
Abstract
Objective: Our objective was to study the long-term prognosis of sporadic hemiplegic migraine (SHM).
Methods: We performed a longitudinal follow-up study in 18 patients who were diagnosed with SHM between 1993 and 1996. Follow-up time between the first and second survey ranged from nine to 14 years. These patients were included as part of a genetic study in which we systematically analysed the role of the three known familial hemiplegic migraine (FHM) genes.
Results: In 12 out of 18 patients the clinical diagnosis was unchanged. In two of the six remaining patients the attacks were no longer associated with hemiplegia; one of them had an ATP1A2 gene mutation (E120A). In the four other patients, the diagnosis changed into FHM, because a family member had developed hemiplegic migraine since the initial diagnosis was made. In two of the four patients a mutation was demonstrated (CACNA1A [R583Q] and ATP1A2 [R834X]).
Conclusion: This study shows that the diagnosis of SHM changes into FHM in a considerable percentage of patients (22% [4 of 18]), almost a decade after the initial diagnosis. This indicates that a careful follow-up of SHM patients and their families is advisable for optimal care and counseling. Diagnostic screening of FHM genes in SHM patients can be of value. Our genetic and clinical follow-up studies reinforce the evidence that FHM and SHM are part of the same spectrum of migraine.
Introduction
Hemiplegic migraine (HM) is a rare subtype of migraine with aura that is characterized by the presence of reversible motor weakness during attacks. Two different types of HM are recognized (1). In familial hemiplegic migraine (FHM) there is at least one first- or second-degree family member with attacks of HM. Three genes have been identified for FHM; the CACNA1A gene (FHM1), the ATP1A2 gene (FHM2) and the SCN1A gene (FHM3) (2–4). In sporadic hemiplegic migraine (SHM) the family history is negative for subjects with hemiplegic attacks. Interestingly, the majority of SHM patients have at least one first-degree relative who experiences migraine headache with or without aura (5).
FHM and SHM attacks are clinically identical (6), suggesting a common pathophysiological basis. Understanding the genetic background of SHM clearly would be of help to clinicians in diagnostic and therapeutic decision making and may prevent misdiagnosis and mistreatment of patients. Earlier, we cross-sectionally studied the involvement of the three FHM genes in pure SHM (i.e. without cerebellar signs). Scanning of the FHM2 (ATP1A2) gene had the highest likelihood of finding a causal mutation (5,7). We hypothesized that because SHM and FHM share, at least to some extent, the same genetic background, a clinical follow-up study of SHM patients and their relatives might show that SHM families eventually develop FHM. To test this idea, we performed a longitudinal follow-up study in 18 SHM patients and their families.
Material and methods
Between 1993 and 1996, a diagnosis of SHM according to the International Headache Society (IHS) criteria (8) was made in 19 Dutch patients at our department. In order to make the diagnosis, a standardized personal history of the patient and all available first-degree family members was taken. Second-degree family members were not interviewed personally, but the SHM patients and their interviewed family members did not report hemiplegic attacks in other family members at baseline or after follow-up.
The total cohort at baseline consisted of 19 patients and 74 first-degree family members. At follow-up, all living individuals received an information letter to ask for cooperation, and a standardized questionnaire, with questions aimed at headache diagnosis and aura symptoms, especially hemiplegia. Follow-up time between the first and second surveys ranged from nine to 14 years. After the questionnaire, a structured telephone interview was performed by a specifically trained medical student (MAL), who was blinded for the previous diagnosis and for the mutation status of the patients and their relatives. The final diagnosis at follow-up was made according to the IHS criteria (1) in cooperation with a physician specializing in headache (AHS) and a neurologist (GMT), who were also blinded for the previous diagnosis. Approval was obtained from the local ethical committee in accordance with national legislation; all participating subjects gave written, informed consent for this study.
Results
Detailed clinical information about all patients and their relatives


F = female. M = male. MO = migraine without aura. MA = migraine with aura (patients who had attacks of MO as well as attacks of MA are categorized as MA). SHM = sporadic hemiplegic migraine. FHM = familial hemiplegic migraine.
Information on a change of the HM diagnosis in the 18 SHM patients is summarized in Table 2. Of the 18 patients diagnosed as SHM at baseline, 12 still reported attacks that fulfilled the IHS criteria for SHM at follow-up. Two patients diagnosed with SHM at baseline did not report hemiplegia as a part of their attacks any more and—most remarkably—neither recalled ever having had hemiplegic attacks. These two probands reported MA at follow-up (both with visual aura and sensory symptoms). One of these patients had a mutation in the ATP1A2 gene (mutation E120A). Remarkably, still no family members of this proband suffered from HM attacks at follow-up. In the families of four SHM patients, a first-degree family member also had developed HM, and consequently, the diagnosis in these families changed to FHM. At baseline, two of these family members were diagnosed as “no migraine”, and one as having MA. The fourth was too young for a reliable history at baseline (six years). Information on the 67 family members is summarized in Table 3. Two out of these four patients and their family members who developed HM had a mutation in the ATP1A2 (R834X) and CACNA1A (R583Q) gene, respectively. For the other two FHM families, no mutations were found in any of the known FHM genes. Thus, from the group of 67 first-degree family members, four individuals from four different families also had developed hemiplegic attacks. Age at onset of HM in these four individuals ranged from 12 to 15 years. Pedigrees of these four families are provided in Figure 1.
Pedigrees of the four familial hemiplegic migraine cases. The pedigrees show the diagnosis after follow-up. Family numbers are similar to proband numbers in Table 1. The following symbols are used to indicate the diagnosis: filled lower half = familial hemiplegic migraine; right upper quadrant = migraine with aura; left upper quadrant = migraine without aura. Circle = female; square = male. Arrows indicate probands. Individuals heterozygous for a DNA variant are indicated by the respective variant. Fam = family. Diagnoses in SHM patients at baseline and after follow-up
a
MA = migraine with aura. SHM = sporadic hemiplegic migraine. FHM = familial hemiplegic migraine. If a mutation in the CACNA1A or ATP1A2 gene was identified this is indicated between brackets. Diagnoses in family members at baseline and after follow-up MO = migraine without aura. MA = migraine with aura (patients who had attacks of MO as well as attacks of MA are categorized as MA). HM = hemiplegic migraine.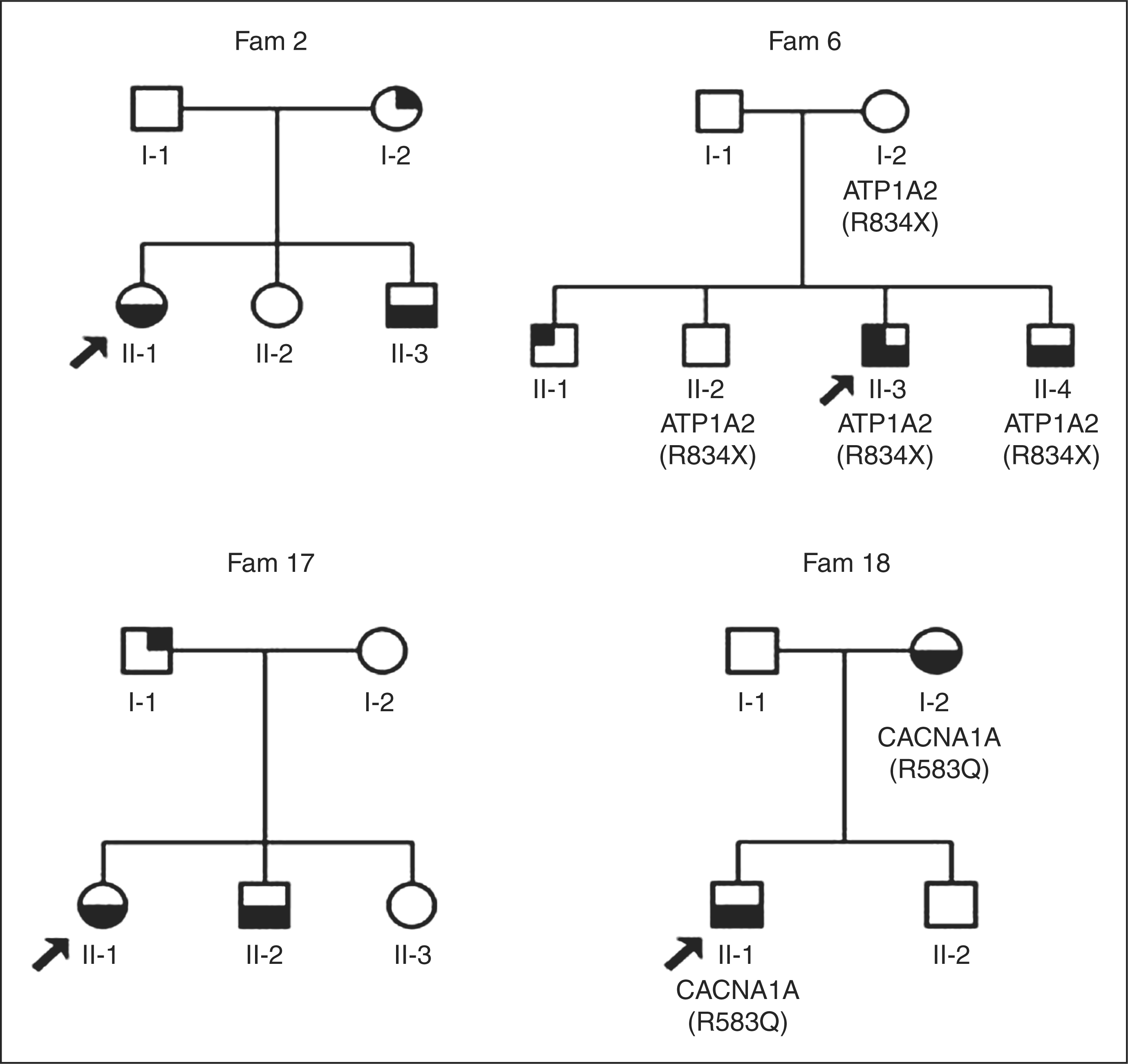


Discussion
We longitudinally studied the diagnostic evolution of 18 SHM patients and their family members and showed that SHM changed into FHM in four of 18 probands with SHM after a follow-up of nine to 14 years. This suggests that the diagnosis of SHM can precede that of FHM in a considerable number of patients (in our study 22%). In half of these patients, an FHM gene mutation was present.
Indeed, a genetic relation between SHM and FHM was recently confirmed in a study performed by our group in which the three known FHM genes (CACNA1A, ATP1A2 and SCN1A) were analyzed in 39 SHM patients (7). Among the 39 patients that were investigated in that study, one CACNA1A mutation and five ATP1A2 mutations were identified, showing that a considerable proportion (i.e. 15%) of SHM patients have a causal FHM gene mutation. Another study reported a considerable lower percentage of functional CACNA1A and ATP1A2 mutations in 100 SHM patients (1% for both genes) (9).
Because SHM patients with de novo mutations may represent the founder of a new family with disabling FHM, genetic conformation of the diagnosis may have consequences for genetic counseling. The current study, which shows that in a clinical follow-up a diagnosis of SHM can change to FHM, also in patients with no mutations in one of the known FHM genes, gives support to the previously stated hypothesis that FHM and SHM share neurobiological mechanisms (10).
Surprisingly, two of the 18 patients with SHM attacks at baseline did not recall having had HM attacks in the past at follow-up. One of these SHM patients has a E120A mutation in the ATP1A2 gene, but as this patient was an 83-year-old woman, it is well conceivable that she simply forgot about the attacks she had when she was younger. This indicates that recall bias, forgetting attacks at younger age, thus may play a role. This may in fact prevent diagnosing FHM in SHM patients as elderly may have forgotten the precise symptoms of their migraine attacks, even when these have been severe, with hemiparesis. This is the more intriguing because in this particular SHM case a mutation in one of the FHM genes has been found but even after a long clinical follow-up still no other family members report hemiplegic attacks. Several steps have been taken to evaluate causality of the E120A mutation (7). In conclusion, the mutation was considered to be a causal mutation, but had apparently milder functional consequences effect than some other ATP1A2 variants. The other SHM patient who did not recall having had HM attacks reported disturbances of coordination at follow-up, but no paresis.
It is noteworthy that of 18 patients with HM four also have attacks of MA. One patient with HM also has attacks of MO. Of 67 first-degree family members 22 have attacks of MA. It is well established that MA occurs more frequently in FHM patients and their first-degree relatives than in the general population (11–13). Based on the very similar aura features of HM and MA (except for the motor component), it could be speculated that MA attacks in HM are in fact abortive HM attacks. The high prevalence of MA in our group of first-degree relatives also gives support to the idea that HM and MA could be part of the same genetic spectrum.
It might be noted that the fact that some of the SHM patients have an FHM mutation can have an effect on the outcome of the follow-up. However, the aim of this study is to follow the clinical evolution of all SHM patients, independent of their genotype. Therefore, we think it is reasonable to keep the SHM patients with FHM mutations in this study. Information about the presence of FHM mutations in these SHM patients is of interest after the follow-up diagnosis was made, to see whether a SHM patient with an FHM mutation might have a different prognosis. So the follow-up diagnosis was made by a clinician who was blinded for the previous diagnosis and for the mutation status of the patient. Only after having diagnosed all patients and their relatives, this information was unblinded. We feel that this also relates better to clinical practice, where neurologists encounter patients with HM often without knowledge on the mutation status of their patients. Furthermore, familial occurrence is often not clear at first visit and may change during the follow-up time, as we observed in our study.
Taken together, our findings stress the importance of a thorough history and follow-up of SHM patients and their families. Even in the absence of family members with hemiplegic attacks, genetic screening of SHM patients can be of value. The occurrence of attacks with hemiplegia in a patient without a positive family history of HM may provide a diagnostic challenge for clinicians. Screening for mutations in patients with attacks of hemiplegia can be helpful to confirm the diagnosis HM, and by finding a mutation other causes can be excluded which can be reassuring for both patient and doctor. Although attacks of HM can be quite worrisome, they are usually relatively benign depending on the type of mutation and FHM gene involved. Furthermore, a negative mutation analysis of the known FHM genes in SHM patients does not exclude a genetic cause for SHM and thus a transmission to the next generation. This may be of importance for an early recognition and treatment of HM attacks in family members. Our genetic and clinical follow-up studies reinforce the evidence that FHM and SHM are part of the same spectrum of migraine.
Footnotes
Disclosures
Anine H. Stam has received independent support from Netherlands Organization for Scientific Research NWO. Mark A. Louter reports no disclosures. Joost Haan has in the past three years received consultancy fees from Merck and Janssen-Cilag. Boukje de Vries reports no disclosures.
Rune R. Frants reports no disclosures. Arn M. J. M. van den Maagdenberg reports no disclosures. Michel D. Ferrari has in the past three years received grants and consultancy or industry support from Almirall, Coherex, Colucid, Eisai, GlaxoSmithKline, Linde, MAP, Medtronic, Menarini, Merck, Minster, Pfizer and and St Jude. Gisela M. Terwindt received consultancy or industry support from Merck and Janssen-Cilag as well as independent support from NWO.
Acknowledgements
This work was supported by grants of the NWO (903-52-291, MDF, Vici 918.56.602, MDF, 907-00-217 GMT and 920-03-473, AHS), and by a grant from the Centre for Medical Systems Biology (CMSB) established by the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (NGI/NWO).