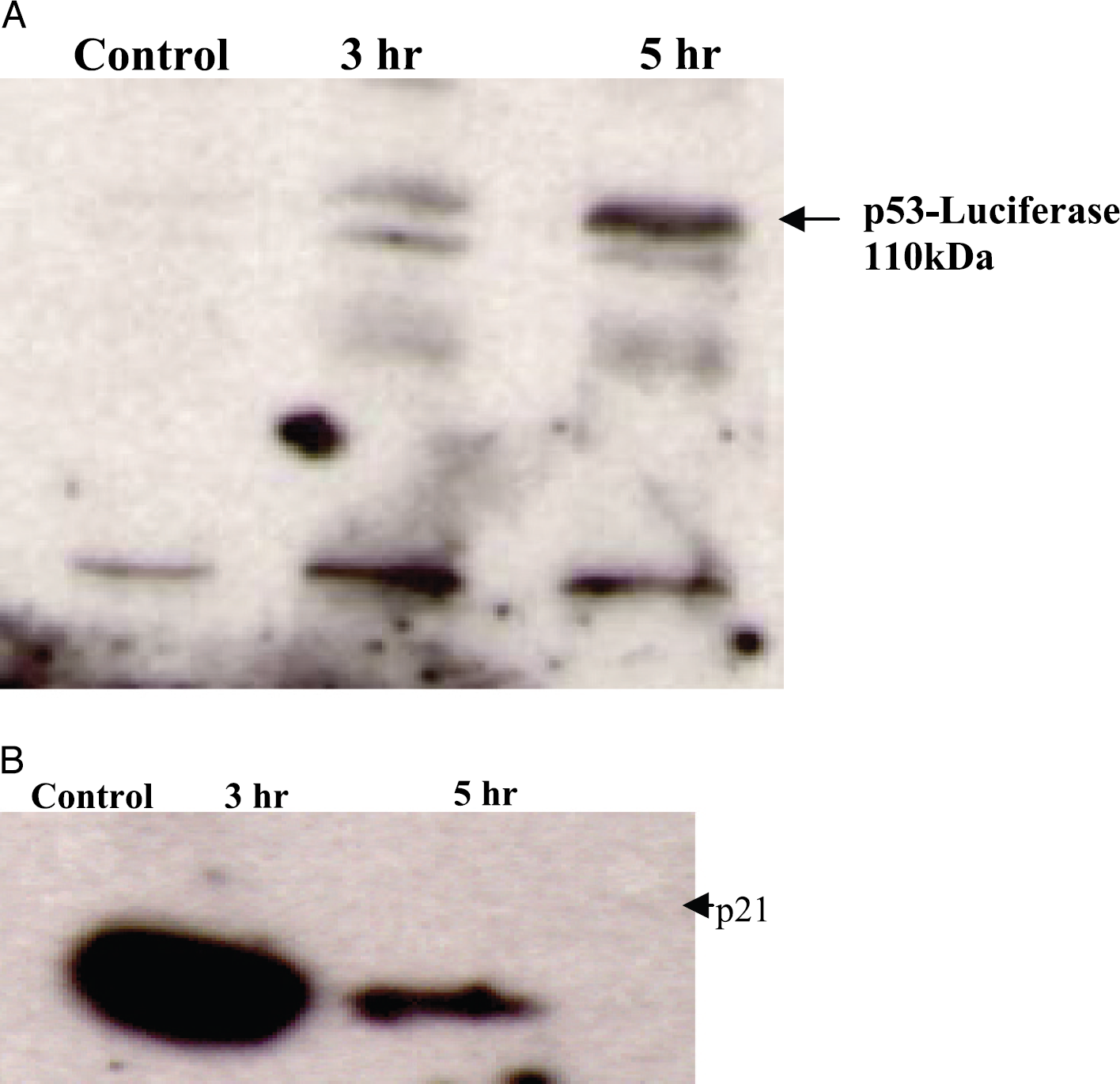
DNA damage-mediated accumulation of the p53-Luc fusion in HCT116 cells. The p53-Luc expression plasmid was stably transfected into HCT116 p5300/– cells. One of the resulting clones was analyzed for the expression of the fusion in the absence and presence of 10 Gy of ionizing radiation at 24 and 48 hr postirradiation using a p53 specific antibody (A). The same samples were also analyzed for expression of p21 (B).
Technologies involved in molecular imaging applications span many modalities including MRI [7–10], PET [11,12], and optical techniques [13–17]. p53-dependent gene expression has been previously imaged in vivo with PET and by in situ fluorescence [18]. Using a retroviral vector (Cis-p53/TKeGFP) generated by placement of HSV1-tk and enhanced GFP fusion gene (TKeGFP, a dual-reporter gene) under control of a p53-specific response element, DNA damage-induced up-regulation of p53 transcriptional activity was imaged and correlated with the expression of p53-dependent downstream genes (including p21). Similarly, using HCT116 colon cancer cells that were stably transduced with PG13-luc, a p53 reporter with a Firefly luciferase gene under the control of 13 p53 response elements, Wang and El-Deiry [19] reported noninvasive imaging of p53 activation, both in vitro and in vivo. The imaging strategy described here is unique in that it would not only enable noninvasive detection of p53 activation (which correlates with accumulation of p53 protein), but it would also enable noninvasive detection of cells wherein p53 has undergone a mutagenic event (which also results in accumulation of the p53 protein) in a variety of cell lines. The results presented here validate recent studies [20] demonstrating that a p53-GFP fusion protein retains the properties of wild-type p53. This fusion protein is degraded with similar kinetics to wild-type p53, is recognized by the same p53-reactive antibodies, transactivates transcription from a p53-responsive element comparably to wild-type protein, and is able to suppress tumor cell growth. Irradiation of cells expressing p53-GFP resulted in a 50–100-fold increase in p53-GFP levels. Due to the fact that noninvasive imaging of GFP is limited to superficial tissues, the use of luciferase in our studies provides a distinct advantage for in vivo imaging of p53 within deep tissues. Recent work from the Vogelstein group emphasizes the need for methods to study p53 activation in vivo in response to therapeutic interventions [21]. They found that isogenic tumor cell lines differing only in their p53 status responded to drugs differently in culture than when grown in subcutaneous xenografts. Specifically, p53-knockout lines were potently sensitized to γ-irradiation and adriamycin in vitro, but not in vivo. These results underscore the difficulty in extrapolating between in vitro and in vivo observations.
Induction of Ionizing Radiation-Induced Apoptosis in HCT116 in the Presence and Absence of p53-Luc Expression

The percentage of cells undergoing apoptosis upon 10 Gy irradiation was determined by morphological methods (Material and Methods). HCT116 p53–/– cells as well as a derivative cell line expressing the p53-Luc fusion were tested and the data presented are an average of three independent experiments.
Activation of the p53 protein protects the organism against the propagation of cells that carry damaged DNA with potentially transforming mutations. MDM2, a p53-specific E3 ubiquitin ligase, is the principal cellular antagonist of p53, acting to limit the p53 growth-suppressive function in unstressed cells. In unstressed cells, MDM2 constantly monoubiquitinates p53 and thus is the critical step in mediating its degradation by proteasomes. Disruption of the p53-MDM2 complex by multiple routes is the pivotal event for p53 activation, leading to p53 induction and its biological response. Because the p53-MDM2 interaction is structurally and biologically well understood, the design of small molecules that disrupt or prevent it has become an important target for cancer therapy. The imaging paradigm described here would not only facilitate the in vitro discovery of compounds that disrupt p53-MDM2 function using high throughput screening, but would also be useful in validating lead compounds developed using other assays.
Footnotes
Acknowledgments
This work was supported in part by the following NIH/NCI grants: R24CA83099 and P50CA93990.