
Abstract
A possible relation between migraine and epilepsy has been a matter of debate for many decades. Clinical, epidemiological and therapeutic similarities may be coincidental and are no proof of a common aetiological background. However, a genetically determined dysfunction of ion channels seems to point to a common underlying mechanism for both paroxysmal disorders. For example, mutations in the three known genes for familial hemiplegic migraine can cause epilepsy. It is likely that the development of specific drugs aimed at restoring ion-channel function and/or related cellular signalling pathways might benefit patients with epilepsy as well as those with migraine. This review will briefly summarize the clinical, epidemiological, pathophysiological and therapeutic similarities between migraine and epilepsy. Most attention will be paid to the genetic relationship between these two paroxysmal disorders.
Introduction
Migraine and epilepsy are both characterized by paroxysmal cerebral dysfunction. Therefore, a possible relationship between migraine and epilepsy has been debated for decades (1–4). There are authors who have pointed at similarities and who consider migraine and epilepsy as two strongly related brain diseases. Historical examples of this viewpoint are Hughlings Jackson, Charcot and Dejerine. Others have stressed the differences between the two syndromes, which has resulted—in its most extreme form—in the famous phrase by William Gowers: ‘The most frequent relation between migraine and epilepsy is the wrong diagnosis’ (5). Recent discoveries, especially in the field of genetics, have shed new light on this debate.
In this review we will summarize the clinical, epidemiological and therapeutic aspects of the relationship between migraine and epilepsy. Most attention, however, shall be given to the recent genetic discoveries regarding the two syndromes.
Clinical similarities
Migraine and epilepsy are paroxysmal disorders, both with temporary dysfunction of the cerebral cortex during attacks. In spite of this similarity, in most cases the clinical differences between attacks of migraine and attacks of epilepsy are not difficult to recognize. This is of practical importance, as the clinical work-up, prognosis and treatment of both syndromes differ considerably.
However, there may be several diagnostic problems. Attacks of epilepsy can be accompanied or followed by migraine-like headache (6–11), and attacks of migraine can lead to unconsciousness (12), especially in the case of basilar migraine (13). Migraine attacks can cause EEG abnormalities, resembling epilepsy (14–17). Acute confusional migraine combines EEG abnormalities with epilepsy-like clinical features (18, 19). In so-called ‘migralepsy’, an attack of migraine with aura provokes an attack of epilepsy (20–23). Epilepsy can follow a migraine attack with reversible cerebral magnetic resonance imaging abnormalities (24, 25). Attacks of migraine and of epilepsy also have various trigger factors in common (such as hormonal factors, sleeping disturbances or brain trauma) (4). In both syndromes a status can occur, a status epilepticus, a status migrainosus or a migraine aura status (14).
Separate migraine and epilepsy within one individual can of course be coincidental when one considers the high prevalence of migraine in the general population, and consequently also in the population of patients with epilepsy. However, there are some epilepsy syndromes that are associated more frequently with migraine than can be explained by chance alone. Examples are occipital lobe epilepsy (26–29), Rolandic epilepsy (30, 31), adult onset myoclonus epilepsy (32), petit mal epilepsy (33) and temporal lobe epilepsy (34).
Finally, epilepsy can also be provoked by cerebral damage caused by migrainous infarction (35), and migraine and epilepsy can both be caused by an underlying (structural) brain disease, such as cerebral trauma (36), arteriovenous malformation (37), mitochondrial disorders (38) or Sturge–Weber syndrome (39).
Epidemiological studies
Earlier epidemiological studies of the relationship between migraine and epilepsy were unreliable, as they lacked strict criteria for the diagnosis of migraine and epilepsy, as well as a good control group [reviewed in (40)]. The first state-of-the-art epidemiological studies to use the International Headache Society (HIS) criteria for migraine were performed in the 1990s. Marks and Ehrenberg found migraine in 20% of patients with epilepsy (21), and in 3% of these patients the attacks of epilepsy occurred in a close temporal relationship with the attacks of migraine. In a population-based study of almost 2000 patients with epilepsy, Ottman and Lipton found concomitant migraine in 24% (36, 41). Of the family members with epilepsy, 26% also had migraine, whereas only 15% of the family members without epilepsy had migraine. Comparable results have been found in another population-based study (42), and in children with epilepsy (43). Indeed, in a recent retrospective study from Iceland, children aged 5–15 years with epilepsy had an almost fourfold higher chance of also having migraine, especially migraine with aura (44).
Genetics of migraine and epilepsy
Migraine and idiopathic epilepsy are complex genetic disorders. In both, multiple genetic and environmental factors play a role. In the last decade, intensive research has clarified part of the genetic background of monogenic forms of migraine and epilepsy. However, much remains unknown, especially of the common multifactorial forms. It is remarkable that the more that is discovered about the genetic background of both disorders, the greater the overlap between migraine and epilepsy appears to be.
Migraine genes often also cause epilepsy, but there have been few reports of epilepsy genes causing migraine. This may be due to research bias, as epilepsy in a patient or family with migraine will probably receive attention, whereas migraine in a patient or family with epilepsy will often be seen as coincidental because of the high incidence of migraine in the general population.
The genes and mutations in many of the complex forms of epilepsy remain undiscovered (45–47). Most known genes are involved in rare monogenetic forms of epilepsy and code for ion channels, such as those for sodium, potassium and chloride (45–47). Mutations in such genes cause abnormal synchronization or increased excitability of cortical neurons (46).
As in epilepsy, present molecular knowledge of the common forms of migraine is limited (48). Only three of the genes that play a role in a rare autosomal dominant subtype of migraine with aura, familial hemiplegic migraine (FHM), are known, called FHM1, FHM2 and FHM3. In FHM attacks, unilateral motor weakness occurs during the migraine aura (IHS, 2004) (49).
Fhm1
The FHM1 gene CACNA1A codes for the pore-forming subunit of Cav2.1 P/Q-type calcium channels and is located on the short arm of chromosome 19 (50). Mutations in this gene are present in more than half of all FHM families, including those with cerebellar ataxia. At present, 17 different mutations in this gene are known, some of which also cause epilepsy (Table 1) (50–61). In most cases the seizures only occur during severe hemiplegic migraine attacks (58), but a family has been recently described in which epileptic seizures occurred independently of FHM attacks (59). The proband of the family, with a de novo CACNA1A I1710T mutation, had no epilepsy, but her two children had complex partial seizures, one of whom had additional (febrile and afebrile) generalized tonic-clonic seizures. Both children suffered from cerebellar ataxia and FHM and carried the I1710T mutation. The same I1710T mutation has been reported to cause status epilepticus during FHM attacks in another family (54).
Mutations in the FHM1 CACNA1A gene associated with epilepsy
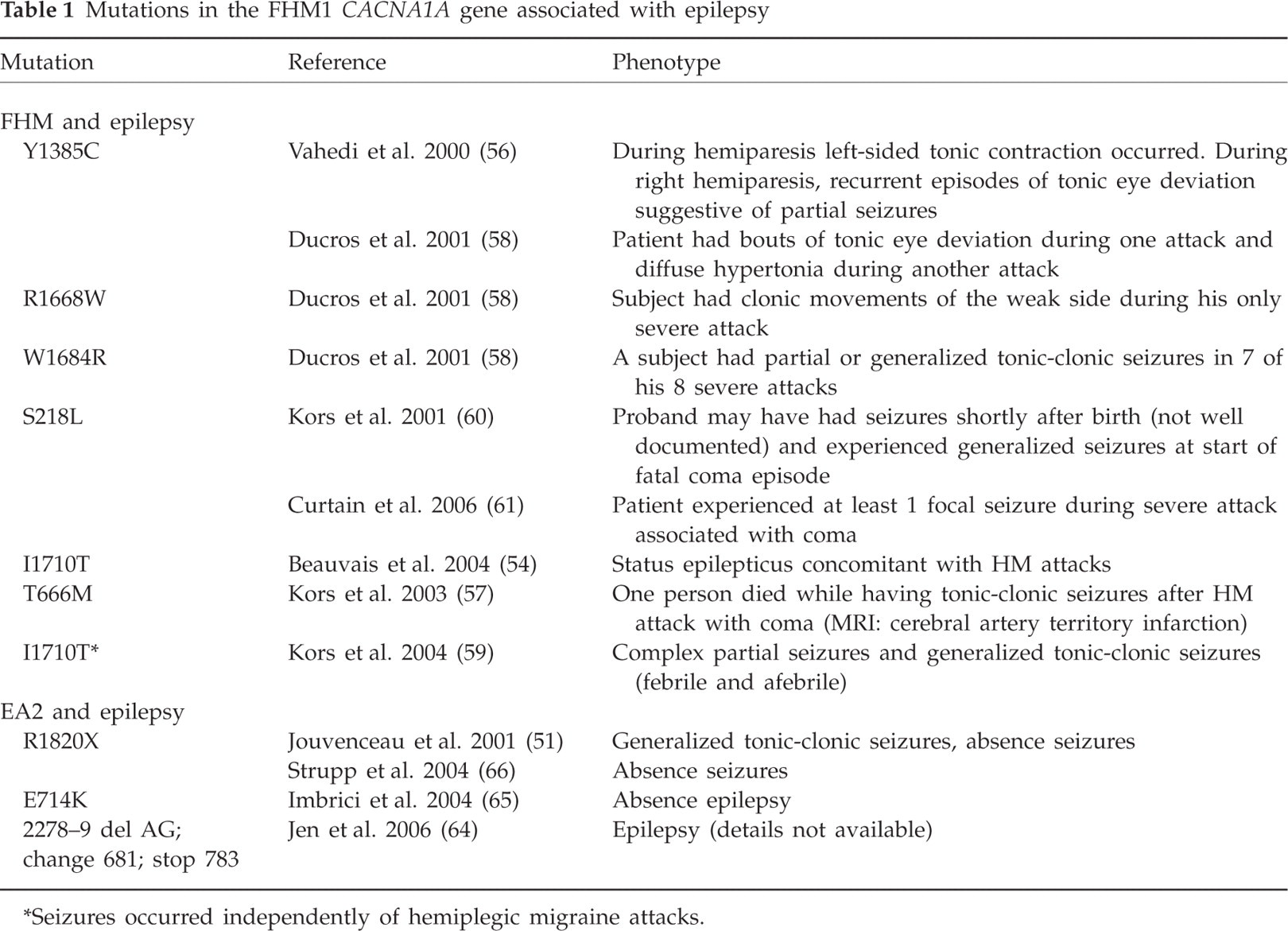
Seizures occurred independently of hemiplegic migraine attacks.
Affected members of families with a CACNA1A S218L mutation have a severe phenotype with recurrent atypical migraine attacks, often triggered by trivial head trauma (60, 61). The proband of one of the families died after a period of coma following a symptom-free period of several hours after a mild head trauma. She had never experienced an attack of (hemiplegic) migraine, but this attack might have been the first symptom of this disorder. Two carriers of the S218L mutation, from two different families, had generalized epileptic seizures (60, 61).
CACNA1A mutations also cause spinocerebellar ataxia type 6 (SCA6) and episodic ataxia type 2 (EA2). SCA6 is characterized by a late-onset, slowly progressive cerebellar ataxia, with oculomotor abnormalities, gait ataxia, mild upper limb ataxia and dysarthria, but not by epilepsy (62, 63). EA2 patients present with early-onset attacks of imbalance and/or vertigo, lasting hours to days (64). Patients with EA2 can have interictal permanent cerebellar ataxia, and the attacks can be associated with symptoms of (basilar-type) migraine (64). Several EA2 patients with epilepsy have been reported (51, 64–66).
The involvement of the CACNA1A gene in epilepsy has been further supported by an association study reporting a link between this gene and idiopathic epilepsy (67, 68). Of four single nucleotide polymorphisms (SNPs) and one microsatellite marker located within the CACNA1A gene, one SNP in exon 8 showed a significant association. A follow-up study showed that two SNPs in the immediate vicinity of exon 8 were responsible for the association with epilepsy (69). The association was not limited to a specific epileptic syndrome or subgroup. An effect of these SNPs on expression or alternative splicing of the protein has been suggested but not further investigated. An independent study has not confirmed the association of epilepsy with the CACNA1A gene (70), leaving the true value of these findings uncertain.
The involvement of the CACNA1A locus in epilepsy in humans is of interest in view of the epileptic phenotypes observed in natural mutant mice with mutations in the autologous Cacna1a gene. Several natural mouse Cacna1a mutants displayed ataxia and epilepsy. The main effect of tottering, leaner and rolling Nagoya mutated Cav2.1 calcium channels appears to be reduction of calcium current density (71–74). CACNA1A mutations might very well influence cortical spreading depression, since P/Q-type calcium channels mediate glutamate release in cortical neurons (48). Experimentally induced cortical spreading depression has revealed an increased threshold in leaner, tottering mice with concomitant reduced glutamate release (75). A knock-in mouse model has recently been generated, carrying the human pure FHM1 R192Q mutation (76). Unlike the natural Cacna1a mutant mouse models, transgenic R192Q mice exhibit no overt phenotype. Multiple gain-of-function effects were found, including increased Ca2+ influx in cerebellar neurons, increased release of neurotransmitters at the neuromuscular junction and, in the intact animal, a reduced threshold and increased velocity of cortical spreading depression.
FHM2
The FHM2 gene ATP1A2 is located on the long arm of chromosome 1 (77, 78). It codes for the α2 subunit of sodium/potassium ATPase, responsible for pumping potassium ions in the cell and sodium ions out of the cell. Disturbance of the sodium/potassium balance can cause cortical spreading depression. As in FHM1, various mutations in the ATP1A2 gene have been reported (79), many of which also cause epilepsy (Table 2). In two Italian families with a ATP1A2 mutation, five patients had epilepsy together with FHM (77, 80). The L764P mutation mainly caused benign childhood epilepsy and the W887R mutation caused partial seizures. In a Dutch-Canadian family with FHM2 and a R689Q ATP1A2 mutation, seven family members had a benign form of childhood epilepsy [benign familial infantile convulsions (BFIC)] (78). BFIC is a rare autosomal dominant benign form of epilepsy with strictly partial non-febrile convulsions that begin between 3 and 12 months of age and disappear after the first year of life. In the R689Q family, all seven BFIC persons tested had the missense mutation, but BFIC and FHM only partially cosegregated. It seems that, in this family, migraine and epilepsy had partially overlapping mechanisms related to dysfunction of ion transport. However, in 12 Italian BFIC families (without FHM or other type of migraine), no ATP1A2 mutations were found (81). No association was found between the ATP1A2 gene and temporal lobe epilepsy (82) or idiopathic generalized epilepsy (83).
Mutations in the FHM2 ATP1A2 gene associated with epilepsy

Seizures occurred independently of hemiplegic migraine attacks.
AHC, alternating hemiplegia of childhood; FHM, familial hemiplegic migraine.
A further association of an ATP1A2 mutation (G301R) and epilepsy has bben found by an Italian group in a family with FHM, seizures, coma, sensory deficits and transient and permanent cerebellar signs (84). Epileptic seizures were also present in German D718N and P979L ATP1A2 mutation carriers (85), in French R583Q and M829R mutation carriers (79) and in a child with a very severe phenotype including mental retardation with a G615R mutation (86). Finally, in a family combining clinical features of FHM and alternating hemiplegia of childhood, four carriers of a T378N mutation had generalized tonic-clonic seizures (87).
In Atp1a2 mutant mouse models, homyzygous Atp1a2-null 18.5-day-old fetuses have shown selective neuronal apoptosis in the amygdala and piriform cortex in response to neural hyperactivity (88). Atp1a- null mice died immediately after birth because of severe motor deficits that also abolished respiration (88, 89). Interestingly, in line with the observed epilepsy in patients, Atp1a2 null mice on 129sv genetic background display frequent and generalized seizures, but die within 24 h after birth (88). Heterozygous Atp1a2 +/– mice have revealed enhanced fear/anxiety behaviour after conditioned fear stimuli, probably because of the observed neuronal hyperactivity in the amygdala and piriform cortex (88).
FHM3
Mutations have recently been found in FHM families in the SCN1A gene located on 2q24, already known to be associated with epilepsy (90). Three of the 18 patients with FHM3 had had concomitant epilepsy in their youth, in two consisting of benign febrile convulsions and in the third of benign focal epilepsy (Table 3).
Mutation in the FHM3 SCN1A gene associated with epilepsy

More than 150 different mutations in SCN1A have been previously described in families with epilepsy (91). SCN1A mutations can cause generalized epilepsy with febrile convulsions (GEFS), severe myoclonus epilepsy of infancy (SMEI), and some other rare epilepsy syndromes. An association with migraine had not been reported before, but this may be due to the above-mentioned research bias. Homozygous Scn1a knockout mice develop ataxia and have a reduced life span, whereas heterozygous Scn1a mice exhibit spontaneous seizures and sporadic deaths starting at postnatal day 21 (92).
Therapeutic consequences
In attacks of migraine and epilepsy a paroxysmal change in cortical neuronal activity occurs, in epilepsy hyperexcitation and in migraine hyperexcitation or hypoexcitation (spreading depression). Much is still unknown about the pathophysiology of both diseases, but it is clear that in both there are disturbances of glutamate metabolism (93), serotonin metabolism (94, 95), dopamine metabolism (96) and the function of ion channels (45–48). It is therefore not surprising that the preventive treatment of migraine and epilepsy overlaps. In epilepsy, different types of preventive medication have been specifically developed based on pathophysiological considerations, but the discovery of preventive treatment in migraine has often depended on coincidence. For example, migraine can be treated with antihypertensive (e.g. propranolol), antidepressive (amitriptyline) and antiepileptic drugs. Of the latter category, only sodium valproate and topiramate have proven efficacy in double-blind placebo controlled trials (97). Gabapentine seems also to be effective, but this has not been sufficiently investigated. All other antiepileptic drugs have been tried only in open trials, in small numbers of patients.
Conclusion
Migraine and epilepsy are two paroxysmal neurological disorders of which an association has been made very likely by genetic discoveries. Mutations in all three known FHM genes can cause epilepsy. A genetic disturbance in the function of ion channels therefore seems to be a shared pathophysiological mechanism of migraine and epilepsy. Of course, a broader focus on pathophysiology of common forms of migraine and epilepsy is possible. Voltage-gated ion channels are part of extensive signalling pathways. In addition to affecting neuronal excitability, they affect intracellular signalling pathways through various mechanisms (98, 99). Channels can directly activate enzymes linked to signalling pathways and serve as cell adhesion molecules or components of the cytoskeleton, and their activity can alter the expression of other specific genes (98). Although it is likely that voltage-gated ion channels are critically positioned in the pathways associated with migraine and epilepsy, the contribution of numerous other genes/proteins in the pathways merit future attention.
Although there are many FHM patients with epilepsy, it is remarkable that most patients with FHM ion channel mutations do not have epileptic manifestations. There are probably additional genetic or environmental factors that determine the occurrence of epilepsy. This seems an obvious explanation for the observed incomplete penetrance of epilepsy.
An important task for migraine researchers is to prove that a disturbance in ion channel function also plays a role in ‘normal’ migraine with and without aura, and not only in extremely rare phenotypes such as FHM. The development of specific compounds aimed at the normalization of ion channel functioning will probably also benefit the ‘normal’ migraine patient (100).