
Abstract
Background
Familial hemiplegic migraine (FHM) is a rare type of migraine with aura that is characterized by transient hemiparesis. Mutations in three genes (CACNA1A, ATP1A2, and SCN1A) have been found to cause FHM. Among these, nine SCN1A gene mutations were reported to cause familial hemiplegic migraine type 3 (FHM3). However, none of them was reported in China.
Method
The clinical manifestations of a Chinese FHM family were recorded and all coding exons and flanking intronic regions of the CACNA1A, ATP1A2, and SCN1A genes were tested for mutations.
Results
All FHM patients in the investigated family have typical hemiplegic migraine attacks characteristic of FHM. We identified a novel mutation (p.Leu1670Trp) of the SCN1A gene. The affected amino acid is highly conserved across different species and therefore likely plays an important role in SCN1A gene function.
Conclusion
The identification of a novel mutation in the SCN1A gene in the Chinese population may further aid in the understanding of FHM genetics.
Introduction
Familial hemiplegic migraine (FHM) is a rare type of migraine with aura, characterized by transient attacks with motor aura (1). FHM follows autosomal dominant inheritance patterns. So far, mutations in three genes, CACNA1A, ATP1A2, and SCN1A have been found to be responsible for familial hemiplegic migraine type 1 (FHM1), familial hemiplegic migraine type 2 (FHM2), and familial hemiplegic migraine type 3 (FHM3), respectively (2–4).
More than 20 different missense mutations of CACNA1A (FHM1) (5) and over 30 different missense mutation of ATP1A2 (FHM2) have been identified as causing FHM (6). However, only nine missense mutations of the SCN1A gene have been reported to cause FHM3 (2,5,7–11), and none of them have been reported in China. The aim of this study was to further investigate the genetics of FHM in the Chinese population. Here we report a Chinese FHM3 family with a novel mutation in the SCN1A gene.
Subjects and methods
Patients
The proband (patient III-20), a 27-year-old Chinese male, had recurrent headache attacks from 16 to 27 years of age. These attacks were of moderate intensity, and usually originated in the bilateral temple. Routine physical activity aggravated headaches. The total number of headache attacks was five. Two out of the five attacks were accompanied by nausea and/or vomiting. The first attack happened at the age of 16, the second episode at the age of 21, and three more episodes occurred at 27 years of age. These headache attacks were preceded by aura symptoms, including visual disturbances, speech problems, sensory disturbances, and motor weakness. In general, the aura symptoms lasted 2–3 hours and then the patient gradually recovered. The visual disturbances always lasted at least 20 min and were described as increased scotomata and filled squares in the bilateral visual field. The speech problems were described as the inability to form a coherent sentence while being able to fully comprehend sentences. Sensory problems were unilateral or bilateral and were described as a spreading numbness of the left or right limbs, whereas motor weakness always located unilaterally and was described as movements out of control. For instance, the hand could not hold items steadily and there was a drag to the gait while walking. The patient presented with prolonged symptoms for 30–40 hours during the last episode. Neurological examinations were unremarkable. Cerebral magnetic resonance imaging (MRI) and Magnetic Resonance Angiography (MRA) showed no abnormalities.
A neurologist interviewed all family members. The proband's mother (patient II-9) had suffered from typical migraine attacks (bilateral moderate pulsatile headache aggravated by walking, accompanied by nausea and vomiting) for 37 years, and with five attacks per year on average. The aura symptoms were similar to the proband’s symptoms, including bilateral visual symptoms (scotomata), speech difficulties (aphasia), and one-side sensory and motor disturbance. The patient also suffers from depressive disorder and hypotension.
Clinical features of FHM patients in this family. NK: not known; NA: not applicable.

Patient II-5 (Figure 1, Table 1), a 59-year-old maternal uncle, had headache attacks with nausea, vomiting, visual symptoms (black spots), and weakness in one arm and leg. These attacks lasted 2–4 hours.
Family tree of a Chinese FHM family. Males and females are represented by squares and circles, respectively. The arrow indicates the proband. The diagonal line indicates a deceased family member. Filled black symbols represent FHM patients. “+” represents a carrier of the SCA1N mutation (p.Leu1670Trp); “−” represents family members without the SCN1A mutation.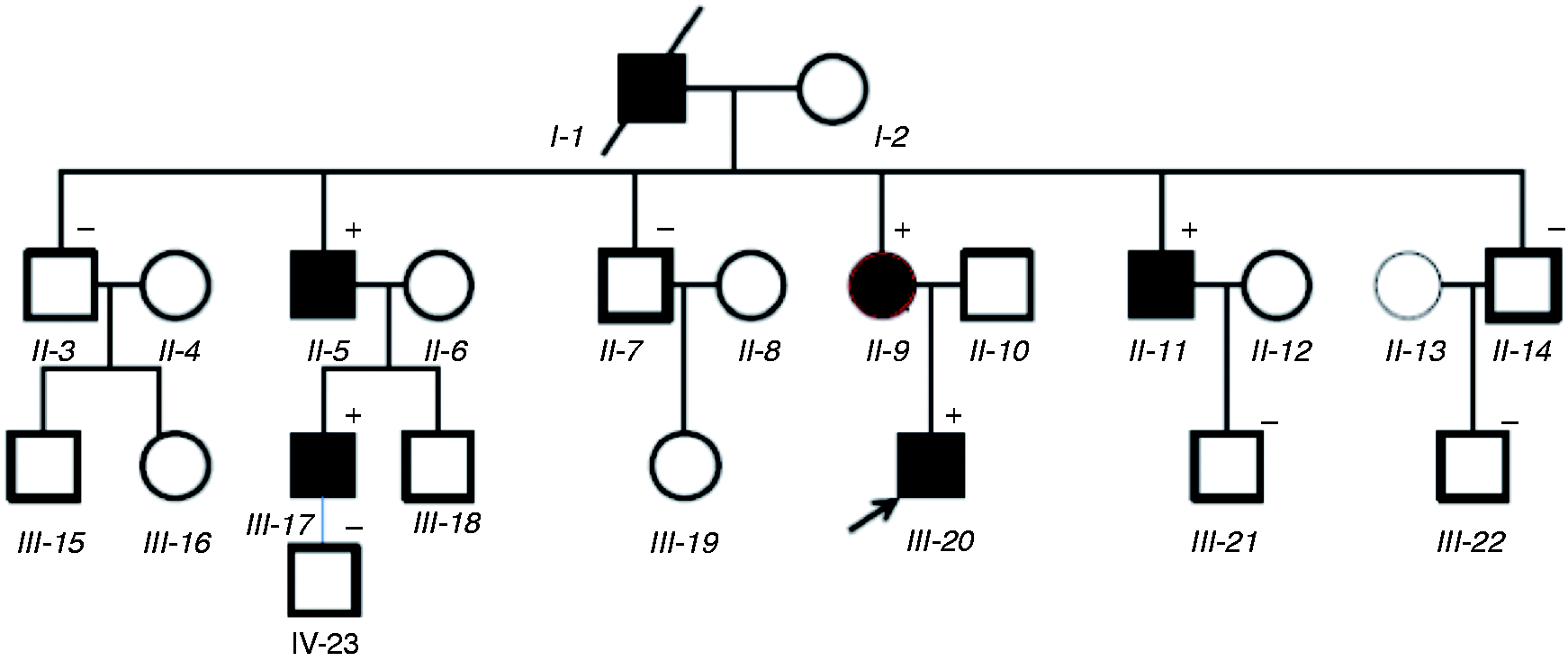
Patient II-11, a 46-year-old maternal uncle (Figure 1, Table 1) had suffered from migraine with nausea and vomiting since youth. Aura symptoms included visual (black spots), speech (aphasia), and sensory, with motor weakness in one arm and leg.
Patient III-17, a 38-year-old maternal cousin (Figure 1, Table 1), had migraine attacks with visual symptoms, speech difficulties, and motor weakness in one arm.
None of the family members reported childhood epilepsy or other SCN1A associated syndromes, such as cerebellar ataxia and elicited repetitive daily blindness.
Genetic analysis
Genomic DNA from family members was extracted from peripheral blood leukocytes according to standard procedures (12). The FHM genes CACNA1A, ATP1A2, and SCN1A were analyzed by direct sequencing of polymerase chain reaction. Causal mutations in the CACNA1A and ATP1A2 gene were excluded by testing all coding exons and flanking intronic regions. All 26 exons and flanking intronic sequences of the SCN1A gene were analyzed.
Results
A novel heterozygous point mutation in exon 26 (c.5009T > G, p.Leu1670Trp) of the SCN1A gene was identified by mutation scanning of the proband and his family. The mutation was found in the proband’s genome and all his relatives who were affected with FHM. It was absent in other unaffected family members (Figure 1). This mutation has not been described in the literature and is not reported in the Single Nucleotide Polymorphism Database (http://www.ncbi.nlm.nih.gov/snp).
The SCN1A gene encodes for the α1 subunit of the NaV1.1 sodium channel, containing four homologous domains (DI-DIV). Each domain consists of six transmembrane segments. Leucine1670 is located at the intracellular loop between the S4 and S5 segment in domain IV. The missense mutation leads to the replacement of leucine by tryptophan. Although no missense mutations were reported at this location, various known mutations near leucine1670, for example missense mutation p.Pro1668Ala and p.Met1664Lys, have been identified as causing SMEI (severe myoclonic epilepsy of infancy, also known as Dravet's syndrome) or GEFS+ (generalized epilepsy with febrile seizure plus) (13,14).
Moreover, leucine1670 is highly conserved between different species (Figure 2). Different bioinformatics prediction programs, such as SIFT (Deleterious (−5.216)) and PolyPhen2 (probably damaging (1.0)) indicate that the Leucine1670 mutation is likely pathogenic.
Protein alignment of different SCN1A sequences from several species. Arrow indicates the location of the p.Leu1670Trp mutation with highly conserved leucine1670.
Discussion
Here we report a novel missense mutation, p.Leu1670Trp, in a FHM3 family in China. The FHM3 patients in this family display very similar migraine attack symptoms. The attack frequency varies from four episodes during an 11-year period to 5–6 episodes per year. Seizures or other SCN1A-associated syndromes were not observed. To our knowledge this is the first SCN1A gene mutation reported in China and the mutation itself has not previously been reported in the literature. Although no functional tests were performed for this novel mutation, several lines of evidence indicate that the mutation likely has an impact on gene function. The mutation co-segregated with FHM in this family. Similar nearby missense mutations present with epileptic phenotypes. Leucine1670 is a highly conserved amino acid in sodium channel α1 subunits across different species (Figure 2), and two bioinformatics prediction programs both indicate that the mutation is most likely pathogenic.
Previous studies have reported three FHM patients in China. Regretfully, only our study performed mutational analysis of the patient’s and the family members’ genes. Review of the clinical data of the previous three FHM cases in China revealed that their clinical manifestations did not involve seizures or other syndromes (15–17). However, only limited information is available for these FHM cases.
The p.Leu1670Trp mutation is the sixth mutation found to cause pure FHM3. The other five mutations are Q1489K, L1649Q, L1624P, I1498M, and F1661L (2,7,10,11), while other SCN1A mutations are associated with epileptic seizures or other symptoms. Our study indicates that screening for the SCN1A gene mutation should be considered in FHM when no mutation is found in the CACNA1A and ATP1A2 gene (7).
In conclusion, we identified a novel SCN1A gene mutation in a Chinese FHM3 family. Our case report can contribute to a better understanding of the genetic underpinnings of FHM, in particular in China.
Clinical implications
A new mutation point (p.Leu1670Trp) for of FHM3 was identified that can be used for gene testing and consultation of FHM patients. This the first report of a SCN1A gene mutation in a Chinese FHM family. Our new finding may focus more attention on FHM and genetic research in China.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.