
Abstract
We studied the safety and efficacy of 0 U, 50 U, 100 U, 150 U (five sites), 86 Usub and 100 Usub (three sites) botulinum toxin type A (BoNTA; BOTOX®; Allergan, Inc., Irvine, CA, USA) for the prophylaxis of chronic tension-type headache (CTTH). Three hundred patients (62.3± female; mean age 42.6 years) enrolled. For the primary endpoint, the mean change from baseline in the number of TTH-free days per month, there was no statistically significant difference between placebo and four BoNTA groups, but a significant difference favouring placebo vs. BoNTA 150 was observed (4.5 vs. 2.8 tension headache-free days/month; P = 0.007). All treatment groups improved at day 60. Although efficacy was not demonstrated for the primary endpoint, at day 90, more patients in three BoNTA groups had ≥50± decrease in tension headache days than did placebo (P ≤ 0.024). Most treatment-related adverse events were mild or moderate, and transient. BoNTA was safe and well-tolerated in the study.
Introduction
Tension-type headache (TTH) is one of the primary headache disorders—having an overall annual prevalence as high as 38%—with 2% of patients affected on a daily or almost daily basis (1). Sensitization of afferents within peripheral tissues (e.g. muscles) by local nociceptive mediators can give rise to increased nociceptive afferent fibre activity, leading to central sensitization, and may be a pathogenic marker in chronic tension-type headache (CTTH) (2–8). The exact role of peripheral and central sensitization in CTTH is still unclear (8–10).
At motor neurons, botulinum toxin type A (BoNTA; BOTOX®; Allergan, Inc., Irvine, CA, USA) inhibits exocytosis of acetylcholine from presynaptic nerve endings through proteolytic cleavage of a specific target protein, SNAP-25 (synaptosomal protein with a molecular weight of 25 kDa), that is essential for vesicle fusion with neuronal membranes leading to decreased muscle contractions (11). In preclinical studies, BoNTA has also been shown to block the release of nociceptive mediators such as substance P, glutamate, and calcitonin gene-related peptide (CGRP) at non-cholinergic, non-motor neurons via SNAP-25 cleavage (11–14). Cui and colleagues have demonstrated that local peripheral injection of BoNTA significantly reduces formalin-induced nociceptive behaviour in rats in a dose-related manner with an absence of obvious muscle weakness (13). A small, open-label preliminary study of the analgesic effects of BoNTA in humans found that while BoNTA did inhibit peripheral neuropeptide release from nociceptive nerve fibres, its analgesic effects were reported by the authors to be limited (15). These results may have been influenced by the model used in this study since an electrical stimulus was applied directly to stimulate the pain nerve. The BoNTA pretreatment was only able to inhibit the peripheral nociceptive peptide release (measured as neurogenic flare) in the volunteers. Since BoNTA does not cross the blood–brain barrier and is not transported centrally by the nerve, there was no effect of BoNTA on hyperalgesia or allodynia elicited by electrical stimulation. Therefore, BoNTA would not have the ability to block electrically induced pain. In the preclinical studies, BoNTA was effective in situations where sufficient peripheral sensitization (i.e. formalin or capsaicin) could be achieved, so BoNTA may inhibit the peripheral release of nociceptive signals and thus reduce sensory input into the central nervous system (CNS) (12). Further clinical studies of this effect in humans are needed to confirm the results of preclinical studies.
BoNTA modification of pain pathways may potentially inhibit the pain associated with headache disorders. The results of double-blind, placebo-controlled trials, open-label trials and retrospective chart reviews show mixed evidence of efficacy that focal injections of BoNTA significantly reduce the frequency, severity and disability associated with migraine, TTH and other types of headaches (16–30). The varying results may be due to patient selection, the number of BoNTA treatments administered, the injection protocol employed or the dosage range used.
The purpose of this trial was to evaluate the safety and efficacy of a single BoNTA treatment in five active treatment groups compared with placebo for the prophylactic treatment of CTTH.
Methods
Objective
The objective of this study was to examine the safety and efficacy of a single treatment of BoNTA for the prophylactic treatment of CTTH.
Study design
This was a double-blind, randomized, placebo-controlled, parallel-group, multicentre study conducted from 4 January 2000 to 28 February 2001. There were 22 North American study centres (one in Canada and 21 in the USA) and seven European study centres (three in Germany, two in the UK, one in Belgium and one in Denmark). Prior to enrolment and subsequent randomization to BoNTA or placebo, patients entered a 30-day baseline period during which they were screened for eligibility. During the baseline period patients were required to record data capturing specified characteristics of their headache episodes and headache medication use using a paper diary. All eligible subjects who continued to meet eligibility criteria at the end of a 30-day screening baseline period were randomized to one of six treatment groups.
Participant characteristics
Eligible participants were men or women aged 18–65 years with a history of CTTH as defined by International Headache Society criteria (31) (Key inclusion and exclusion criteria are outlined in Table 1).
Key inclusion and exclusion criteria

The investigator could discontinue participants prematurely from the study for adverse events, protocol violations, lack of efficacy, or administrative reasons (e.g. inability to continue, lost to follow-up).
Patients taking prophylactic headache medications were required to have been on a stable dose for at least 3 months prior to enrolment and had to maintain a stable dose throughout the study. The use of any concurrent prescription or over-the-counter medication and the reason for taking it were recorded.
Intervention and protocol
Each vial of BOTOX® (Allergan, Inc.) was stored in a freezer between −20°C and −5°C before use and contained 100 U of botulinum toxin type A, 0.5 mg albumin (human) and 0.9 mg sodium chloride in a sterile, vacuum-dried form without a preservative. BOTOX was reconstituted with 3 ml of the diluent, 0.9% sterile saline (without preservative). Reconstituted solutions were stored in a refrigerator until use (within 4 h). To maintain the blinding of the study and the investigator (injector) and other staff involved in assessing or reporting on the subject, an individual not involved with these activities was identified at each study centre to perform the dilutions and draw up the study medication.
At the injection visit (day 0), patients received a single treatment of 10 intramuscular injections of BOTOX, placebo or both in five muscle groups (Fig. 1, Table 2). Depending on the muscle area, 0.20–0.80 ml (3.3–40 U) was injected. Three treatment groups received BOTOX (50 U, 100 U or 150 U) distributed in all five muscle groups; two treatment groups received BOTOX (either 86 U or 100 U) distributed in three muscle groups (and placebo in two muscle groups); and one group received placebo in all five muscle groups (Table 2). Total BOTOX doses were 50 U, 86 Usub, 100 U, 100 Usub and 150 U (‘sub’ identifies the groups in which only three muscle groups received active treatment); the number of BOTOX U per muscle for each treatment group was fixed. Post-injection follow-up evaluations were scheduled on days 30, 60, 90 and 120.

Location of injection sites. Adapted, with permission, from Nucleus Medical Art (http://www.nucleusinc.com).
Injection protocol

At each study visit, participants were given a 4-week paper diary and instructed to complete it on a daily basis throughout the study. Patients recorded the occurrence, severity, maximum severity and location of TTH; associated headache symptoms (nausea, vomiting, increased sensitivity to light and/or noise, and worsening head pain with physical activity); percentage of each day with headache, as calculated from the hours awake with headache as a proportion of those hours; headache medications with dose and time of dose; and the occurrence of non-TTH. Patients were administered the Headache Pain Specific Quality of Life Questionnaire, the Tension-Type Headache Impact Questionnaire and the SF-36 Health Survey to evaluate the impact of treatment. The diaries were collected at the scheduled monthly study visits.
This study was conducted in compliance with institutional review board regulations in each participating country, the Declaration of Helsinki, informed consent regulations of each participating country, the recommendations of the International Headache Society for studies in the prophylactic treatment of CTTH, and the International Conference on Harmonization guideline for Good Clinical Practice.
Outcome measures
Efficacy
The primary efficacy variable was the mean change from baseline in the number of TTH-free days per month for the 30 days prior to day 60 (postinjection) recorded by the patient in the paper diary. The clinical criterion for effectiveness was a 3 days/month difference between treatment groups in the mean change from baseline for the number of TTH-free days.
Secondary efficacy measures included the percentage of patients reporting at least a 50% decrease in TTH days; percentage of the day with headache, as calculated from the hours awake with headache as a proportion of the hours awake; average of headache severity, as recorded in the patient diary; and concurrent headache medication usage.
Information from measures collected during office visits included the subject's response to treatment [rated on a 9-point Global Assessment Scale from −4 (very marked worsening) to +4 (clearance of signs and symptoms)]; the subject's rating of worst, least and usual TTH-associated pain as measured on an 11-point scale (0 = no pain to 10 = pain as bad as can be); and the subject's score on a Beck Depression Inventory. Other efficacy measures documented in the patient's diary included days of current acute headache medication usage per 30-day interval, average maximal headache severity per 30-day interval, presence/absence of associated headache symptoms and non-TTH days per 30-day interval.
Safety
Adverse events.
All reported adverse events were recorded, with information regarding the date of onset, resolution date (if applicable), severity (mild, moderate or severe), duration, frequency, relationship to study treatment, action taken regarding study treatment, treatment required (if any), and outcome included. The relationship between an adverse event and study treatment was assessed by the investigator as none, possible, probable or definite. A serious adverse event was defined as one that was fatal, life threatening, or permanently disabling, or that resulted in hospitalization or prolongation of existing hospitalization.
Laboratory variables.
Blood specimens were collected at the first screening visit and the last scheduled visit (day 120 or study exit). Standard blood chemistry, electrolyte panel and haematology analyses were performed and reported.
Physical examination and vital signs.
A physical examination was performed, and height, weight, blood pressure, pulse rate and body temperature were measured at the first screening visit and the last scheduled visit (day 120 or study exit). At all other study visits, pulse rate and blood pressure were measured.
Statistical analyses
Analyses were performed on data from the intent-to-treat population, which included all randomized subjects. Subjects were analysed as randomized, regardless of which treatment they actually received, for all except safety variables. For safety variables, subjects were analysed as actually treated. All tests were two-sided and utilized a type I error of α = 0.05 to determine statistical significance, except that treatment-by-subgroup interactions were examined at the 0.10 level.
A key variable (mean change from baseline in TTH-free days) and visit (day 60) were identified. For the key variable and visit, protection against multiple comparison errors was accomplished by examining P-values for significance between any of the five BoNTA groups and placebo only if the calculated P-value was <0.05/5 = 0.01 (Bonferroni method). Other variables, visits and pairwise treatment comparisons were considered supportive.
Data were summarized with descriptive statistics and/or response frequencies, including sample size, mean, standard deviation, median, minimum and maximum. For variables with ordered response categories and variables with a continuous response range, simultaneous comparisons among all treatments were done with the Kruskal–Wallis test (via analysis of variance of ranks). Comparisons between pairs of treatment groups were done with the Wilcoxon rank sum tests, even if the among-treatment results were not significant. For the continuous variables, if the responses appeared to be normally distributed (via the Shapiro–Wilk statistic), one-way analysis of variance methods were considered to improve the efficiency of the inferential test. If there were significant baseline differences between the treatments in a primary or secondary efficacy variable, a baseline covariate was included in an analysis of covariance of the variable. For binomial data, comparisons between treatment groups were done with χ2 tests, 2 × 6 among-group comparisons, and 2 × 2 pairwise treatment comparisons. If the expected value of any cell in a table was <5, the affected distribution was tested with Fisher's exact test.
Results
Patient demographics
The screening included 429 patients, 300 of whom [187/300 (62.3%) female; mean age 42.6 years (range 18–65 years)] were eligible for participation and enrolled. There were no significant demographic differences between the treatment groups. Forty-nine patients were randomized to BoNTA 150 U, 51 patients to BoNTA 100 U, 52 patients to BoNTA 100 Usub, 51 patients to BoNTA 86 Usub, 47 patients to BoNTA 50 U and 50 patients to placebo (Table 3). A total of 279/300 patients (93%) completed the study. The most common reason for discontinuation was loss to follow-up [13/300 (4.3%)]. Seven BoNTA patients discontinued the trial because of lack of efficacy. Of these patients, three were in the 150 U treatment group, two were in the 100 Usub treatment group, one was in the 86 Usub treatment group and one was in the 50 U treatment group. One patient in the 100 Usub treatment group discontinued participation for other (personal) reasons. No patient discontinued the study due to adverse events.
Tension-type headache (TTH) characteristics at baseline

HA, Headache.
Mean (median) values provided for TTH days/month and TTH duration (hours/day).
Headache history and medication use
All patients had a diagnosis of CTTH. The mean time since onset of CTTH was 14.7 years (range 0–54 years) and the mean age at onset was 28.3 years (range 1–61 years). There were no significant differences between the treatment groups in the CTTH history. Patients recorded a mean frequency of 23.0, 23.9, 23.8, 22.8, 25.5, and 25.2 TTH days per month in the BoNTA 150 U, 100 U, 100 Usub, 86 Usub, 50 U and placebo groups, respectively (Table 3). In addition to having CTTH, 95/300 (31.6%) patients reported having at least one migraine per month. Patients experienced a mean of 0.7 migraines per month with a mean duration of 1.2 days per month. A total of 13/300 enrolled patients had less than 15 headache days per month at baseline (seven in the 150 U group; two each in the 100 U, 100 Usub and 86 Usub groups) and should not have been enrolled as per the protocol criteria. Overall, patients reported a mean headache duration of 12.4 h per day. Most patients (87.9%) reported taking analgesic and/or prophylactic medications including aspirin, ibuprofen, acetaminophen, acetaminophen plus codeine, non-steroidal anti-inflammatory drugs, β-blockers, antidepressants and anticonvulsants for TTH pain. A total of 98 patients (32.7%) reported taking concomitant prophylactic headache medications. Patients in the BoNTA 150 U, 100 U, 100 Usub, 86 Usub, 50 U and placebo groups used acute headache pain medication for a mean of 8.7, 10.3, 9.8, 11.9, 8.8 and 8.5 days, respectively, during the 30-day baseline period. There were no statistically significant differences between the treatment groups in their use of acute headache pain medication at baseline. There were 111/300 patients (37%) who used headache pain medication for 16 or more days during the baseline period. At baseline, most patients [195/300 (65%)] did not report associated symptoms with their headaches; however, the rest [105/300 (35%)] reported one or more associated symptoms with some of their headaches. These symptoms included sensitivity to light [68/300 (22.7%)], sensitivity to sound [49/300 (16.3%)], nausea [15/300 (5%)] and vomiting [2/300 (0.7%)]. Pain associated with TTH was most often reported as bilateral [182/300 (60.7%)] and localized to either the front of the head [202/300 (67.3%)] or the back of the head [161/300 (53.7%)]. According to the investigator assessment, patients in the 100 U group (43.1%) had a significantly higher incidence of pain in the cervical region compared with patients in the other groups (14.0–29.8%; P = 0.015).
Efficacy
Primary variable: mean change from baseline in the number of TTH-free days
At baseline, two of the five BoNTA treatment groups, BoNTA 100 U (6.5 days; P = 0.001) and 86 Usub (5.3 days; P = 0.008), had a significantly higher mean frequency of headache-free days (3.3 days) than the placebo group (Table 4). For the primary endpoint, there was no difference between placebo and four of the BoNTA groups (50 U, 100 U, 86 Usub and 100 Usub) but a statistically significant difference favouring placebo compared with the BoNTA 150 U group was observed (4.5 vs. 2.8 tension headache-free days per month) (Fig. 2). All groups demonstrated improvement at the day 60 primary endpoint.

Mean change from baseline in the number of tension-type headache-free days at day 60.
Tension-type headache-free days at baseline
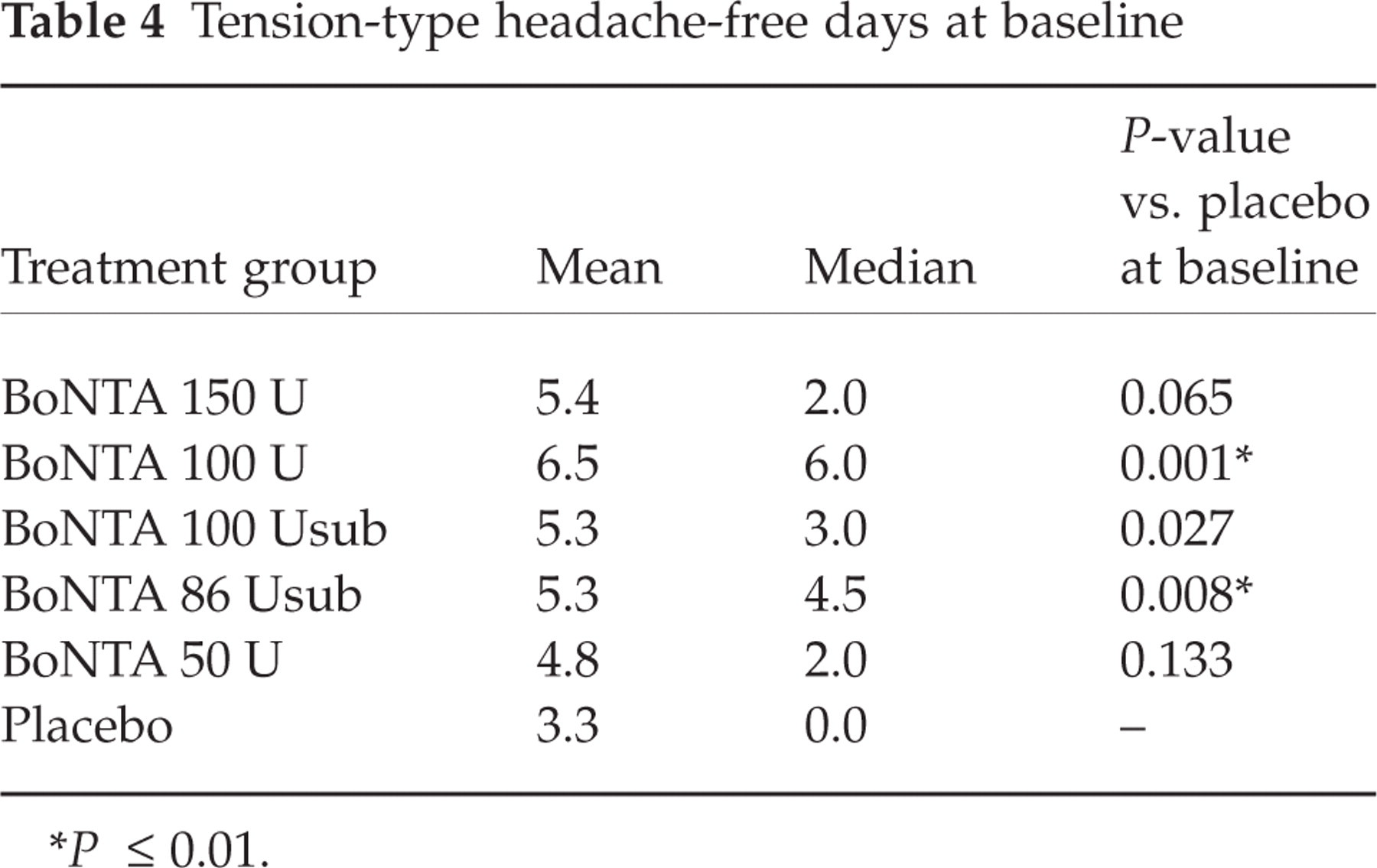
P ≤ 0.01.
Secondary variables
Reduction of ≥50% of TTH days.
No significant differences were found in the number of patients reporting a decrease of 50% or more headache days at day 60, the primary endpoint. However, at day 90, significantly larger percentages of patients in the BoNTA 100 U group [15/47 (31.9%); P = 0.017], the 100 Usub group [15/49 (30.6%); P = 0.024] and the BoNTA 86 Usub group [15/47 (31.9%); P = 0.017] reported a 50% decrease in TTH days compared with the placebo group [6/50 (12.0%)] (Fig. 3).

Percentage of patients with a decrease of 50% or more in tension-type headache days.
Percentage of the day with headache.
There were no significant differences at day 60 in the percentage of the day with headache, calculated from hours awake and hours awake with headache as reported in the patient diary. The mean decrease from baseline was −6.8%, −3.0%, −6.5%, −4.0%, −5.0% and −4.7% for BoNTA 150 U, 100 U, 100 Usub, 86 Usub, 50 U and placebo groups, respectively.
Average usual headache severity.
There were no significant differences in the average usual headache severity at any time point. At day 60, the mean decrease was −0.1, −0.1, −0.2, −0.2, −0.2 and −0.1 for BoNTA 150 U, 100 U, 100 Usub, 86 Usub, 50 U and placebo groups, respectively.
Other efficacy measures
At day 60, fewer patients in the BoNTA 150 U group reported a decrease of three or more TTH days compared with the BoNTA 86 Usub group [13/48 (27.1%) vs. 27/47 (57.4%); P = 0.003]. There were no significant differences between placebo and any BoNTA group in the other efficacy measures (subject's rating of headache pain, treatment assessment, global assessment, Beck Depression Inventory, days of concurrent headache medication usage, average maximal headache severity, associated headache symptoms and non-TTHs) at day 60 or any other follow-up visit. A decrease in phonophobia, photophobia (except BoNTA 50 U) and headache symptoms worsening with physical activity was noted for all groups. There were no significant differences between the groups in the Headache Pain Specific Quality of Life Questionnaire, the Tension-Type Headache Impact Questionnaire, and the SF-36 Health Survey (at day 60). There were no notable findings between the groups in the subgroup efficacy analyses.
Safety
Adverse events were reported for 61.7% (29/47) of BoNTA 150 U patients, 64.7% (33/51) of BoNTA 100 U, 63.5% (33/52) of 100 Usub, 54.9% (28/51) of BoNTA 86 U, 51.0% (25/49) of BoNTA 50 U and 52.0% (26/50) of placebo patients. The most frequently reported adverse events across all treatment groups were respiratory infection [10.7% (32/300)], neck pain [8.0% (24/300)] and muscular weakness [7.7% (23/300)]. Among 300 patients there were 41 adverse events reported as severe. Severe adverse events included seven, 11, five, four, seven and seven for the BoNTA 150 U, 100 U, 100 USub, 86 USub, 50 U and placebo groups, respectively.
Treatment-related adverse events were reported for 32.0% (96/300) of patients overall. There were no differences among the groups in the incidence of subjects reporting treatment-related adverse events (Table 5). The most frequently reported treatment-related adverse events were muscular weakness [20/300 (6.7%)] and neck pain [16/300 (5.3%)]. There were no significant differences between any BoNTA group and placebo in the incidence of treatment-related adverse events except the overall incidence of the 100 U group [23/51 (45.1%) vs. 11/50 (22.0%); P = 0.014]. A consistent dose–response for these treatment-related adverse events was not apparent, but the BoNTA 100 U group did report the greatest incidence of muscular weakness, neck rigidity, neck pain, headache and pain. Most treatment-related adverse events were either mild or moderate in severity and transient in nature, and most patients had recovered without sequelae at the time that the study ended.
Treatment-related adverse events reported in at least 3% of patients in any treatment group, by number (%) of subjects

There was no significant difference in the analysis of all BoNTA groups combined vs. placebo in the incidence of all adverse events or treatment-related adverse events. There were no clinically relevant differences among the groups in changes from screening to study exit for laboratory variables or vital signs. No subject died during the study. The five serious adverse events were worsening of headache, re-dislocation of left shoulder, chest pain, palpitation (100 U group) and emotional lability (50 U). None of the serious adverse events was considered treatment related.
Discussion
The results of this clinical study suggest that BoNTA is a safe, well-tolerated intervention in patients with CTTH. No discontinuations due to adverse events occurred, nor were any treatment-related serious adverse events reported. The BoNTA patients, especially those treated with 100 U (but not those treated with 150 U) did have a greater percentage of transient, treatment-related, muscular weakness and neck pain, which are known to be associated with BoNTA injections.
As a result of this intervention most patients evaluated in this study had more TTH-free days. The efficacy of BoNTA in the prophylactic treatment of CTTH compared with a placebo treatment was not confirmed in this study at day 60, the primary efficacy endpoint. However, at day 90 there was a significant effect of BoNTA treatment on the percentage of patients reporting a 50% decrease in headache days for several BoNTA dose groups, suggesting that perhaps a longer primary endpoint than 60 days may be warranted. The results of secondary and other efficacy measures did not show that BoNTA was significantly more effective than placebo, although improvement in most secondary and other efficacy measures was observed. The ability to differentiate BoNTA from placebo may have been confounded due to the uneven randomization (32) or due to the fact that patients were allowed to take acute headache pain medication as needed to treat their headaches, thereby potentially aborting or diminishing their headache characteristics.
BoNTA has demonstrated antinociceptive activity in patients with cervical dystonia, spasticity and chronic musculoskeletal pain disorders in clinical trials (33–37), suggesting that BoNTA may have analgesic effects. BoNTA inhibition of the release of nociceptive molecules has been demonstrated in several preclinical studies (12–14) and BoNTA administration has been shown to attenuate pain behaviour in a rat model (13). These effects have yet to be demonstrated in humans (15). BoNTA efficacy as prophylactic treatment of TTH has been assessed in several small controlled trials, but the evidence of a treatment effect has thus far been mixed or lacking (17, 23, 25, 27, 30, 38). The reasons for the lack of efficacy of BoNTA treatment of CTTH in this trial may be related to the injection protocol, the BoNTA dosage range, the patient population or ineffectiveness in this patient population.
Injection protocol
The injection protocol of this trial followed a fixed-site approach for TTH. Blumenfeld and colleagues (39) recommend a follow-the-pain approach for BoNTA treatment of TTH injecting the frontalis (2.5 U each, five sites on each side), temporalis (2.5 U each, four sites on each side), occipitalis (2.5–5 U on each side), trapezius (7.5–15 U on each side), semispinalis capitis (7.5–15 U on each side) and/or splenius capitis muscles, as deemed appropriate (21).
The current study protocol did not include occipitalis injections and it did not use as many sites for BoNTA in various muscle groups (21, 26) (Table 2). With 60–75% of patients describing frontal head pain and 50–60% of patients describing pain in the back of the head, the addition of more frontalis injection sites, the addition of occipital injection sites and an increase in the total BoNTA U injected in these sites might also have had an impact on BoNTA efficacy. Only 147/300 patients (the BoNTA 50, 100 and 150 U groups) received BoNTA injections into all five muscle sites.
BoNTA dosage range
Recent large, randomized, placebo-controlled BoNTA studies in patients with chronic headache disorders suggest that repeated BoNTA treatment between 105 U and 260 U, a higher dose range than those used in this study, may lead to better results (28, 29), although there are no such indications in the present study as fairly low doses of 100 U or 86 U demonstrated a better treatment effect than 150 U.
Patient population
Given that a large number of patients in this study reported concurrent migraine symptoms, such as sensitivity to light and sound, nausea and vomiting, and the numbers of patients reporting migraine headaches (95/300), it is likely that the patients in this study were not suffering exclusively from CTTH but also had coexisting infrequent migraine headaches similar to other TTH populations. In addition, the patients in this study were required to be on stable doses of prophylactic headache medications for at least 3 months prior to the initiation of BoNTA treatment and could continue their prophylactic regimen throughout the course of the trial. The additional effect of concurrent prophylactic headache medication could have confounded the ability to assess a BoNTA treatment effect.
Conclusions
BoNTA treatment of CTTH in a dose range of 50 U to 150 U was shown to be safe and well tolerated. For tension headache-free days per month, all groups improved at the day 60 primary endpoint. There was no statistically significant difference between placebo and four BoNTA groups, but a significant difference favouring placebo vs. BoNTA 150 U was observed. At day 90, significantly more patients reported a 50% decrease in headache days in several BoNTA groups, suggesting that a longer period of evaluation may be needed to see a treatment effect. The failure of BoNTA to demonstrate efficacy in this trial may be related to the injection protocol, the BoNTA dosage range, the patient population, the use of concomitant prophylactic headache medication or a lack of a clinically relevant effect in CTTH. BoNTA may not be an effective prophylactic treatment of CTTH, but further study may provide insight into appropriate study parameters and a possible treatment effect.
Dosing and results reported in this study are specific to the formulation of botulinum toxin type A manufactured by Allergan, Inc. (Irvine, CA, USA). The Allergan, Inc. formulation is not interchangeable with other botulinum toxin products and cannot be converted by using a dose ratio.
BOTOX CTTH Study Group
Andrew Blumenfeld, MD (California); Roger Cady, MD (Missouri); Joanna Cooper, MD (California); James Couch, MD (Oklahoma); Seymour Diamond, MD (Illinois); Hans-Christoph Diener, MD (Germany); Marek J. Gawel, MD (Canada); Peter Goadsby, MD, PhD (UK); Jerome Goldstein, MD (California); V. Daniel Kassicieh, DO (Florida); Jack Klapper, MD (Colorado); Ninan Mathew, MD (Texas); Alexander Mauskop, MD (New York); Volker Pfaffenrath, MD (Germany); Sid Rosenblatt, MD (California); Joel Saper, MD (Michigan); Jack Scariano, MD (Tennessee); Jean Schoenen, MD (Belgium); Elliott Schulman, MD (Pennsylvania); Fred Sheftell, MD (Connecticut); Timothy Smith, MD (Missouri); Egilius Spierings, MD, PhD (Massachusetts); Stuart Stark, MD (Virginia); Timothy Steiner, MB, PhD (UK); Paul Winner, DO (Florida).