
Abstract
Background
Migraine is a common neurological disorder with a strong genetic component, yet the precise mechanisms underlying its genetic susceptibility remain largely unknown. Genome-wide association studies (GWAS) have identified multiple risk loci, but monogenic forms of migraine, particularly Familial Hemiplegic Migraine (FHM), have provided deeper insights into the pathophysiology of migraine. Studying monogenic disorders that present migraine as a symptom could help identify novel therapeutic targets by uncovering shared molecular pathways.
Methods
A narrative literature review was conducted using a stepwise approach in the PubMed database. Reviewers were divided into three groups, each focusing on different aspects of migraine genetics. The first group analyzed monogenic migraine syndromes, including FHM and related ion-channelopathies. The second group examined clinical manifestations and phenotypic spectrum of FHM-related genes. The third group expanded the search to other monogenic disorders associated with migraine, such as Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), and Familial Advanced Sleep Phase Syndrome (FASPS). Additional searches were conducted using the Compendium of Causative Genes for Monogenic Disorders.
Results
The review identified multiple monogenic disorders associated with migraine, revealing distinct but interconnected mechanisms. Ion-channel dysfunction (CACNA1A, SCN1A, ATP1A2), vascular impairment (NOTCH3, TREX1), mitochondrial dysfunction (MT-TL1), and circadian dysregulation (CSNK1D) emerged as critical contributors to migraine pathophysiology. These findings highlight the roles of neuronal excitability, cortical spreading depression, trigeminal sensitization, and neurovascular dysfunction in migraine.
Conclusions
Monogenic migraine disorders offer valuable insights into migraine pathogenesis, emphasizing the importance of ion homeostasis, vascular function, and circadian regulation. Although genetic studies have not yet directly translated into new therapeutic targets, the study and knowledge of these rare conditions is pivotal for neurologists and migraine specialists, as it might improve diagnosis and care, and provide new insights into migraine pathophysiology that may ultimately inform future treatments.
This is a visual representation of the abstract.
Introduction
Genetic diseases encompass a group of pathologies that arise due to alterations in the genetic material. Monogenic diseases result from mutations in a single gene, whereas polygenic or complex diseases develop due to the interaction between various genetic variants and environmental factors.
A complex interplay between genetic and environment shapes the clinical phenotype of migraine, a disabling and multifaceted neurological disorder (1,2) affecting almost 15% of the population worldwide (3) and with an estimated heritability of 37–54% (4). A growing body of evidence has identified multiple genetic variants associated with migraine (1,5–8). Populational genomics, specifically, genome-wide association studies, have identified more than 100 susceptibility loci (5,9), affecting genes involved in neuronal excitability, neurotransmission, the vascular system and genes related to migraine-specific drug targets (Calcitonin-Related Polypeptide Alpha (CALCA) / Calcitonin-Related Polypeptide Beta (CALCB), Serotonin Receptor 1 (HTR1F)). Recent advances in polygenic risk scores (PRS) have provided a means to quantify an individual's genetic predisposition to migraine by aggregating the effects of these susceptibility loci. PRS models have shown promise in identifying high-risk individuals and may aid in patient stratification for targeted preventive treatments. However, their clinical utility remains an area of ongoing research, particularly in integrating PRS with environmental and lifestyle factors for a more comprehensive risk assessment (10,11). However, the study of monogenic migraine forms, such as Familial Hemiplegic Migraine (FHM) (5,6), has also proven useful in understanding aspects of migraine pathophysiology, including in developing animal models of migraine. In line with this, the study of other monogenic disorders reporting migraine as a common condition can unravel potential shared pathophysiological mechanisms.
The aim of this review is to describe monogenic disorders that involve migraine as one of their symptoms to provide an important source of knowledge for the understanding of migraine pathophysiology, and to uncover novel potential targets for migraine therapeutics. It must be recognized that, despite more than three decades of genetic research, no therapeutic targets have yet been directly identified through genetics alone. Nevertheless, monogenic conditions are important for clinicians to know about, not only to improve diagnosis and management of affected patients, but also to enhance understanding of migraine mechanisms.
Search strategy and selection criteria
This is a narrative literature review. The reviewers were divided into three groups and worked independently, conducting an extensive literature search in the PubMed database. A stepwise approach was applied to ensure a comprehensive selection of relevant studies. The first group focused on identifying genetic disorders associated with migraine, particularly Familial Hemiplegic Migraine (FHM), by searching for terms related to monogenic conditions, chromosomal aberrations, and genetic mechanisms in migraine. Additionally, specific queries were designed to explore the role of CACNA1A, ATP1A2, SCN1A, and PRRT2 in migraine pathophysiology. The second group examined the clinical spectrum of FHM-related genes, using search terms that combined genetic variants with key phenotypic descriptors such as clinical presentation and disease spectrum. This allowed for the identification of studies detailing the neurological and systemic manifestations associated with mutations in these genes. The third group expanded the search to include other monogenic disorders linked to migraine, using a combination of genetic and clinical search terms. Additionally, this group utilized the Compendium of Causative Genes for Common Monogenic Disorders (12) to systematically explore genes associated with migraine-related conditions. No publication date restrictions were applied, but preference was given to original research articles published in the last ten years, the final search was conducted between November 2024 and February 2025. Reviews were considered when a broader discussion was necessary. Letters to editors, and articles in languages other than English were excluded. As this is a narrative review based primarily on PubMed, we acknowledge the potential for selection bias; however, given PubMed's coverage of neurology/genetics and our citation-tracking steps, we believe the likelihood of missing pivotal studies is low. Each group independently screened relevant articles, resolving conflicts through consensus, and two senior reviewers (E.C., P.P.-R.) supervised the selection process. After an initial screening, all researchers reviewed the full texts of the selected studies, refining the reference list based on research quality and relevance. The final reference selection was approved by the senior reviewers after resolving any remaining conflicts.
Familial hemiplegic migraine (FHM) and other monogenic disorders with mutations in the FHM-associated genes
FHM is a monogenic, autosomal dominant subtype of migraine with aura, clinically characterized by hemiparesis or hemiplegia during the aura phase, often accompanied by visual, sensory, speech, or language disturbances (7,13). Affected individuals frequently have a first-degree or second-degree relative with hemiplegic migraine. Genetically, FHM is caused by single-gene mutations in CACNA1A, ATP1A2, SCN1A, and PRRT2 (6–8). The functional consequences of these mutations typically differ by gene: gain-of-function mutations are most often observed in CACNA1A and SCN1A, whereas ATP1A2 mutations generally lead to loss-of-function (6–8). Beyond migraine, mutations in these genes are implicated in other neurological disorders, highlighting their broader significance in neuronal excitability and synaptic function (Table 1).
Other monogenic disorders involving mutations in FHM-associated genes

All references for studies in this table are listed in the online supplementary references.
*Cytogenetic location refers to the chromosomal band where the gene is mapped, based on OMIM/HGNC nomenclature.
CACNA1A
The CACNA1A gene is located at 19p13 and encodes the alpha 1A subunit of P/Q-type calcium channels (CaV2.1) found on presynaptic neurons in the brain and cerebellum (Online Mendelian Inheritance in Man, OMIM: 601011) (5,7,8,14). Mutations in the CACNA1A gene can either result in a gain or a loss of function. With gain-of-function mutations, there is prolonged opening of the affected calcium channels with an increased influx of calcium ions and neuronal excitability (15). Conversely, with loss of function, there is impaired or improper opening of the calcium channels, leading to less influx of calcium ions and decreased neuronal excitability. Several mutations in the CACNA1A gene with variable phenotypic expressions have been identified (15). Of the more than 25 different CACNA1A mutations associated with FHM1, T666 M and S218L are the most common (6). Clinically, patients with T666 M and S218L mutations typically present with severe hemiplegic migraine attacks, accompanied by coma and fever. Cerebellar degeneration has also been reported in FHM1 (16). Mutations of the CACNA1A gene have been identified in other monogenic disorders, including episodic ataxia type 2 (EA2), developmental and epileptic encephalopathy 42 (DEE42), and spinocerebellar ataxia type 6 (SCA6) (15,17). EA2 is a monogenic disorder with an autosomal dominant pattern of inheritance. Patients with EA2 present clinically with recurrent episodes of imbalanced vertigo and ataxia. Attacks or episodes are usually precipitated by physical or emotional stress (18). Most cases of CACNA1A mutations associated with EA2 are loss-of-function point mutations, which result in decreased neuronal excitability and progressive loss of Purkinje cells in the cerebellum (18). Spinocerebellar ataxia type 6 is a subtype of spinocerebellar ataxia (autosomal dominantly inherited progressive neurodegeneration of the cerebellum) and is characterized by adult-onset, progressive cerebellar-type ataxia, dysarthria, and nystagmus (19,20). DEE42 is characterized by neonatal-onset refractory seizures of various types, which typically begin in the first hours or days of life and are associated with significant developmental delay and intellectual impairment (21). Affected infants may also present hypotonia, tremor, ataxia, and abnormal eye movements. Most patients with DEE42 have heterozygous mutations in the CACNA1A gene (21).
ATP1A2
ATP1A2, located on chromosome 1q23.2 (OMIM 182340), encodes the catalytic α2-subunit of the Na+/K + -ATPase (14,22). The α2-subunit is abundantly expressed in muscle tissue (skeletal, cardiac and vascular smooth muscle) and in the brain, where it is primarily found in cortical astrocytes, though neuronal expression has been described during developmental periods (23). The Na+/K + -ATPase regulates electrochemical Na + and K + gradients across the plasma membrane, which are essential for cellular resting potential and excitability, and activity of secondary active transporters (23). Dysfunction of neuronal and astrocytic Na+/K + -ATPases results in hyperexcitability due to extracellular K + and intracellular Ca2 + accumulation, and failure to clear neurotransmitters such as glutamate from the synaptic cleft, which increases susceptibility to CSD(23).
ATP1A2 mutations in FHM2 typically cause partial or complete loss of function. However, certain variants that subtly affect the voltage dependence of the pump may exhibit voltage-dependent functional effects, resulting in loss of function at some membrane potentials and gain of function at others (23). ATP1A2 mutations have also been implicated in sporadic hemiplegic migraine (SHM2) and common forms of migraine (i.e., migraine with and without aura); familial migraine with brainstem aura (24); FARIMPD, an autosomal recessive syndrome with fetal akinesia, respiratory insufficiency, microcephaly, polymicrogyria, and dysmorphic facies, typically resulting in death in infancy (25); developmental and epileptic encephalopathy-98 (26); and alternating hemiplegia of childhood 1 (27). Recently, an association between a novel heterozygous missense mutation in ATP1A2, resulting in a depolarizing inward leak current in skeletal muscle under hypokalaemic conditions, and hypokalemic periodic paralysis (HypoPP) with CNS involvement was suggested. HypoPP is a rare neuromuscular disease, usually caused by an aberrant depolarizing leak current through mutant sarcolemmal voltage-gated calcium or sodium channels. Mutations in ATP1A2 have not previously been linked to muscle dysfunction (28). Other pathologies have also been reported, often in relation to the above-mentioned disorders (see Table 1). Phenotypic overlap can present a diagnostic challenge.
SCN1A
SCN1A, located on chromosome 2 (OMIM 182389) (14), encodes the alpha subunit of the sodium voltage-gated channel Nav1.1. This channel plays a critical role in generating and propagating action potentials in cortical neurons, especially in GABAergic inhibitory interneurons (29).
Mutations in SCN1A are implicated in numerous monogenic disorders besides FHM3: in particular, SCN1A is one of the most important epilepsy genes, resulting in a wide phenotypic and prognostic spectrum ranging from self-limited and pharmacoresponsive epilepsies to developmental and epileptic encephalopathies (DEEs).
SCN1A is most notably associated with Dravet syndrome (DS), a refractory DDE in which about 80% of cases are attributed to more than 1800 pathogenic variants (30). Additionally, about 20% of families with Genetic Epilepsy with Febrile Seizures Plus (GEFS+) harbor mutations in this gene (30).
Other epileptic encephalopathies linked to SCN1A include Epilepsy with Myoclonic Atonic Seizures (EMAtS), Epilepsy of Infancy with Migrating Focal Seizures (EIMFS), West syndrome, Lennox-Gastaut syndrome, Rett syndrome and Nonsyndromic Epileptic Encephalopathies (NEE) (31).
Beyond epileptic disorders, SCN1A mutations are also associated with non-epileptic conditions, such as autism spectrum disorder (ASD), Sudden Unexpected Death in Epilepsy (SUDEP) and non-epileptic sudden deaths (31); rarely, they have been linked to Arthrogryposis Multiplex Congenital (AMC) and Melkersson-Rosenthal syndrome (MRS) (32).
The diversity in phenotypes is partly explained by the functional effects of SCN1A mutations. The mutational spectrum of FHM3 is limited to missense mutations, typically resulting in a gain of function and enhancing NaV1.1 activity and triggering cortical spreading depression (CSD) (33). In contrast, epileptic phenotypes often arise from missense or truncating mutations that cause loss-of-function, consistent with haploinsufficiency, leading to reduced GABAergic interneuron inhibition, network hyperexcitability, and thus seizures.
PRRT2
PRRT2, located on chromosome 16p11.2 (OMIM 614386), encodes the Proline Rich Transmembrane Protein 2 and is highly expressed in the human brain, particularly in the cerebral cortex, basal ganglia and cerebellum (34,35). PRRT2 localizes to presynaptic terminals, especially at glutamatergic synapses, where it interacts with and modulates different ion channels, e.g., GRIN1A, a glutamate receptor and member of the AMPA receptor family, and voltage-gated Nav1.2 and Nav1.6 channels (34,35). Additionally, PRRT2 interacts with proteins of the exocytosis complex, e.g., the synaptic t-SNARE protein Synaptosomal-Associated Protein 25 (SNAP25), indicating that PRRT2 has a possible role in synaptic endocytosis, vesicle docking, and Ca2+-mediated neurotransmitter release (34,35). PRRT2 also plays an important role in brain development and synaptogenesis (34,35). Mutations in PRRT2 have recently been associated with hemiplegic migraine, and due to its prevalence, it has been suggested as the fourth HM gene (FHM4) (36). FHM4 associated mutations typically involve loss-of-function caused by nonsense/missense mutations, deletions, duplications, frameshift mutations leading to premature stop codons or read-through mutations resulting in longer transcripts, which result in synaptic dysfunction and increased excitability of neural networks (34–36). Mutations in the gene have also been implicated in benign familial infantile seizures (BFIS2) (14,34,35); Episodic kinesigenic dyskinesia 1 (EKD1) and Familial Infantile Convulsions with Paroxysmal Choreoathetosis (ICCA), a syndrome with overlapping features from BFIS2 and EKD1 (14,34,35). Other pathologies relating to paroxysmal movement disorders, seizures, and intellectual disability and/or developmental delay have also been reported (see Table 1). Different phenotypic presentations can occur from the same mutations in PRRT2, and overlap can happen (14,34–36).
Potential genes involved in FHM and associated disorders
While CACNA1A, ATP1A2, SCN1A, and PRRT2 are the primary genes implicated in FHM, their mutations do not fully explain the full spectrum observed in clinical practice, suggesting additional genetic contributors (37). Several other genes, including PNKD, SLC4A4, SLC1A3, SLC2A1, ATP1A3, and ATP1A4, have been linked to FHM and related neurological disorders, influencing synaptic function, ion transport, and neurotransmitter regulation (14,38–40). A detailed overview of these genes, their functions, and associated phenotypes is provided in Table 1.
Monogenic disorders reporting association with migraine
The literature review showed different monogenic disorders reporting significant association with migraine (see Table 2).
Monogenic disorders associated with increased migraine prevalence

All references for studies in this table are listed in the online supplementary references.
Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
CADASIL is one of the most common causes of inherited monogenic cerebral small-vessel disease characterized by severe leukoencephalopathy and high risk of recurrent ischemic events and vascular dementia in adults. The prevalence of CADASIL is estimated to be 2–5 cases per 100,000 individuals (41). It is caused by mutations in the NOTCH3 gene (>95% of cases) (42), which encodes a transmembrane receptor primarily expressed in vascular smooth muscle cells. More than one hundred mutations are reported, all occurring in exons 2–24, with strong clustering in exons 3 and 4 which encode several EGF-like extracellular domains. The majority are missense mutations involving a cysteine residue, others are small-in frame deletions or splice-site mutations. The histopathological hallmark is the degeneration of vascular smooth muscle cells, characterized by local accumulation of the Notch3extracellular domain, aggregation, and impaired endocytosis. This prevents protein ubiquitination and degradation and leads to the formation of extracellular deposits of granular osmiophilic material and fibrous thickening of the arterial walls (43,44). Immunofluorescence techniques reveal the accumulation of NOTCH3 protein in the vessel walls (42). Although these histopathological findings are systemic (45), in the cerebral vessels they can lead to cerebral dysfunction.
The main clinical features include migraine with aura, recurrent subcortical/lacunar ischemic events, mood disturbances, apathy, cognitive impairment, and seizures (46–49). Brain MRI shows classical white matter hyperintensities in typical sites: anterior temporal pole lobe, periventricular white matter, internal and external capsule, pons, multiple lacunar infarcts and microbleeds. Also, lacunar infarcts are frequent, with association with cognitive dysfunction.
Migraine prevalence in CADASIL patients ranges from 14 to 72% (50) with variability in different geographic areas (higher in occidental countries compared to Asian countries) and a pooled prevalence of 38% (95% CI of 33–42%). The prevalence of migraine with aura is higher in CADASIL patients (51), compared to the general population, and is more frequent in females who presented the first attacks at younger age than men.
Migraine with aura is the most frequent and with an earlier clinical presentation occurring in the second-third decade of life, also migraine without aura and tension type headache are reported. The aura is typical visual or sensory aura in almost half of the cases (52), while the other half could present with atypical features: aura without headache, hemiplegic auras, brainstem aura, prolonged aura, and acute onset aura. Rarely patient with CADASIL may present with acute status migrainosus with persistent aura or with prolonged and persisting headache, acute altered mental status (visual hallucinations, behavioral disturbances), speech difficulties and memory deficits that could also evolve to coma (53–56).
The exact mechanism linking CADASIL to migraine manifestation is not fully understood. However, it is hypothesized that the vascular abnormalities caused by NOTCH3 mutations may lead to chronic subcortical ischemia, which can trigger cortical spreading depression, a phenomenon associated with migraine aura. No association between onset and severity of migraine presentation and the MRI signs of microvascular damage in both white matter and basal ganglia (52) was found nor association with CGRP levels (57).
Another known associated factor that could modulate migraine susceptibility is the hyperhomocysteinemia that could lead to a younger age of onset (58). Different NOTCH3 gene variants may influence the presentation and severity of migraine attacks in CADASIL patients. Some variants have been linked to variable headache courses, as shown in a recent study identifying specific pathogenic mutations (e.g., p.Tyr189Cys, p.Arg153Cys, p.Cys144Arg). Additionally, certain benign mutations are associated with specific migraine phenotypes, such as aura type (59). However, these findings are limited by small sample sizes and geographically restricted cohorts. There is no specific targeted treatment for CADASIL, nor for migraine associated with CADASIL, clinicians should also consider other comorbidities and eventual symptom worsening (e.g., neuropsychiatric manifestations). No clinical trials are available for this rare condition, but according to an individual case level meta-analysis beta-blockers and simple analgesics are the most commonly prescribed drugs (60).
However, the authors stated that beta-blockers should be avoided in CADASIL patients due to the potential for adverse events, possibly related to impaired cerebral vasoreactivity and reduced perfusion reserve (61–63). In contrast, acetazolamide and valproate showed neutral results compared to other preventive treatments and did not exhibit adverse effects. However, they warrant further investigation, as their potential efficacy is supported by specific mechanisms of action on smooth muscle vessels and anecdotal evidence from small cohorts and case series (61,62,64,65).
Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S)
RVCL-S is an autosomal dominant systemic small-vessel disease, caused by mutations in the TREX1 gene (66). Clinically, RVCL-S is characterized by progressive white matter lesions, focal neurological deficits, migraine, cognitive decline, psychiatric disturbances, and seizures. Systemic features include vascular retinopathy, liver dysfunction, nephropathy, hypertension, anemia, Raynaud's phenomenon, and gastrointestinal bleeding. The disorder typically manifests around the age of 40, with a median survival age of 53 years (67,68).
TREX1 encodes a 3′-5′ DNA exonuclease that plays a crucial role in clearing cytosolic DNA and preventing activation of the innate immune response (67). Mutations in TREX1 result in the accumulation of cytosolic DNA, triggering chronic inflammation via the STING pathway, which underpins the systemic vasculopathy observed in RVCL-S (1,68). Migraine is a common feature in TREX1-associated disorders, affecting 42–59% of individuals with mutations (67,68). Migraine attacks often present with aura and may precede other systemic or neurological symptoms, complicating early diagnosis (1). The pathophysiology linking TREX1 mutations to migraine is not fully understood, but vascular dysregulation and chronic inflammation likely lower the threshold for migraine initiation. The overlap between migraine aura phenomena and other neurological symptoms of RVCL-S, such as visual disturbances and cognitive changes, further highlights the complexity of this relationship.
Current research on TREX1-related disorders faces several limitations. Most studies involve small sample sizes or retrospective data collection. Furthermore, migraine progression in relation to other RVCL-S symptoms is poorly documented due to a lack of longitudinal studies (1,67,68).
KCNK18 related disorders
Mutations in the KCNK18 gene, encoding the TWIK-related spinal cord potassium channel (TRESK), are associated with familial migraine with aura. KCNK18 mutations were first linked to migraine through the identification of a frameshift mutation (F139WfsX24) in a multigenerational family, where it segregated with typical migraine with aura in an autosomal dominant pattern (69–71). TRESK is a two-pore domain potassium channel highly expressed in trigeminal and dorsal root ganglion neurons. It regulates resting membrane potential and reduces neuronal excitability, thus acting as a brake on nociceptive signaling. Loss-of-function mutations in TRESK increase the excitability of trigeminal nociceptors, enhancing susceptibility to migraine attacks. In experimental models, TRESK-deficient neurons exhibit increased calcitonin gene-related peptide (CGRP) release, which contributes to central and peripheral sensitization observed in migraine (69–71).
Beyond migraine, KCNK18 mutations have also been implicated in intellectual disability and neurodevelopmental disorders. Biallelic variants affecting TRESK function have been reported in individuals with global developmental delay, cognitive impairment, and seizures, suggesting a broader role of KCNK18 in neuronal homeostasis and brain function (72).
Individuals carrying the F139WfsX24 mutation typically report severe migraine, accompanied by visual aura, photophobia, and nausea, often beginning in adolescence. However, inconsistent findings regarding the pathogenicity of some KCNK18 variants in sporadic migraine suggest that additional genetic or environmental factors modulate this relationship (73,74).
Current research on KCNK18 and its role in migraine faces notable limitations. While the F139WfsX24 mutation is well-characterized, other missense variants demonstrate variable effects on channel function, complicating genotype-phenotype correlations. Studies often involve small cohorts, with limited representation of diverse populations, reducing the generalizability of findings. Hence, despite evidence linking TRESK dysfunction to trigeminal hyperexcitability, the precise molecular pathways connecting KCNK18 mutations to migraine remain inadequately understood (69,75).
Familial advanced sleep phase syndrome (FASPS)
FASPS is a rare, autosomal-dominant circadian rhythm disorder characterized by early sleep onset and early morning awakening, often beginning in childhood or adolescence. This condition is associated with mutations in the CSNK1D gene, encoding casein kinase Iδ (CKIδ), a critical regulator of circadian rhythms through phosphorylation of core clock proteins such as PER2 (76,77). The CKIδ-T44A mutation results in reduced kinase activity, which shortens the circadian period in humans and animal models, leading to the FASPS phenotype (76). Headaches, including migraine with aura, are frequently observed in FASPS-affected individuals. In one study, all CKIδ-T44A mutation carriers experienced migraine, predominantly with aura, characterized by visual disturbances and heightened sensitivity to light and sound (77). Experimental models have shown that CKIδ mutations increase cortical spreading depression susceptibility (78), a key mechanism underlying migraine aura, and enhance nitroglycerin-induced hyperalgesia, further supporting a mechanistic link between circadian dysregulation and migraine pathophysiology. Moreover, some studies described the role of CK1δ in regulating astrocytic connexin-43 (Cx43) forming gap junctions between astrocytes (79). Mice lacking Cx43 in astrocytes showed an increased velocity of hippocampal spreading depolarization waves (79). Although evidence remains limited to single reports and pre-clinical studies, the FASPS model offers valuable insights into how circadian rhythms interact with migraine (80).
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS)
MELAS is a mitochondrial disorder that manifests with migraine or migraine-like features (81,82). The prevalence of MELAS based on specific mutation is estimated to be 236 per 100,000 (0.24%;95% CI 0.10–0.49%) in Caucasian populations (83). This syndrome is typically maternally inherited and occurs in children and young adults (84,85), although adult-onset (86,87) cases have been reported.
MT-TL1 is the implicated gene in MELAS syndrome (84). This gene, located at position 12 on the short arm of the mitochondrial DNA, facilitates the transfer of the amino acid leucine to the polypeptide chain during translation. The most common variant accounting for approximately 80% of cases is the A3243G mutation which involves substitution of Adenine(A) to Guanine(G) in the nucleotide of the transfer RNA (tRNA) (81,87). Other mutations include m.3271T > C, m.3252 A > G (88). This point mutation causes defective mitochondrial translation and protein synthesis resulting in energy production deficiency. This energy deficiency leads to abnormal proliferation of mitochondria, which manifests in different organ-systems like myopathy, cardiomyopathy, and angiopathy and impairs blood perfusion to major organs. Clinical features of MELAS include stroke-like episodes, ataxia, intellectual disability, hearing loss, seizures, cortical blindness, hypertrophic cardiomyopathy, renal failure, diabetes mellitus, hypothyroidism or hyperthyroidism and psychiatric disorders (82,84,89). Headache prevalence ranges from 35.5% to 70% depending on the population (82,87,90). Headaches may present as recurrent migraine attacks, migraine-like headaches that resemble migraine in their clinical features – such as unilateral throbbing pain with photophobia – but do not meet the full diagnostic criteria. These migraine-like episodes may also overlap with or be precipitated by stroke-like events (82,85,86). The frequency and duration of headaches are variable, with no reported racial predilection. Although the exact mechanism of MELAS and migraine is not known, neuronal hyperexcitability and trigeminovascular activation have been suggested (91). The severity of migraine headaches may be associated with defects in the respiratory chain (82).
Treatment is primarily symptomatic and multidisciplinary, depending on the affected organs or systems. An open-label clinical trial reported that taurine combined with L-arginine reduced the frequency of stroke-like episodes (92). L-arginine is effective for acute episodes, and oral supplementation reduces stroke-like episodes (93). Additionally, erenumab at a dose of 70 mg was reported to be effective and well tolerated in a patient with MELAS with chronic treatment-refractory migraine (94). Although clinical trials are available, there is a lack of consensus on medications to treat MELAS Syndrome.
COL4A1 related disorder
Mutations in the COL4A1 gene, responsible for type IV collagen synthesis, are associated with a variety of clinical phenotypes, including juvenile stroke, leukoencephalopathy and ocular abnormalities (95). These mutations can cause vascular fragility and cerebral microangiopathy. A history of migraine has been reported in several studies, often involving familial cases with documented genetic mutations (96,97). Available evidence is limited by small sample sizes and non-standardized methodologies.
Therapeutic considerations in FHM and other monogenic causes of migraine
Since controlled trials are not available, current evidence is based on single reports and case series. An 11-case study involving four FHM patients and seven Sporadic Hemiplegic Migraine patients demonstrated that onabotulinumtoxinA treatment reduced both headache intensity and aura severity and frequency in most participants. The majority of patients received a 150-U modified PREEMPT protocol under the 12-week cycle which resulted in a typical “wear-off” period between weeks 9 and 10 (98). The anti-CGRP monoclonal antibodies have demonstrated therapeutic effects across different genetic backgrounds; galcanezumab reduced headache days and acute medication use and MIDAS scores and attack severity in PRRT2-associated FHM patients in a small family study (99) and a sporadic HM patient with SCN1A variant mutation showed positive response to an anti-CGRP mAb after standard preventive treatments failed (100). Observational studies of FHM1 patients with CACNA1A T666 M mutation show that calcium antagonist medications like verapamil and flunarizine provide long-term benefits, while studies suggest acetazolamide and lamotrigine could work for specific patient phenotypes (101).
Two patients with chronic migraine in CADASIL showed enduring benefits from erenumab treatment without any adverse effects according to research (102). However, caution has been raised regarding the use of CGRP blockade in small-vessel disease, as CGRP might exert vasoprotective functions during ischemia, and its inhibition might theoretically worsen cerebrovascular outcomes (103). No published evidence is currently available on the use of gepants in FHM or CADASIL. Overall, the reported benefits of onabotulinumtoxinA and peripherally acting CGRP-targeted therapies suggest that such approaches may have a role in managing disorders characterized by Central Nervous System dysfunction, although further research is warranted to clarify safety and efficacy across genetic subtypes.
Insights
Monogenic diseases provide crucial insights into migraine pathophysiology and may help identify new therapeutic targets. Investigating these genetic conditions enhances our understanding of both migraine aura and headache mechanisms, paving the way for innovative treatment strategies. The genetic landscape of monogenic migraine disorders mirrors the complexity observed in GWAS studies, where multiple genes with diverse functions—vascular regulation, inflammation, and neuronal excitability—contribute to disease susceptibility. This supports the notion that migraine is a multifactorial disorder influenced by various genetic pathways. (Figure 1) A key feature of monogenic migraine disorders is the disruption of ion homeostasis in both neuronal and glial cells. In FHM type 1 and 3, gain-of-function mutations in ion channels lead to excessive glutamatergic transmission and an increased susceptibility to cortical spreading depression (CSD), a critical event in migraine aura. Meanwhile, FHM type 2, associated with ATP1A2 mutations, highlights the role of glial cells in synaptic ionic balance, where loss-of-function mutations result in neuronal hyperexcitability and heightened susceptibility to CSD (22,104). These findings align with GWAS research identifying migraine risk loci within glutamate receptor genes, reinforcing the central role of synaptic excitability in migraine pathogenesis (9). Beyond ion homeostasis, monogenic disorders such as KCNK18-related disorders may be linked to increased excitability of trigeminal neurons, which could trigger migraine attacks. CADASIL exemplifies how vascular dysfunction can predispose individuals to migraine. The vascular abnormalities in CADASIL likely trigger ischemic events and facilitate CSD, reinforcing the vascular component in migraine pathophysiology (46). On the other hand, FASPS highlights the potential role of circadian biology and hypothalamic function in migraine. The mutation alters the regulation of core clock proteins, which not only affects sleep-wake cycles but also increases susceptibility to CSD in preclinical models (77). These findings suggest that circadian misalignment and hypothalamic dysregulation may act as migraine triggers, offering potential pathways for interventions targeting circadian biology and hypothalamic pathways. Among the different migraine phenotypes, migraine with aura appears to have a stronger genetic basis than migraine without aura. This is supported by its predominant presence in monogenic disorders, likely due to the direct impact of genetic mutations on neuronal excitability and CSD. The link between aura and these mutations provides a crucial perspective on the underlying biological processes that distinguish migraine subtypes. Further reinforcing the genetic connection, the genes implicated in FHM are often associated with other episodic neurological disorders, such as epilepsy and ataxias. While some of these conditions are neurodegenerative, most involve transient dysfunction rather than progressive deterioration, suggesting shared genetic pathways between migraine and other paroxysmal disorders. A particularly illustrative case is MELAS, where mitochondrial dysfunction, particularly affecting the respiratory chain and energy metabolism, plays a central role (84). This highlights the importance of cellular energy homeostasis in migraine pathogenesis and suggests potential therapeutic avenues aimed at improving mitochondrial function. In parallel, therapeutic observations from case reports highlight that peripherally acting treatments such as onabotulinumtoxinA and anti-CGRP monoclonal antibodies can reduce attack frequency and severity in FHM, while preliminary data in CADASIL suggest possible benefits but also raise safety considerations due to the vasoprotective role of CGRP. Although anecdotal, these findings provide practical insights for clinicians managing patients with genetic migraine syndromes and underscore the need for systematic studies. By understanding these genetic pathways, we could not only enhance migraine treatment but also improve care for a range of related neurological and systemic disorders providing broad therapeutic benefits across neurological and systemic disorders.
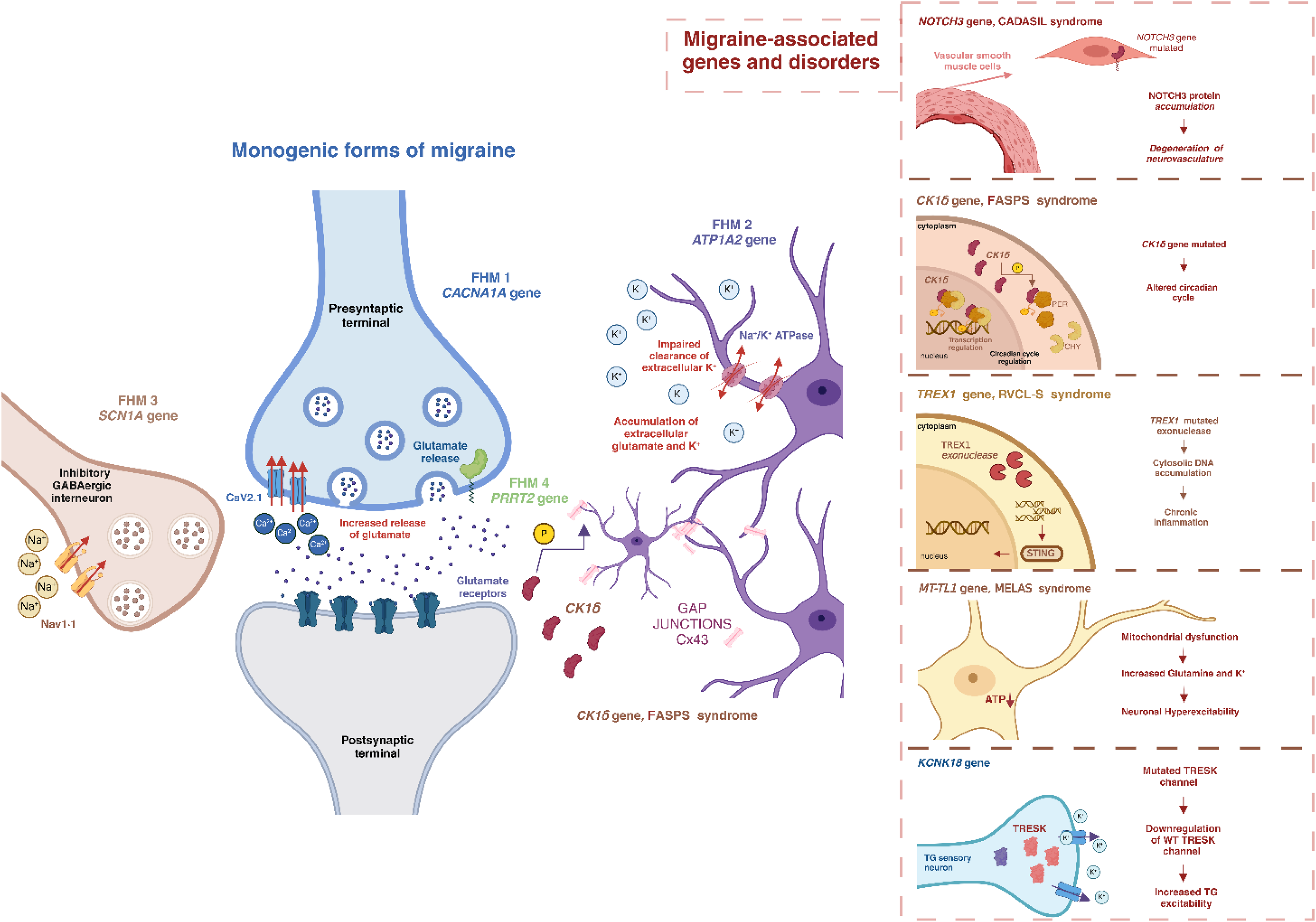
Genes involved in monogenic forms of migraine and migraine-associated disorders. Representation of the function and localization of genes involved in FHM in an excitatory presynaptic and postsynaptic terminal, an inhibitory GABAergic interneuron and astrocytes. CACN1A gene encoding for the voltage-gated calcium channel CaV2.1, and responsible for FHM1, regulates the calcium influx and the release of glutamate in the synaptic cleft. ATP1A product is localized in astrocytes and is involved in the clearance of extracellular K+ and glutamate. Located in a GABAergic interneuron can be found represented the gene responsible for FHM3, SCN1A, which encodes a Na+ channel and regulates the ion influx. FHM4 involves the PRRT2 gene, which has been associated with the regulation and fusion of synapse vesicles to the plasma membrane. All the genes implicated in FHM contribute to an increase in extracellular K+ and glutamate, which results in increased hyperexcitability and susceptibility to CSD. NOTCH3, CK1δ, KCNK18, TREX1 and MT-TL1 are some of the genes that have been associated with migraine and are responsible for other monogenic disorders. NOTCH3 is a gene expressed in vascular smooth muscle cells and accumulates in the cerebral vasculature causing neurovascular degeneration. CK1δ gene encodes a kinase which plays a key role in regulating the circadian cycle by interacting with core clock proteins PER and CRY. In addition, CK1δ interacts with astrocytic Connexin-43. KCNK18 gene encodes for a two-pore domain potassium channel named TRESK, whose mutated form interacts with the wild-type protein and causes its downregulation resulting in an increased excitability. TREX1 mutated exonuclease, related to RVCL-S syndrome, leads to an accumulation of cytosolic DNA which results in chronic inflammation through STING pathway. MT-TL1 mutated gene, which is responsible for MELAS syndrome, results in increased neuronal hyperexcitability due to mitochondrial dysfunction. ATP1A = ATPase Na+/K+ transporting subunit alpha 1. CACN1A = calcium voltage-gated channel subunit alpha1. Cav2·1 = calcium voltage-gated channel subunit α1 A. CRY = cryptochrome proteins. CK1δ = casein kinase I isoform delta. Cx43= Connexin 43 protein. FHM = familial hemiplegic migraine. KCNK18 = potassium two pore domain channel subfamily K member 18. Nav1·1 = sodium voltage-gated channel α subunit 1. MELAS = maternally inherited mitochondrial disorder. MT-TL1 = mitochondrially encoded tRNA-Leu (UUA/G) 1. NOTCH3 = notch receptor 3. PER = period circadian protein homolog 1. PRRT2 = proline rich transmembrane protein 2. SCN1A = sodium voltage-gated channel alpha subunit 1. STING = cGAMP–stimulator of interferon genes pathway. TREX1 = three prime repair exonuclease 1. RVCL-S = Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations. Figure created with BioRender.com
Conclusions
Migraine is a complex genetic disorder that involves various biological pathways. The study of monogenic forms of migraine and of monogenic disorders reporting strong association with migraine can help in the identification of pathophysiological mechanisms of migraine, including ion homeostasis, vascular function, and circadian regulation. Future studies will be essential to better characterize these pathways. Even if direct genetic-to-therapy translation has not yet occurred, knowledge of monogenic conditions remains vital for accurate diagnosis, optimized patient care, and deeper pathophysiological understanding that could eventually guide therapeutic innovation.
Key findings
Monogenic disorders offer a natural model to dissect migraine pathophysiology, particularly in relation to Migraine with Aura
Genes such as CACNA1A, ATP1A2, and SCN1A are central to our understanding of familial hemiplegic migraine and highlight the role of ion channel dysfunction.
Overlap with other rare genetic syndromes (e.g., PRRT2-related paroxysmal disorders, mitochondrial and vascular diseases) suggests shared complex and multifaced neuronal mechanisms.
Insights from these disorders may guide precision medicine, identifying potential targets for future disease-modifying therapies.
This review offers a comprehensive, updated synthesis of migraine-related monogenic conditions.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251389241 - Supplemental material for Migraine in monogenic disorders: Shedding light on new therapeutic targets
Supplemental material, sj-docx-1-cep-10.1177_03331024251389241 for Migraine in monogenic disorders: Shedding light on new therapeutic targets by Davide Mascarella, Zixuan Alice Zhuang, Otilia Gliga, Federico De Santis, Chiara Rosignoli, Austeja Dapkute, Mundih Noelar Njohjam, Elena Mazzotta and Funmilola Eunice Adewinle, Edoardo Caronna, Patricia Pozo-Rosich, in Cephalalgia
Footnotes
Author contributions
D.M.: literature research and data curation, writing original draft, editing and reviewing manuscript, project supervision. Z.A.Z.: Literature research and data curation, writing original draft, subgroup supervision. O.G.: literature research and data curation, writing original draft, subgroup supervision, visualization. F.D.S., C.R., A.D., M.N.N., E.M., F.E.A.: literature research and data curation, writing original draft. E.C., P.P.-R.: conceptualization and methodology, editing and reviewing manuscript. project supervision.
Data availability statement
Complete search methods and articles found and selected are available upon request.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: D.M has received educational and travel grants from Eli Lilly, Organon, Abbvie, Teva. E.C. has received honoraria from Novartis, Chiesi, TEVA, Lundbeck, Medscape. P.P.-R. reports that within the prior 36 months, having received honoraria as a consultant and speaker for: AbbVie, Amgen, Biohaven, Chiesi, Eli Lilly, Lundbeck, Medscape, Novartis, Pfizer and Teva. Her research group has received research grants from Novartis, Teva, AbbVie, EraNET Neuron, RIS3CAT FEDER, AGAUR, ISCIII, International Headache Society; has received funding for clinical trials from Alder, Amgen, Biohaven, ElectroCore, Eli Lilly, Lundbeck, Novartis, Teva. She is the Honorary Secretary of the International Headache Society. She is a member of the Clinical Trials Guideline Committee of the International Headache Society. She serves as an associate editor for Cephalalgia, Headache, The Journal of Headache and Pain, Neurologia, and Revista de Neurologia. She is the founder of ![]() . P.P.-R. does not own stocks from any pharmaceutical company. All remaining authors have nothing to declare.
. P.P.-R. does not own stocks from any pharmaceutical company. All remaining authors have nothing to declare.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.