
Abstract
The importance of reversible phosphorylation for neuronal signaling and cell survival is well recognized. Knowledge in vertebrates, however, is so far limited to O-phosphates from serine, threonine, and tyrosine. The authors describe an enzyme acting on N-phosphates. It is the first protein histidine phosphatase identified in vertebrates. This histidine phosphatase is ubiquitously expressed in mammalian tissues including brain. Characterization and sequencing showed a yet unknown protein with no similarity to other phosphatases. In Caenorhabditis elegans, the homolog of this histidine phosphatase was exclusively expressed in neurons, suggesting a distinct role of reversible histidine phosphorylation in neuronal functions.
Signal transduction is vitally important for growth and cell survival. Reversible phosphorylation of eukaryotic proteins takes place on serine, threonine and tyrosine (for review see Graves and Krebs, 1999). The kinases and phosphatases involved are regulated by different mechanisms (Zolnierowicz and Bollen, 2000; Dombradi et al., 2002). As exemplified with protein kinase B (= Akt) signaling or the mitogen-activated protein kinase cascade, kinases themselves are often subject to phosphorylation. Defects in such signaling have been found as the underlying basis for a number of human diseases, including neurodegeneration.
With the focus on amino acids containing hydroxyl groups, other phosphorylation sites on proteins may have been overlooked in vertebrates. For instance, bacteria rely primarily on histidine and aspartate signal transduction systems (Hess et al., 1988; West and Stock, 2001). The common feature among this large family of proteins is kinases that autophosphorylate on histidine residues and subsequently transfer the phosphoryl group to aspartate on a cognate response regulator. Such histidineaspartate phospho-relay systems play an important role in chemotaxis. In general, reversible phosphorylation of histidine is known to influence enzymatic, mechanistic, and transcriptional properties of bacterial proteins.
Phosphohistidine has also been detected in many eukaryotic proteins. Vertebrate histidine kinases and histidine phosphatases, however, remain unknown. Furthermore, only a few of the vertebrate phosphohistidine-containing proteins were identified, that is, histone H4 (Fujitaki et al., 1981) and annexin I (Muimo et al., 2000). There is additional evidence for one or more phosphohistidine signaling pathways in mammalian cells: stimulation of platelets with thrombin or collagen led to phosphorylation of P-selectin on a histidine residue (Crovello et al., 1995). These are examples of covalent modification of the histidine residues by phosphorylation—not the formation of intermediates as suggested for the G protein β-subunit (Wieland et al., 1993) and known for long for some metabolic enzymes such as glucose-6-phosphate phosphatase.
It has been reported that serine–threonine protein phosphatases type 1, 2A, and 2C are capable to some extent of dephosphorylating [32P-his]histone H4 in vitro (Kim et al., 1995). Phosphatases specific for phosphohistidine, however, have not been identified in vertebrates. Here we describe a novel protein acting as protein histidine phosphatase (PHP) in mammalian cells. Although originally purified from rabbit liver, this enzyme is also present in brain tissue and in neurons, thus raising the possibility of neuronal functions with participation in yet unidentified signaling pathways.
MATERIALS AND METHODS
Measurement of protein histidine phosphatase activity
The substrate used for assaying protein histidine phosphatase activity was recombinant bacterial histidine autokinase CheA (chemotaxis protein A) labeled at histidine-48 by [γ-32P]ATP (Swanson et al., 1993). Dephosphorylation reactions were carried out in the presence of protein containing PHP activity, in 25 mmol/L triethanolamine, 10 mmol/L Mg2+, and 0.1% β-mercaptoethanol, pH 7.5, at 37°C for 30 minutes. For determination of the specific activity, 40-μL dephosphorylation reactions were stopped by adding 10 μL 0.5 mol/L ethylene diamine tetraacetic acid (EDTA) and 150 μL methanol/acetone (1+1), centrifuged at 14,000g for 5 minutes and the supernatant quantified for [32Pi] content by liquid scintillation counting. For alternative visual detection, 10-μL dephosphorylation reactions were stopped with 5 μL Laemmli sample buffer, applied to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) minigels and autoradiographed.
Preparation and isolation of protein histidine phosphatase
Tissues from mice, rats, and rabbits were homogenized in buffer (2 to 4 mL/g tissue) containing 20 mmol/L triethanolamine pH 7.5, 1 mmol/L EDTA, 300 mmol/L sucrose, 0.1 mmol/L phenyl-methyl-sulfonyl-fluoride, 1 mmol/L benzamidine, 0.1% β-mercaptoethanol, and centrifuged at 17,300g for 30 minutes.
Protein histidine phosphatase was isolated from soluble extracts (additional centrifugation at 48,000g for 1 hour) from rabbit liver. The buffer used for all chromatographic purification steps consisted of 20 mmol/L triethanolamine pH 7.5, 1 mmol/L EDTA, 0.1% β-mercaptoethanol, and 0.02% NaN3. Purification by fast protein liquid chromatography included the anion exchange material Source 30Q (V = 12 mL; elution with buffer containing 200 mmol/L NaCl) and gel filtration on Superdex 75 (V = 320 mL). Final purification was achieved using affinity chromatography on Blue Sepharose (V = 1 mL) operated with the same buffer except for 10 mmol/L Mg2+ (instead of EDTA) for loading, and 200 mmol/L NaCl, 0.1 mmol/L EDTA for elution. Glycerol (5%) was added to PHP-containing fractions after gel filtration and Blue Sepharose, to avoid loss of enzyme activity on storage.
Antibody generation and immunoblotting
The peptide QIPDVDIDSDGVFKYC corresponding to the N-terminal amino acids 8 through 22, plus an extra cysteine for keyhole limpet hemocyanin coupling, was used for generating antibodies in rabbits. Antisera were purified using the antigen immobilized on cyanogen bromide (CNBr)-activated sepharose and stored in 66 mmol/L glycine, 0.33 mol/L arginine, 1.67 mmol/L citric acid, 1.33% mannite, 0.02% NaN3, and 0.003% Tween 80, pH 8. Western blots were performed according to standard protocols using 5% skim milk powder in 10 mmol/L Tris, 137 mmol/L NaCl, 0.1% Tween 20, pH 7.6 as blocking solution.
Sequencing and cloning of protein histidine phosphatase
Purified PHP was cleaved on treatment with trypsin and CNBr, the peptide fragments separated by reversed-phase high-performance liquid chromatography, then automated Edman degradation and MALDI mass spectrometry were performed. A complementary DNA (cDNA) clone was obtained by reverse transcription–polymerase chain reaction from rabbit liver messenger RNA using degenerate primers developed according to the protein sequencing results. Rapid amplification of messenger RNA ends by PCR gave the missing 5′- and 3′-ends to yield the full-length cDNA clone of PHP.
Caenorhabditis elegans and green fluorescent protein expression
Caenorhabditis elegans strains were grown under standard conditions. An expression vector was constructed containing the egfp gene and predicted php coding region (515 bp), under the control of the php promotor. Plasmids were injected into wild-type worms and PHP localization was analyzed by fluorescence in cells expressing the green fluorescent protein (GFP) constructs.
RESULTS
Searching for histidine phosphatases requires easily accessible and pure substrate proteins phosphorylated on histidine residues. Although histidine kinases are expected to be present in vertebrates, they have not been identified. Therefore, we were using the bacterial protein CheA—phosphorylating itself on histidine—as a substrate for potential PHPs in vertebrates.
Detection and purification of protein histidine phosphatase
[32P-his]CheA became dephosphorylated in homogenates of various organs from mice in a time- and protein-dependent manner. Running the assays in the presence of 10 mmol/L Mg2+ showed PHP activity in the centrifugation supernatants (Fig. 1A). This was observed for heart, kidney, and liver (Fig. 1A). Measuring the dephosphorylation of phosphohistidine in the absence of Mg2+, in contrast, showed a different and less uniform distribution pattern (Fig. 1B). Endogenous interfering histidine kinases, some of which are active in the absence of Mg2+ (Noiman and Shaul, 1995), may be the reason for this observation.
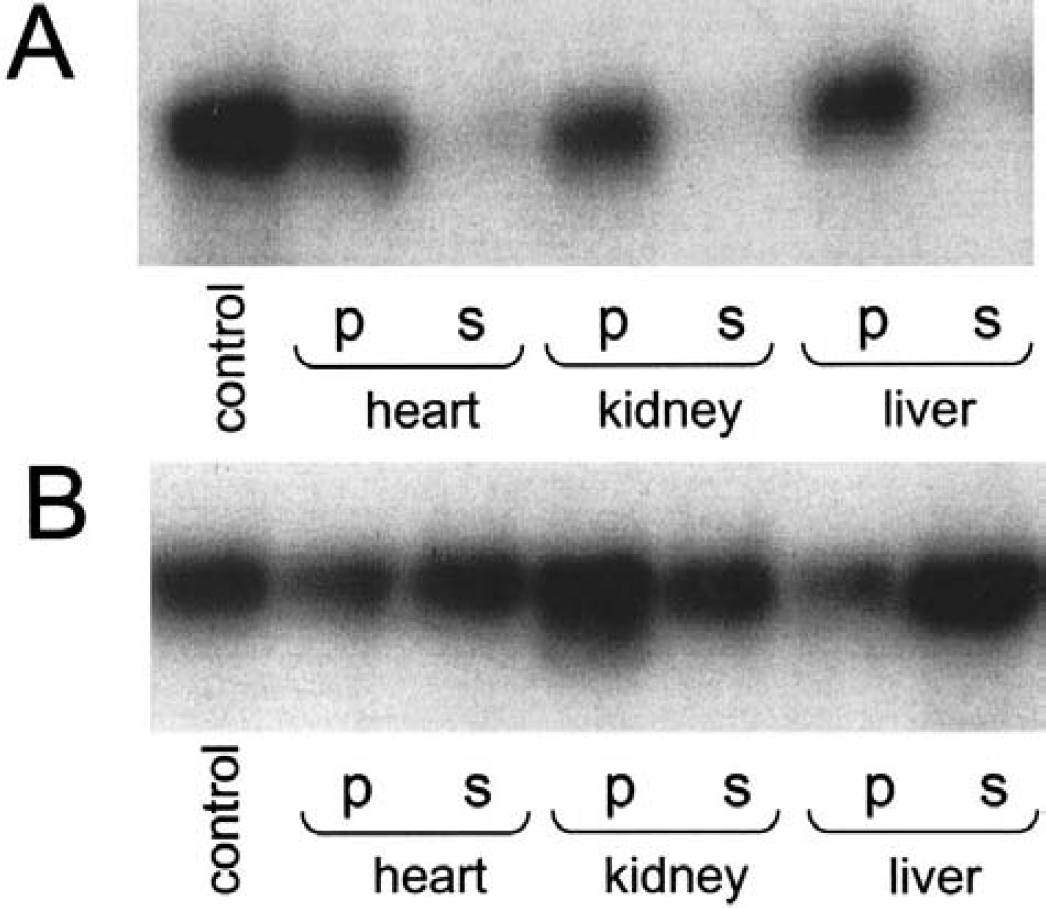
Dephosphorylation of [32P-his]-labeled chemotaxis protein A ([32P-his]CheA). An autoradiogram after sodium dodecyl sulfate–polyacrylamide gel electrophoresis is shown.
Protein histidine phosphatase was purified from soluble extracts from rabbit liver (Table 1). This enzyme was retained by anion exchange columns. It eluted as a single peak at 200 mmol/L NaCl, with a yield of 70%. Fractions containing PHP activity were pooled and concentrated by 90% (NH4)2SO4 precipitation. Subsequent gel chromatography on Superdex 75 proved the most powerful purification step, removing 99% of contaminating proteins. Protein histidine phosphatase activity eluted, corresponding to an apparent molecular mass of 14 to 16 kd. Final purification was achieved using affinity chromatography on Blue Sepharose. The enzyme, provided 10 mmol/L Mg2+ was added, was retained by this resin. It eluted in the absence of Mg2+, but the presence of 200 mmol/L NaCl. After this step there was only one protein of 14 kd visible on silver-stained SDS-PAGE (Fig. 2). Overall, PHP was purified about 12,000-fold with a yield of 12% (Table 1).
Summary of the purification of protein histidine phosphatase from rabbit liver
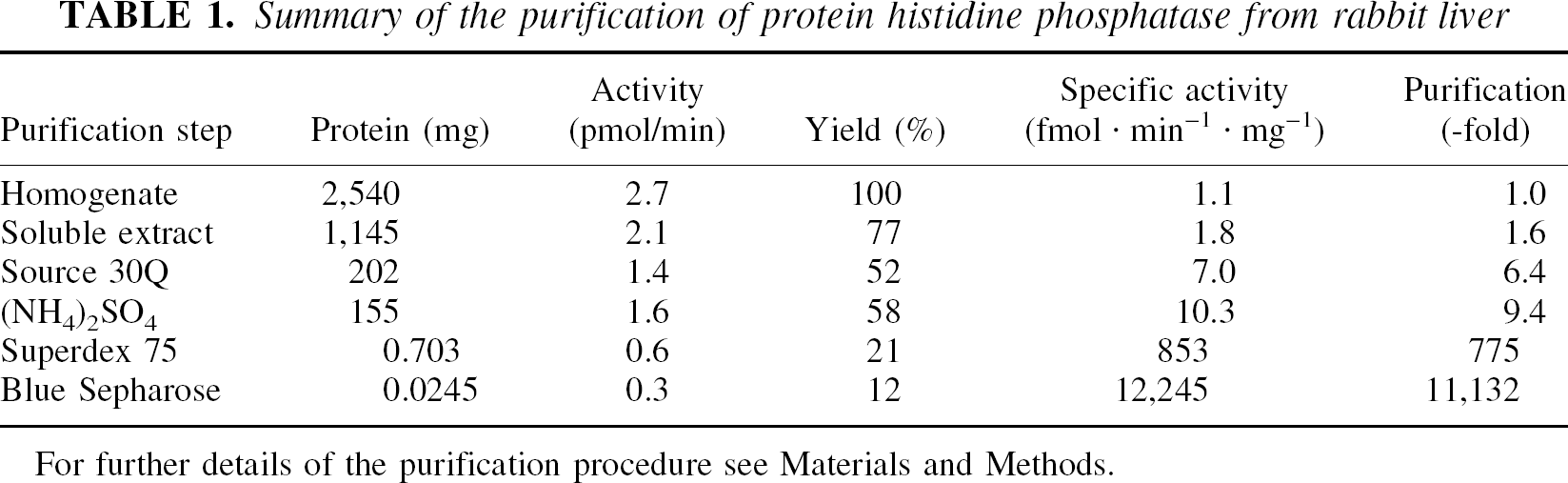
For further details of the purification procedure see Materials and Methods.
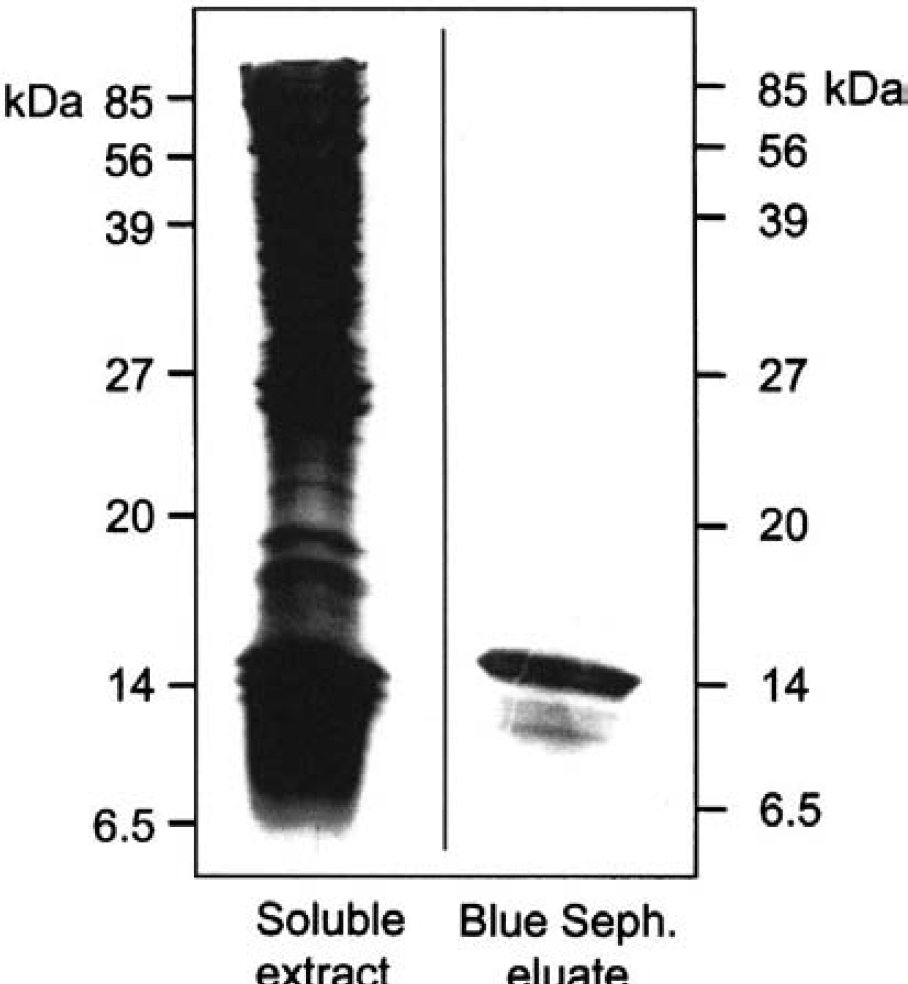
Protein purification. Fractions were analyzed on 17.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and silver stained. The lanes shown contained 14 ug protein of the soluble extract from rabbit liver and 2 μg protein from the Blue Sepharose eluate containing protein histidine phosphatase activity.
Characterization of protein histidine phosphatase
Low molecular-weight protein tyrosine phosphatases are in the same kilodalton range as the PHP purified here (Ramponi and Stefani, 1997). Enzyme characterization, however, showed that we were dealing with a novel type of protein phosphatases: (1) 100 μmol/L vanadate, as universal inhibitor of tyrosine phosphatases (Gordon, 1991), and (2) 1 μmol/L okadaic acid, known to block serine–threonine phosphatases except for PP2C (Cohen et al., 1990; Fernandez et al., 2002), did not inhibit PHP activity. Sequencing yielded final proof that a yet unknown protein was discovered. CNBr and trypsin digestion made 77% of the primary structure accessible for microsequencing (Fig. 3; amino acids underlined). Missing amino acids were deduced from the cloned gene for PHP. Rabbit liver PHP had a molecular mass of 13.7 kd (Fig. 3). It showed no sequence similarity to known phosphatases. The amino acid sequence of PHP from other tissues and species, however, was present already in the databases. It had been submitted by others, not knowing that the protein does have histidine phosphatase activity.

Amino acid sequence of protein histidine phosphatase 1 from rabbit liver (Genbank accession number P83468). Underlined amino acid sequences were obtained by microsequencing of respective peptides. The N-terminal boxed amino acid sequence represents the peptide used for the generation of polyclonal antibodies.
With regard to substrate specificity, PHP did not hydrolyze [32P-serine–threonine]casein or the [32P-tyrosine] epidermal growth factor receptor (data not shown), suggesting that this enzyme acts specific on phosphohistidine.
Presence of protein histidine phosphatase in brain and its localization in neurons
Antibodies raised against a peptide sequence from rabbit PHP effectively recognized PHP in rodents. Western blots showed the presence of PHP in various areas of rat brain, that is, striatum, hippocampus, cerebellum, and cortex (Fig. 4A). The amount of PHP in those brain tissues exceeded the level of PHP expressed in lung; it was comparable to that in liver. Neuronal cell lines, that is, PC12 cells, also contained PHP (data not shown). In addition to this immunologic approach, enzyme activity was examined as well. Specific activity of PHP in centrifugation supernatants from homogenized brain areas was 5 to 8 fmol Pi released × min−1 × mg−1 (Fig. 4B). This dephosphorylation rate was comparable with, and even somewhat higher than, that in lung and liver. On purification, the specific activity of PHP increased up to 12 to 15 pmol × min−1 × mg−1 (Table 1, Fig. 4B).
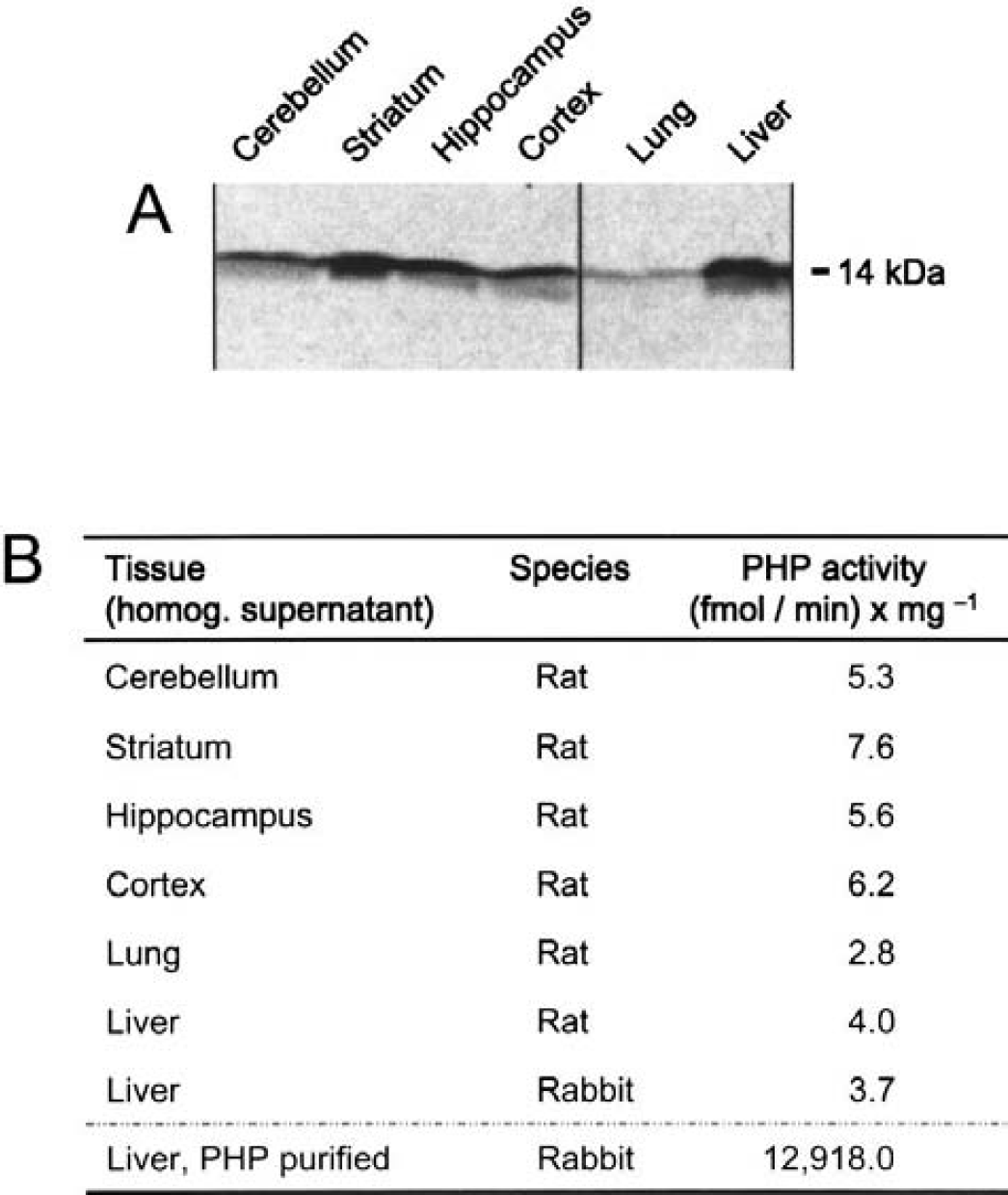
Detection of protein histidine phosphatase (PHP).
The most striking data in context with a potential neuronal involvement of PHP came from studies using nematodes as model organisms. A php::gfp fusion gene was constructed to determine the expression pattern in C. elegans L3 larva. Protein histidine phosphatase was found in 6 pharyngeal sensorimotor neurons (Fig. 5A). In addition, PHP was exclusively localized in all motor neurons along the ventral nerve cord (Fig. 5B).
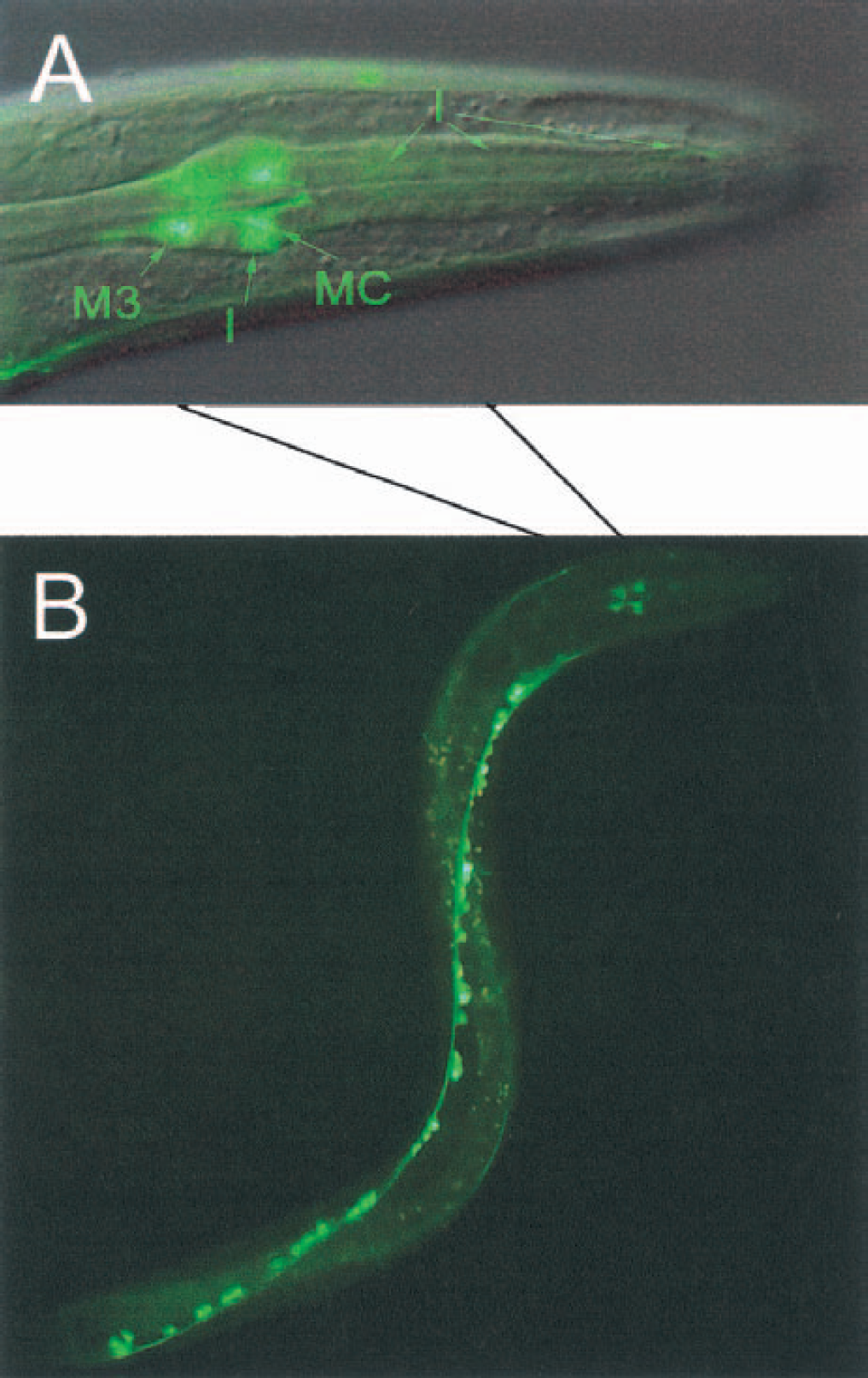
Expression pattern of protein histidine phosphatase (PHP) in Caenorhabditis elegans using a php-1::gfp fusion protein construct.
DISCUSSION
The discovery of the first PHP from vertebrates is described. This enzyme is ubiquitously expressed in mammalian tissues. It is also present in good quantity and high activity in brain tissue and neurons. The implications of these findings are relevant for future studies on signal transduction in neuronal context.
There is an ongoing discrepancy about phosphohistidine in vertebrates: histidine kinases and histidine phosphatases still have not been identified, although knowledge of the presence of phosphohistidine in vertebrate proteins traces back for several decades (Chen et al., 1974). Brain, like any other tissue in the body, also contains phosphohistidine-labeled proteins, which had been identified in synaptic plasma membranes more than 20 years ago (Weller, 1978).
It is not easy to deal with phosphohistidine for a variety of reasons; for example, (1) antibodies against phosphohistidine are not available, thus hampering straightforward detection of phosphohistidine-containing proteins; and (2) standard phosphoamino acid analysis is performed in HCl, thus destroying acid-labile phosphohistidines. There are analytical techniques available, however, using acid stability of phosphoserine, phosphothreonine, and phosphotyrosine versus the alkaline stability of phosphohistidine and phosphotyrosine to distinguish N-phosphates from O-phosphates (Duclos et al., 1991). It is a time-consuming process and requires several steps: [32P]-labeling of proteins, denaturing polyacrylamide gel-electrophoresis, electroblotting to polyvinylidenedifluoride membranes, incubation of those in acid and alkaline solutions one after the other (Kamps, 1991), and autoradiography.
It is too early to speculate on a distinct function of the PHP identified. The findings that this enzyme is present in various areas of the brain (Fig. 4), as well as in neuronal cell lines and primary neurons from vertebrates (data not shown), however, argues in favor of neuronal implications. The most striking localization of PHP in C. elegans—exclusively in the neurons (Fig. 5)—is additional strong evidence for its significant involvement in neuronal function.