
Abstract
Background
Familial hemiplegic migraine (FHM) is a rare monogenic subtype of migraine with aura, characterized by motor auras. The majority of FHM families have mutations in the CACNA1A and ATP1A2 genes; less than 5% of FHM families are explained by mutations in the SCN1A gene. Here we screened two Spanish FHM families for mutations in the FHM genes.
Methods
We assessed the clinical features of both FHM families and performed direct sequencing of all coding exons (and adjacent sequences) of the CACNA1A, ATP1A2, PRRT2 and SCN1A genes.
Results
FHM patients in both families had pure hemiplegic migraine with highly variable severity and frequency of attacks. We identified a novel SCN1A missense mutation p.Ile1498Met in all three tested hemiplegic migraine patients of one family. In the other family, novel SCN1A missense mutation p.Phe1661Leu was identified in six out of eight tested hemiplegic migraine patients. Both mutations affect amino acid residues that either reside in an important functional domain (in the case of Ile1498) or are known to be important for kinetic properties of the NaV1.1 channel (in the case of Phe1661).
Conclusions
We identified two mutations in families with FHM. SCN1A mutations are an infrequent but important cause of FHM. Genetic testing is indicated in families when no mutations are found in other FHM genes.
Introduction
Familial hemiplegic migraine (FHM) is a rare monogenic subtype of migraine with aura (MA) that is characterized by transient hemiparesis during the aura phase. Headache and aura characteristics are identical in FHM and MA patients, apart from the motor auras and possible longer duration of auras in FHM (1). Mutations in three neuronal or glial ion transporter genes (CACNA1A, ATP1A2, SCN1A) are well-known to cause FHM (2). Recently, PRRT2 was presented as the fourth FHM gene. Although the great majority of patients with a PRRT2 mutation, which virtually always affect the same stretch of cytosine residues, suffer from “typical PRRT2-related phenotypes” (i.e. paroxysmal kinesigenic dyskinesia, benign familial infantile seizures, and infantile convulsion choreoathetosis syndrome), a small number of them also have symptoms of hemiplegic migraine, MA or migraine without aura (3–9). Consequently, unlike for the three well-known FHM genes, more convincing evidence for the importance of PRRT2 as a hemiplegic migraine gene is still needed. At present, more than 70 mutations in the CACNA1A (FHM1) (10) and ATP1A2 (FHM2) (11) genes have been identified, and mutations in these genes seem to account for the majority of cases with FHM (12). Only five mutations were reported for the third FHM (FHM3) gene SCN1A (12,13) that encodes the α1 subunit of neuronal voltage-gated NaV1.1 sodium channels that are primarily expressed on inhibitory neurons (14,15). Of these, SCN1A mutations p.Leu1649Gln and p.Gln1489Lys cause pure FHM, that is without additional symptoms such as epilepsy or cerebellar ataxia (13,16). The other FHM3 mutations either cause FHM and seizures occurring separately from the hemiplegic migraine attacks (p.Gln1489His (17) and p.Leu263Val (18)) or FHM and “elicited repetitive daily blindness” (p.Gln1489His and p.Phe1499Leu mutations (17)). Functional analyses of pure FHM3 mutations predict a loss-of-function effect on inhibitory neurons, thereby leading to overall neuronal hyperexcitability (19), although this is debated by others (20). Notably, SCN1A is a well-known epilepsy gene with several hundred mutations causing various forms of childhood epilepsy: Dravet syndrome (also known as severe myoclonic epilepsy of infancy (SMEI)) and the milder generalized epilepsy with febrile seizures (GEFS+) (21). Here we report the identification of two novel FHM3 mutations in two families with pure hemiplegic migraine.
Subjects and methods
Patients
Family 1
This four-generation Spanish family includes four hemiplegic migraine patients (see Figure 1, Family 1 and Table 1). The family members were interviewed and neurologically examined (ODF). Diagnoses were established according to the International Classification of Headache Disorders, second edition (ICHD-2) (1). The diagnosis of the proband’s deceased father was based on information provided by the proband.
Pedigrees of FHM families. The arrow indicates the proband. Symbols: black lower-half: hemiplegic migraine; right upper square: migraine with aura (MA); left upper square: migraine without aura (MO). WT: homozygous carrier of the wild-type allele. p.Ile1498Met and p.Phe1661Leu: heterozygous carrier of the respective SCN1A mutations. Characteristics of familial hemiplegic migraine patients in Family 1 and Family 2. MA: migraine with aura; MO: migraine without aura; WT: wild-type allele; p.Ile1498Met and p.Phe1661Leu: heterozygous carrier of the respective SCN1A mutations.?: This information was unavailable.
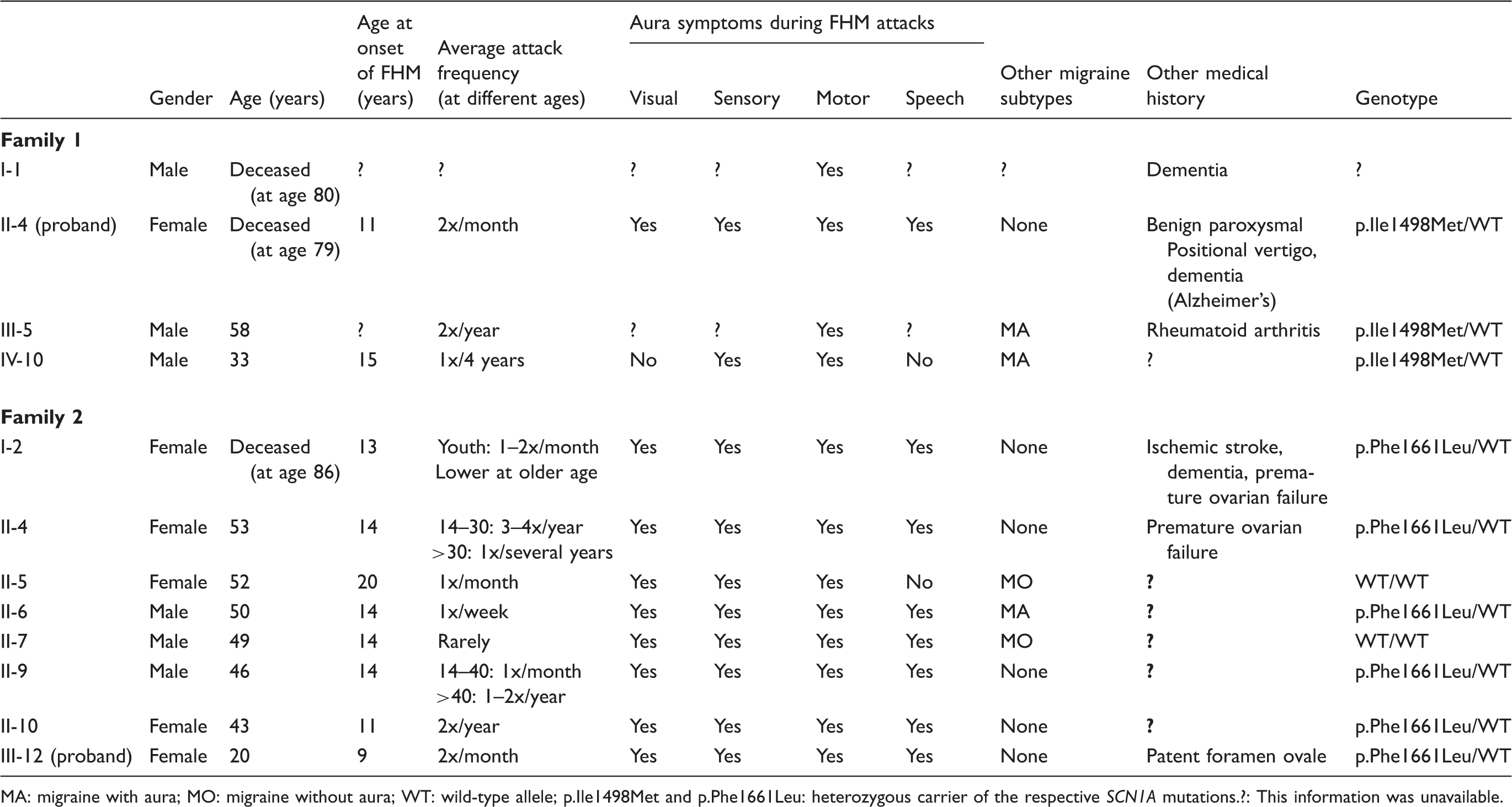
The proband (II-4) had her first migraine attack at age 11 and continued to have on average two attacks per month until the age of 73. These hemiplegic attacks were usually preceded by flashing lights or scotoma, speech problems and/or unilateral sensory symptoms. Auras developed gradually during five to 15 minutes, but on several occasions hemiplegia occurred instantaneously, causing her to fall. In general, hemiplegia lasted 30 to 60 minutes and then diminished gradually. After these auras, she suffered from a typical migraine headache, i.e. unilateral pulsatile headache that increased with movement and was best relieved by sleep, accompanied by photophobia and phonophobia. At the age of 69 years, she experienced an episode of benign paroxysmal positional vertigo that lasted for 15 days and then resolved gradually over the course of two weeks. At age 70, she presented with prolonged hemiplegia for 10 days, which completely recovered during the subsequent three weeks. Cerebral magnetic resonance imaging (MRI) (see Figure 2) and single photon-emission computed tomography (SPECT) (data not shown) during the hemiplegic phase showed vasogenic edema and cortical hypoperfusion in the right hemisphere. Further extensive diagnostic workup for cerebrovascular disease and epilepsy, including electroencephalography, cerebrospinal fluid analysis, echography of the heart and carotid arteries, laboratory analysis of coagulation factors and autoimmune titers, and skin biopsy for excluding cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), showed no abnormalities. From age 72, she showed progressive cognitive deterioration and was diagnosed with Alzheimer’s disease. She died at the age of 79 years of urinary sepsis. Post-mortem examination of the brain showed multiple senile plaques and confirmed the diagnosis of Alzheimer’s disease. No ischemic lesions were found.
Cerebral MRI of the proband of Family 1 during prolonged aura and after full recovery. (a) and (b) MR FLAIR T2-weighted image in axial plane and gadolinium-enhanced T1-weighted image showing a subcortical area of white matter in the perirolandic region on the right is hyperintense on image (a) and relatively hypointense on image (b) with blurring of the cortical sulci adjacent to the convexity. (c) T1-weighted MR image showing the resolution of the radiological abnormalities, in agreement with the patientãs clinical recovery.
The proband’s father (I-1) had similar FHM attacks. He suffered from dementia from age 70 and died at age 80. Patient III-5, now 58 years old, suffered from MA and FHM with an attack frequency of two per year until he was 44. No hemiplegic attacks have occurred over the last 13 years. Patient IV-10, currently 33 years old, has experienced four hemiplegic migraine attacks from age 15 to 31. His attacks started with hypesthesia and hemiparesis during 20 minutes, followed by migraine headache. An MRI scan performed during one of these attacks showed no abnormalities. He also suffers from migraine with visual aura, with an attack frequency of one per three months.
Family 2
This three-generation family is also of Spanish origin. Eight family members were diagnosed with FHM (see Figure 1, Family 2 and Table 1). Their diagnoses were established according to ICHD-2 criteria (1) and confirmed by a neurologist specializing in headache (JP). After the passing of the proband’s grandmother, additional information was provided by her children.
The proband (III-12) had been suffering from approximately three migraine attacks per month since she was nine years old, with visual, sensory and speech auras. At the age of 16 she presented at the emergency department with right-sided hemiparesis, accompanied by scotoma, photopsia, right-sided hypesthesia, contralateral headache and vomiting lasting for three hours. She was diagnosed with hemiplegic migraine.
The proband’s mother (II-4) had three to four FHM attacks per year from age 14 to 30, later decreasing to one attack per several years. All attacks include visual, sensory, motor and speech symptoms lasting 30–60 minutes. The five affected siblings of the proband’s mother experience multiple aura types, in varying combinations during separate attacks, followed by typical migraine headaches (for summary, see Table 1).
The proband’s grandmother (I-2) had suffered migraine attacks since the age of 13. She always experienced visual, sensory and dysphasic auras. Hemiparesis occurred on most occasions. At age 85 she was diagnosed with dementia. She died one year later after a stroke.
Genetic analysis
Genomic DNA was isolated from peripheral leukocytes according to standard procedures (22). Causal mutations in the FHM genes CACNA1A and ATP1A2 were excluded by direct sequencing of all coding exons (and direct flanking sequences) in the probands of both families. We also excluded a causal mutation in PRRT2 by sequencing all four coding exons (and all direct flanking sequences) in the probands of the families. Subsequently, we sequenced all 26 exons and flanking intronic sequences of the SCN1A gene. Detailed information is available from the authors upon request.
Results
The well-known FHM genes CACNA1A and ATP1A2, as well as the more recently implicated disease gene PRRT2, did not yield a possible causal mutation in either of the two Spanish FHM families. Therefore, we sequenced all 26 exons (and direct flanking intronic sequences) of the SCN1A gene in the probands of both families.
In the proband of Family 1, we identified a novel heterozygous point mutation in exon 24 (c.4460G>C; p.Ile1498Met) of the SCN1A gene. The mutation co-segregated with FHM in this family (Figure 1, Family 1) and was absent in our panel of 161 healthy control subjects. The p.Ile1498Met mutation is located in the intracellular loop between transmembrane domains III and IV (Figure 3(a)) and affects the “IFMT motif.” This motif encodes the hydrophobic latch that is responsible for adequate occlusion of the ion pore, which leads to inactivation of the Nav1.1 channel, thereby regulating the generation and propagation of action potentials (14,15). The IFMT motif is named after the four amino acids encoding it, i.e. isoleucine (I), phenylalanine (F), methionine (M) and threonine (T). Mutation p.Ile1498Met leads to the substitution of isoleucine for a methionine residue at the first position of the IFMT motif. Both are hydrophobic nonpolar amino acids with similar molecular weight. Isoleucine is aliphatic while methionine is a sulfur-containing amino acid and is important for the function of the SCN1A-encoding protein. Analysis of this amino acid in homologous and orthologous sodium channel α1 subunits shows high conservation of isoleucine1498 (Ile1498) (Figure 3(b)) (17). Additional support for pathogenicity of the p.Ile1498Met mutation came from three in silico bioinformatics prediction programs, SIFT (deleterious (0)), PolyPhen2 (probably damaging (0.998), and Mutation Taster (disease causing (0.99)), which all indicated that the mutation is likely pathogenic.
Predicted topology of the SCN1A protein and amino acid sequence alignments. (a) Arrows indicate the location of the novel p.Ile1498Met (I1498M) and p.Phe1661Leu (F1661L) FHM3 SCN1A mutations. Previously identified FHM3 mutations are indicated by numbers 1–5. (b) Alignment of the amino acid sequence of alpha subunits of sodium channels from various species.
Mutation scanning of the SCN1A gene in Family 2 revealed a novel heterozygous point mutation in exon 26 (c.4981C>T; p.Phe1661Leu). This mutation partially co-segregated with FHM in this family as two of the eight tested clinically affected individuals (II-5 and II-7) did not have the mutation and are thus diagnosed as phenocopies (Figure 1, Family 2). The p.Phe1661Leu mutation was not found in the proband’s unaffected aunt nor in our panel of 161 healthy control subjects. Phenylalanine1661 (Phe1661) is located in domain IV, in the intracellular loop between the voltage-sensing S4 segment and part of the transmembrane pore (S5) (Figure 3(a)) (21). It is highly conserved within all orthologues and homologues except for the SCN7A gene, which, notably, has a leucine at the equivalent position (Figure 3(b)). Moreover, the importance of Phe1661 is evidenced by the observation that a change of this residue to a serine results in Dravet syndrome (23) and the fact that the mutation affects NaV1.1 channel functioning (24). Additional support for pathogenicity of the p.Phe1661Leu mutation came from the three in silico bioinformatics prediction programs, SIFT (deleterious (0)), PolyPhen2 (probably damaging (1.0)), and Mutation Taster (disease causing (0.99)), which all indicated that the mutation is likely pathogenic.
Discussion
Here we report on two Spanish families with pure hemiplegic migraine (i.e. without other paroxysmal or permanent neurological symptoms such as epilepsy or ataxia) in which we identified two novel SCN1A mutations: p.Ile1498Met and p.Phe1661Leu. Attacks in both families started at a young age, ranging from age 9 to 20 years. Attack frequency was highly variable, ranging from one attack per week to one attack per several years. Prolonged duration of auras, varying from three hours to three weeks, and co-occurrence of FHM attacks with attacks of MA and migraine without aura were observed in both families, as has been reported for other FHM families (25).
Ictal MRI abnormalities (Figure 2), as observed in the proband of Family 1, have not been reported in FHM3 patients, but are similar to observations made in FHM1 and FHM2 (25). Post-mortem examination revealed that the dementia in patient II-4 of Family 1 was due to Alzheimer’s disease. Dementia in patient I-2 of Family 2 is also unlikely related to the SCN1A mutation, given the age at onset after 80.
Although functional testing of the mutations has not been performed, it is probable that mutations p.Ile1498Met and p.Phe1661Leu are pathogenic. First, p.Ile1498Met is present in the three family members with hemiplegic migraine and absent from a panel of 161 control subjects. Second, Ile1498 is a highly conserved amino acid in α1 subunits of vertebrate sodium channels. Third, the mutation is located in the same intracellular loop as three of the five previously described FHM3 mutations (p.Gln1489Lys, p.Gln1489His, p.Phe1499Leu) (Figure 3(a)) (13,17). Of these, the p.Phe1499Leu mutation is also located in the IFMT motif, which encodes the hydrophobic latch, and is essential for the function of the neuronal voltage-gated NaV1.1 sodium channel (14,15). Finally, additional support for the pathogenicity of the pIle1498Met mutation came from in silico bioinformatics prediction tools that consistently predicted that the mutation is likely pathogenic. Interestingly, none of the more than 600 mutations causing Dravet syndrome or GEFS+ affect the IFMT motif (21,26). It is hypothesized that a defective hydrophobic latch will lead to delayed sodium channel inactivation with concomitant decreased neuronal inhibition.
Several lines of evidence indicate that the p.Phe1661Leu mutation in Family 2 is also pathogenic. The mutated Phe1661 is a highly conserved amino acid across homologues and orthologues, except for SCN7A a gene that shares only 52% overall sequence similarity with the SCN1A gene. All three tested in silico bioinformatics prediction programs indicate that the p.Phe1661Leu mutation likely has a deleterious effect on the SCN1A protein. In addition, the p.Phe1661Leu mutation is absent from our panel of healthy control subjects and co-segregates in six of eight hemiplegic migraine patients. The remaining two hemiplegic migraine patients should be considered phenocopies. Phenocopies have been described before in FHM families (27–29).
The p.Phe1661Leu mutation is the first FHM mutation that affects the same amino acid as a known Dravet mutation, i.e. p.Phe1661Ser. In Dravet, Phe1661 is mutated into a much smaller, nucleophilic serine residue (23). Substitution of the phenylalanine for a leucine apparently has a less detrimental effect on protein function, which seems in line with the much milder phenotype in our family. The importance of Phe1661 for sodium channel function is further illustrated by the fact that p.Phe1661Ser affects NaV1.1 channel functioning (24) and that mutating the corresponding residue in SCN4A, which causes paramyotonia congenita, also showed alterations of the kinetic properties of the sodium channel (30).
In conclusion, we identified two novel SCN1A mutations causing pure FHM with large intra-familial variation in severity and frequency of the attacks. Our findings increase the number of FHM-causing SCN1A mutations to seven and illustrate the importance of this gene for genetic counseling and testing of FHM patients, especially when they test negative for CACNA1A and ATP1A2 mutations.
Clinical implications
Screening of SCN1A should be considered in hemiplegic migraine patients if no mutations are found in the CACNA1A and ATP1A2 genes. Familial hemiplegic migraine 3 (FHM3), similar to FHM1 and FHM2, is characterized by a large intra-familial variability in severity and frequency of attacks and accompanying symptoms. Decreased function of neuronal voltage-gated NaV1.1 sodium channels may be the underlying cause of FHM3.
Footnotes
Funding
This work was supported by grants from the Netherlands Organisation for Scientific Research (NWO) (920-03-473, A.H.S., 903-52-291, M.D.F. and R.R.F.; VICI 918.56.602, M.D.F.; VIDI 91711319, G.M.T.; the European Community (EC); and the Centre for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI) (M.D.F. and A.M.J.M.v.d.M). They had no role in the design or conduct of the study.
Conflict of interest
C.M. Weller reports no disclosures. N. Pelzer reports support for conference visits from Menarini and Allergan UK. B. de Vries reports no disclosures. J. Pascual reports consultancy from Allergan. O. De Fàbregues, M.A. Ramos Arroyo, M. Artigas López report no disclosures. Dr Stam reports independent research support from the NWO. S.C. Koelewijn and J. Haan report no disclosures. M.D. Ferrari reports grants and consultancy or industry support from Medtronic, Menarini, and Merck and independent support from NWO, ZonMW, the United States National Institutes of Health (NIH), the European Community, and the Dutch Heart Foundation. G.M. Terwindt reports grants and consultancy/industry support from Merck and Menarini, and independent support from the NWO. A.M.J.M. van den Maagdenberg reports no disclosures.