
Abstract
Recent studies in a variety of species including mammals showed that resveratrol (trans-3, 5, 4″-trihydroxystibene) treatment and caloric restriction increased silent information regulator 2/sirtuin 1 activity, which mediated increase in life span/cell survival. Resveratrol is a naturally occurring phytoalexin and a well-documented cardioprotective agent. Similarly, ischemic preconditioning (IPC) has been shown to be both cardio- and cerebroprotective against subsequent ischemic insults. A major emphasis in this field is to understand the molecular mechanisms that mediate this phenomenon. The goal of this study was to define whether resveratrol can emulate IPC neuroprotection against cerebral ischemia. Employing an in vitro model of cerebral ischemia, the organotypic hippocampal slice culture, we report that resveratrol pretreatment mimics IPC via the SIRT1 pathway. Blockade of SIRT1 activation by sirtinol after IPC or resveratrol pretreatment abolished their neuroprotection. A better understanding of the mechanisms by which resveratrol induces ischemic tolerance in a prophylactic manner may provide a novel therapy against stroke or neurosurgical procedures.
Introduction
Resveratrol (trans-3, 5, 4″-trihydroxystibene) is a naturally occurring phytoalexin produced by a wide variety of plants in response to stress, injury, ultraviolet irradiation, and fungal infection (Burns et al, 2002; Chen et al, 2002; Lyons et al, 2003; Sobolev and Cole, 1999). Resveratrol has been reported to have cardioprotective, anticancer, antiaging, anti-inflammatory, and neuroprotective properties in a prophylactic manner (Aggarwal et al, 2004; Andrabi et al, 2004; Wang et al, 2002).
A number of different molecular targets have been known to be influenced by resveratrol treatment. Howitz et al (2003) showed either resveratrol treatment or caloric restriction in yeast-activated silent information regulator 2 (Sir2). Yeast Sir2 is a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase (Imai et al, 2000). In mammals, there are seven members of Sir2 family, termed sirtuins (SIRTs), among which sirtuin 1 (SIRT1) is of closest homology to yeast Sir2 (Frye, 2000). Mammalian SIRT1 is located in the nucleus and exerts a regulatory effect on p53 by deacetylation of lysine-382. (Luo et al, 2001; Vaziri et al, 2001). Interestingly, IPC is protective against lethal ischemic insults by deactivating p53 in the heart and brain (Mocanu and Yellon, 2003; Tomasevic et al, 1999).
The phenomenon of IPC was discovered many years ago and was shown to protect prophylactically against lethal ischemic insults in many organs including the brain and heart, which are especially sensitive to ischemia. Ischemic tolerance is achieved when a brief (‘sublethal’) ischemic or stressful episode, followed by a period of reperfusion, increases an organ's resistance to ischemic injury. For example, in the brain, IPC induces robust neuroprotection against ischemia in the CA1 region of the hippocampus in a variety of in vivo and in vitro models (Andoh et al, 2002; Kato et al, 1992; Lange-Asschenfeldt et al, 2004; Perez-Pinzon et al, 1997; Raval et al, 2003; Schurr et al, 1986). Since resveratrol and IPC are protective in a prophylactic manner and both are neuroprotective, we asked the question whether single transient resveratrol treatment could emulate IPC, and if that is the case whether both neuroprotective strategies follow similar signal transduction pathways. For this purpose, we used a model of oxygen glucose deprivation (OGD) in the organotypic hippocampal slices, where we have a proven IPC paradigm (Lange-Asschenfeldt et al, 2004; Raval et al, 2003; Xu et al, 2002).
Materials and methods
Preparation of Organotypic Slice Cultures
All protocols were approved by the Animal Care and use Committee of the University of Miami. Neonatal (9 to 11 days old) Sprague–Dawley rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg). Animals were decapitated and the brains quickly removed. Organotypic hippocampal slice cultures were prepared as described previously (Lange-Asschenfeldt et al, 2004; Raval et al, 2003; Xu et al, 2002). In brief, transverse slices (400 mm) were dissected from the hippocampi and placed in Gey's balanced salt solution (Sigma Chemical, St Louis, MO, USA) supplemented with 6.5 mg/ml glucose at 4°C. After 1 h, two slices were placed onto one 30 mm diameter membrane insert (Millicell-CM; Millipore, Bedford, MA, USA) and inserts were transferred to six-well culture plates with 1 mL of culture medium per well. Culture medium consisted of 50% minimum essential medium, 25% Hank's balanced salt solution, 25% heatinactivated horse serum (all Gibco/Life Technologies, Carlsbad, CA, USA) supplemented with 6.5 mg/mL glucose, and 1 mmol/L glutamine. Slice cultures were incubated (equilibrated at 36°C, 95% O2, 5% CO2, humidity 100%) for 14 to 15 days before experiments were performed.
Experimental Design
The organotypic slices were divided into five major groups (Figure 1).
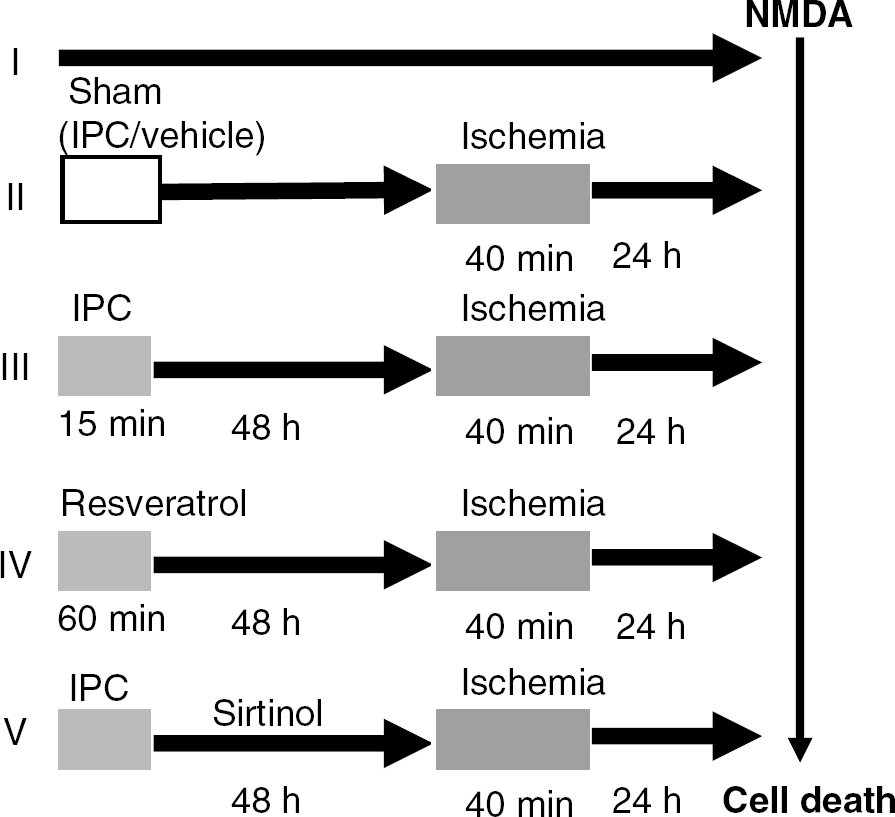
Synopsis of time course and experimental design. The quantification of neuronal death was performed using propidium iodide (PI) fluorescence technique. PI fluorescence images were obtained using a SPOT CCD camera and digitized using SPOT software. The percentage of relative optical intensity was used as an index of cell death.
Group-I: Sham—slices were incubated for 15 mins in Hank's balanced salt solution solution supplied with an equimolar concentration of glucose instead of sucrose (sham–IPC) and were subsequently transferred back to regular media. After 48 h, the same procedure was performed for an extended duration of 40 mins (sham OGD-ischemia).
Group-II: ‘Test’ ischemia—sham IPC was induced to slices as mentioned in group-I followed by ‘test’ ischemia (OGD) after 48 h for 40 mins.
Group-III: IPC—slices underwent IPC (15 mins of OGD) and ‘test’ ischemia (40 mins of OGD) after the 48 or 96 h intervals.
Group-IV: Resveratrol pretreatment—trans-3,4,5-trihydroxystibene was purchased from Sigma and dissolved in dimethylsulfoxide (DMSO). Organotypic slices were incubated at different concentration of resveratrol (75, 100, 200, and 500 μmol/L) for 1 h. The ‘test’ ischemia was induced to these slices 48 h later. To nullify the effects of DMSO or resveratrol, appropriate controls were performed.
Group-V: Blockade of IPC/resveratrol preconditioning with the SIRT1 enzyme inhibitor—organotypic slices were exposed to different concentrations of SIRT1-specific inhibitor—sirtinol (10, 50, and 100 μmol/L)—for 48 h after IPC. Since 10 μmol/L sirtinol was effective in inhibiting IPC-induced neuroprotection, this dosage was used for subsequent experiment. Resveratrol preconditioning blockade was performed by exposing organotypic slices to sirtinol in four different paradigms: (1) sirtinol treatment for 1 h before resveratrol preconditioning, (2) sirtinol pretreatment (1 h) followed by resveratrol preconditioning in the presence of sirtinol, (3) sirtinol treatment before (1 h), during, and after (48 h) resveratrol preconditioning, and (4) sirtinol treatment during and after (48 h) resveratrol preconditioning. To determine that sirtinol had no toxic effects, appropriate controls were performed.
In addition, to provide better controls in these experiments, all six-well plates used contained at least one well for the sham, IPC, and ischemia groups. Also, for statistical purposes, each insert had two slices obtained from two different pups. Thus, every n = 1 (one slice) represents a different animal.
Induction of Ischemia (Oxygen/Glucose Deprivation)
Our ischemia and preconditioning protocols have been defined in the previous studies (Xu et al, 2002; Raval et al, 2003). For OGD, slices were washed three times with aglycemic Hank's balanced salt solution (pH 7.4) of the following constitution: CaCl2 · 2H2O 1.26 mmol/L, KCl 5.37 mmol/L, KH2PO4 0.44 mmol/L, MgCl2 0.49 mmol/L, MgSO4 · 7H2O 0.41 mmol/L, NaCl 136.9 mmol/L, NaHCO3 4.17 mmol/L, Na2HPO4 · 7H2O 0.34 mmol/L, and sucrose 15 mmol/L (all Sigma). The slices were then transferred into an airtight chamber, which was equilibrated with 95% N2/5% CO2 gas (preheated to 37°C and water saturated) blown through the chamber for 5 mins (4 L/min) to achieve anoxic conditions. Then, the chamber was sealed and remained incubated for 10 mins (for a total of 15 mins—preconditioning) or 35 mins (for a total of 40 mins—ischemic insult). After OGD, slices were placed back in the incubator in plates containing the normal culture medium.
Assessment of Neuronal Cell Death by Propidium Iodide Staining Technique
To determine the extent of neuronal damage in the organotypic slice culture, we used the propidium iodide (PI) method, which was described previously (Raval et al, 2003; Xu et al, 2002). Organotypic slices belonging to all the experimental groups were incubated in culture medium supplemented with 2 mg/mL PI (Sigma) for 1 h before imaging. The slices were studied using an inverted fluorescence microscope (Olympus IX 50). Fluorescence digital pictures were taken using a SPOT CCD camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) and SPOT advanced software. Images of cultured slices were taken before the experiment, followed by preconditioning treatment. After preconditioning, the slices were reperfused for 48 h followed by 40 mins of ‘test’ ischemia. The PI fluorescence was taken 24 h after ‘test’ ischemia, and then slices were superfused with N-methyl-D-aspartate (NMDA) (100 mmol/L) for 1 h. The last image was taken 24 h after NMDA treatment. The intensity of PI fluorescence in the CA1 subfield of the hippocampal slices was used as an index of cell death. For quantification, a region of interest (ROI) was selected from bright field images of each slice, using Scion Image software (Windows version, Frederick, MD, USA). Relative cell death was calculated from each ROI as follows:
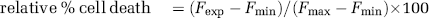
where Fexp is the fluorescence of the test condition, Fmax is the maximum fluorescence (100 mmol/L NMDA treatment for 1 h), and Fmin is the background fluorescence (before preconditioning or OGD). In all groups, experiments (except enzyme activity analysis) were terminated by superfusing slices with an overdose of NMDA 24 h after the end of the experiments to determine total number of cells using the PI technique (Fmax above).
Measurement of Sirtuin 1 Activity in Hippocampal Organotypic Slice Cultures
Organotypic slices, collected after IPC or resveratrol treatment at 30 mins or 48 h of reperfusion, were washed in phosphate-buffered saline (pH 7.4) and homogenized in homogenizing buffer (10 mmol/L 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)/KOH, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol) using pestle (Kimble-Knotes, Vineland, NJ, USA). The homogenate was centrifuged at 1,000g for 10 mins. The resulting pellet was resuspended in high salt buffer (0.35mol/L KCl, 20 mmol/L HEPES, pH 7.9, 25% glycerol, 1.5 mmol/L MgCl2, 0.2 mmol/L ethylenediaminetetraacetic acid, 0.5 mmol/L dithiothreitol, 0.5 mmol/L phenylmethylsulfonyl fluoride) and incubated at 4°C for 30 mins with gentle stirring. The resulting suspension was centrifuged at 100,000g for 30 mins at 4°C. The supernatant obtained at the end of centrifugation was dialyzed using Millipore microcon concentrators (YM-10) (Millipore, Bedford, MA, USA) till 1/5th of the original volume. Four volumes of dialysis buffer was then added to the Millipore microcon concentrator and dialyzed again till 1/5th of the original volume. This step was repeated once more. The resulting dialysate was stored at −80°C until used. This dialysate was used as a source of enzyme for SIRT1 enzyme assay. The SIRT1 enzyme activity was measured using SIRT1 fluorescent activity assay kit (BIOMOL International, Playmouth meeting, PA, USA) based on Fluor de Lys–SIRT1 substrate peptide comprising amino acids 379 to 382 of human p53 (RHKK(ε-acetyl)). Enzyme and time curves were performed from the above-mentioned samples during the pilot study. The enzyme assay was performed at room temperature (25°C). Because of low levels of SIRT1 activity in our samples, the incubation time during the assay was 120 to 300 mins. Fluorescence signal generated ((Ex 350 nm; Em 480 nm) on deacetylation of lysine residue of substrate peptide was recorded. Fluorescent signal at time zero was used as an enzyme blank and was subtracted from the experimental values. Suramine-sensitive (100 mmol/L) and trichostatin-insensitive (500 μmol/L) activities were in close agreement. Suramine-sensitive activity (activity in the absence of suramine minus activity in the presence of suramine) was considered as the SIRT1 enzyme activity.
Statistical Analysis
The results are expressed as mean ± s.d. Statistical significance was determined with an analysis of variance test followed by a Bonferroni's post hoc test.
Results
The first hypothesis that we tested was that resveratrol pretreatment emulated IPC-induced neuroprotection. As in our previous findings, we showed that IPC (15 mins) followed by 48 h of reperfusion induced ischemic tolerance against ‘test’ ischemia (40 mins) in hippocampal organotypic slices (Lange-Asschenfeldt et al, 2004; Raval et al, 2003; Xu et al, 2002) (Figure 2A). Additionally, we also showed that the time window for neuroprotection induction afforded by IPC lasted for 96 h (Figure 2). Next, we tested the efficacy of resveratrol as neuroprotective agent. We exposed organotypic slices to different concentrations of resveratrol (75, 100, 200, and 500 μmol/L) for 1 h, and 48 h later, test ischemia was induced. As compared with test ischemia alone, resveratrol (75 and 100 μmol/L) pretreatment reduced neuronal death in CA1 region of the hippocampus by 43%, which was similar to that achieved by IPC (50%). PI fluorescence in resveratrol-treated (75 and 100 μmol/L), IPC, and ischemic groups were 38.04 ± 8.81 (n =7, P < 0.01 against ischemic group), 37.97 ± 16.73 (n = 13, P < 0.01 against ischemic group), 34.13 ± 19.25 (n = 20, P < 0.01 against ischemic group), and 68.7 ± 15.86 (n = 11), respectively (Figure 2). However, the higher resveratrol dosages could not mimic IPC. The PI fluorescence values of resveratrol (200 and 500 μmol/L) treatment were 60.83 ± 12.53 (n = 13) and 62.19 ± 14.97 (n = 5). To rule out the toxic effects of resveratrol at higher concentrations, control experiments were performed exposing organotypic slices to resveratrol at higher concentrations (200 and 500 mmol/L) without a subsequent ischemic insult. The percentage of cell death in CA1 region was similar to that of the sham group, suggesting resveratrol treatment alone at higher concentrations (1 h) without a subsequent OGD was not causing cell death. PI fluorescence in resveratrol-treated and sham groups were 6.93 ± 1.32 (n = 6; 200 μmol/L), 5.61 ± 1.93 (n = 5, 500 μmol/L), and 6.84 ± 1.63 (n = 5), respectively. However, the higher resveratrol dosages could not mimic IPC. Control experiments were performed exposing organotypic slices to DMSO for 1 h. No significant differences were observed between the DMSO-treated and OGD groups (data not shown).
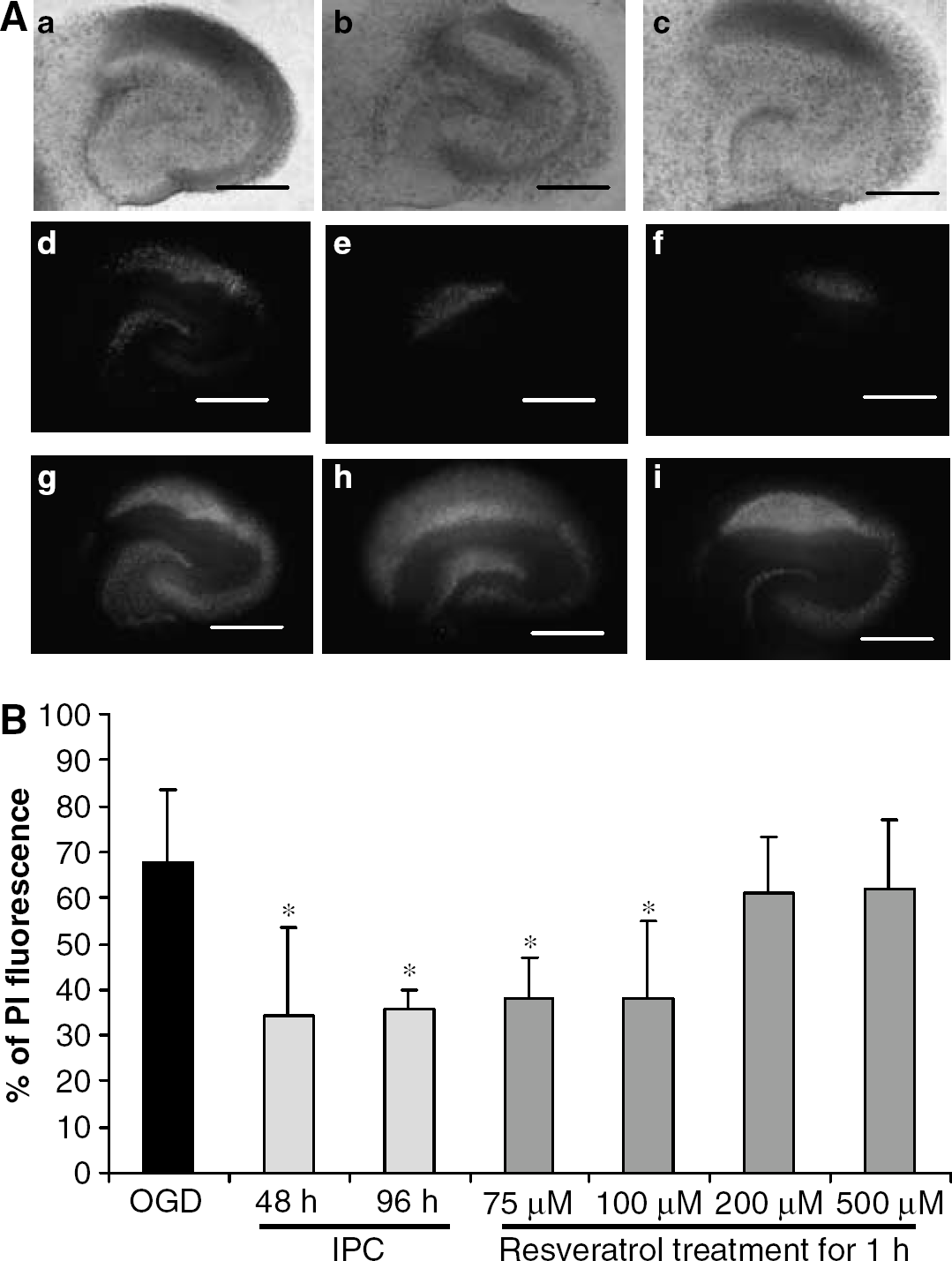
Resveratrol mimics ischemic preconditioning (IPO-induced neuroprotection against ischemia.
These results suggested that resveratrol pretreatment did mimic IPC in the CA1 region of the hippocampus. Thus, we next asked whether both strategies follow similar signaling pathways. A proposed mechanism by which resveratrol protects cell survival is via the SIRT1 pathway (Howitz et al, 2003). Thus, we measured SIRT1 activity in organotypic slices at 30 mins and 48 h after IPC or resveratrol treatment. Resveratrol treatment was able to increase SIRT1 activity by about 85% (n = 4, P < 0.05) at 30 mins after treatment when compared with the vehicle-treated group (n =7) (Figure 3). However, resveratrol-induced SIRT1 activation returned to baseline levels by 48 h after treatment (vehicle treated, n = 7; resveratrol, treated n = 3) (Figure 3). In contrast to resveratrol treatment, SIRT1 activity did not change in the IPC group at 30mins of reperfusion (n = 4) as compared with the sham group (n = 7), but was significantly increased by 81% (n = 4, P < 0.05) at 48 h after IPC, as compared with sham-treated slices.
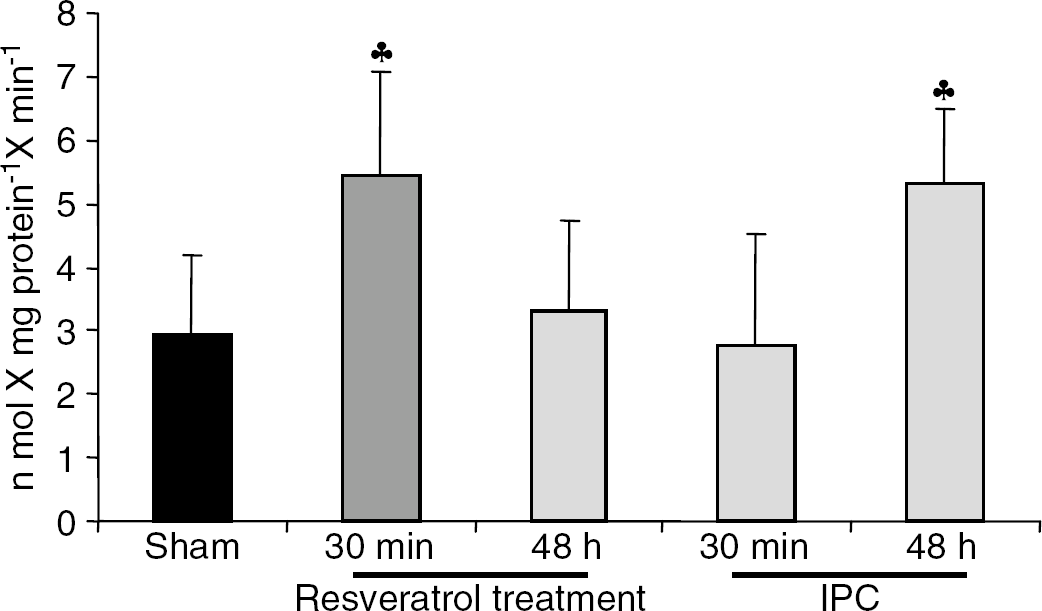
Sirtuin 1 (SIRT1) enzyme activity was measured in organotypic hippocampal slices 30 mins and 48h after IPC/resveratrol pretreatment. The SIRT1 activity levels were increased 30 mins after resveratrol pretreatment and 48 h after IPC. ♣P < 0.05 as compared with sham.
Thus, increased SIRT1 enzyme activity after IPC suggested a key role of SIRT1 enzyme activation in IPC-mediated neuroprotection. To test this hypothesis, we blocked SIRT1 enzyme activity with sirtinol after IPC for the complete duration of reperfusion before exposing cultures to ‘test’ ischemia. Our results showed that sirtinol blocked IPC protection at all concentrations used (10, 50, and 100 μmol/L). The PI fluorescence of sirtinol-treated (10, 50 and 100 μmol/L), IPC, and ischemic groups were 87.12 ± 3.82 (n = 7, P < 0.01 against IPC), 83.96 ± 6.84 (n = 5, P < 0.01 against IPC), 86.88 ± 9.96 (n = 9, P < 0.01 against IPC), 32.74 ± 7.53 (n = 8), and 63.91 ± 6.70 (n = 11; P ± 0.01 against IPC), respectively. Control experiments were performed by exposing organotypic slices to sirtinol alone for 48 h followed by either sham or ‘test’ ischemia. The organotypic slices exposed to sirtinol treatment alone for the duration of 48 h followed by sham OGD did not show any cell death as compared with the sham group. The PI fluorescence values of sirtinol exposure alone for the duration of experiment and sham group were 6.50 ± 2.97 (n = 2) and 5.45 ± 2.85 (n = 8), respectively. However, the PI fluorescence values obtained from slices exposed to sirtinol for 48 h followed by ‘test’ ischemia(67.59 ± 7.05, n = 6) were similar to PI fluorescence values of ischemia group (63.91 ± 6.70, n = 11) (Figure 4).
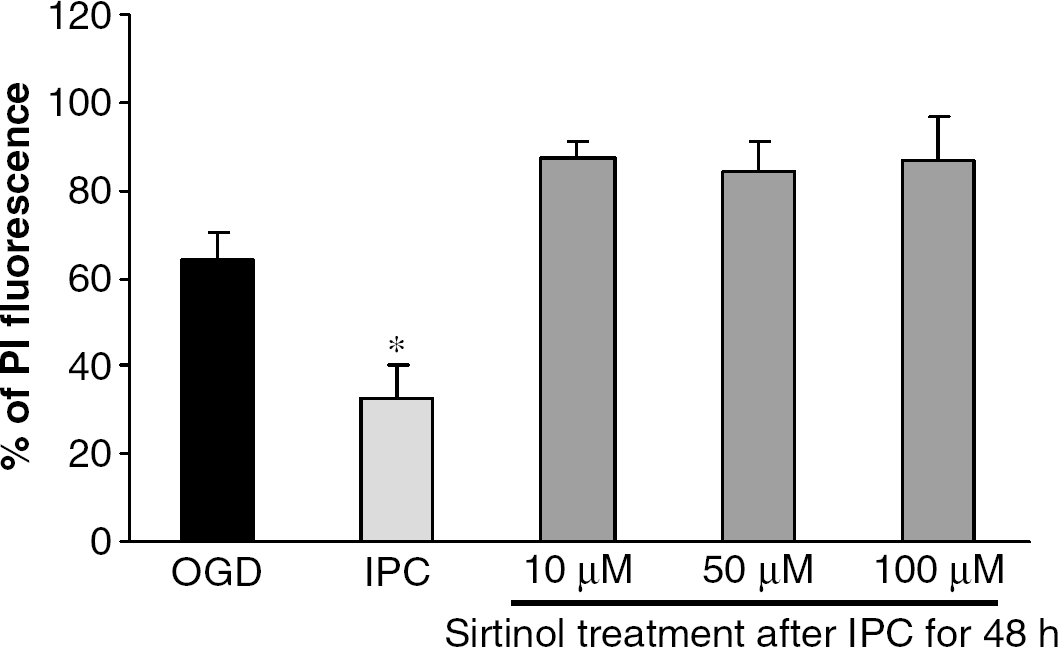
Sirtuin 1 (SIRT1) enzyme inhibitor, sirtinol, abolished IPC-mediated hippocampal neuroprotection. These results confirmed SIRT1 activation is key mediator of neuroprotection after IPC. *P<0.001 as compared with test ischemia.
Similarly, the blockade of SIRT1 activation during 48 h of reperfusion after resveratrol treatment abolished neuroprotection, suggesting key role of SIRT1 activation during reperfusion phase in mediating hippocampal CA1 protection. The PI fluorescence of sirtinol-treated and ischemic groups were 66.13 ± 8.38 (n = 11; sirtinol treatment prior, during and after resveratrol treatment), 67.94 ± 9.51 (n = 8; sirtinol treatment after resveratrol treatment), and 63.22 ± 5.08 (n = 9), respectively (Figure 5). However, sirtinol treatment only before or prior and during resveratrol treatment could not block neuroprotection (Figure 5).
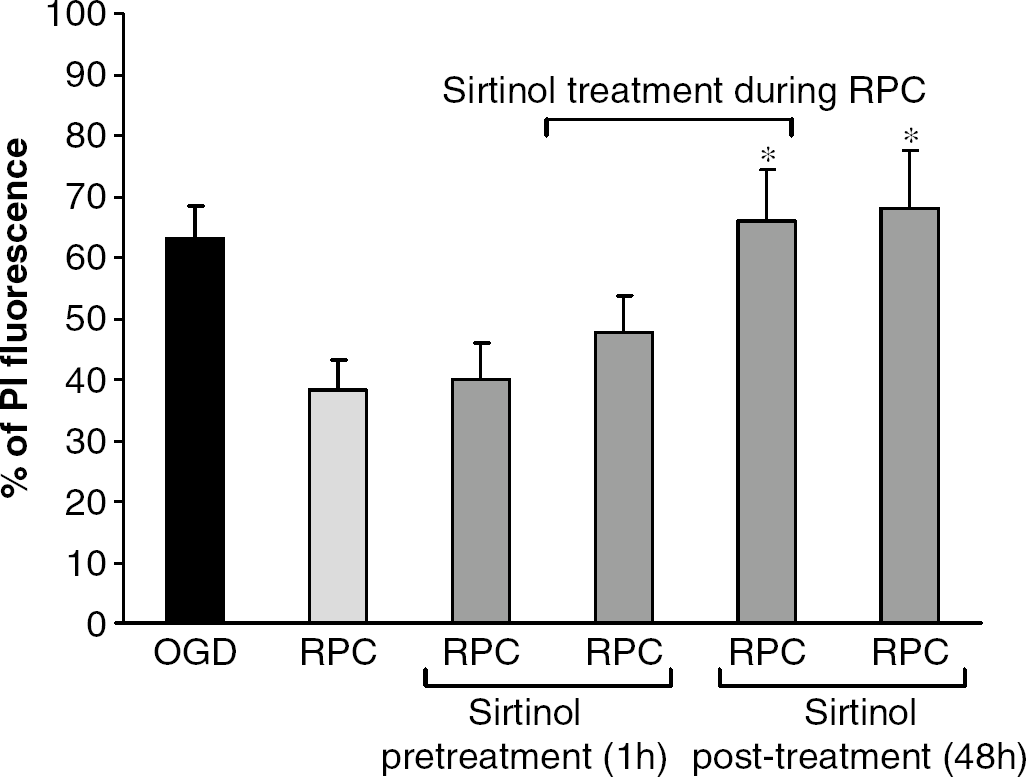
SIRT1 enzyme inhibitor, sirtinol, during 48 h of reperfusion after resveratrol treatment abolished resveratrolmediated hippocampal neuroprotection. *P < 0.001 as compared with resveratrol preconditioning.
Discussion
In the present study, we present convincing evidence that resveratrol administered 48 h before an ischemic insult is neuroprotective as in IPC, but also that IPC increases the activity of one of resveratrol's main cellular targets for protection (SIRT1). Inhibition of SIRT1 enzyme activation during reperfusion either after IPC or resveratrol treatment abolished neuroprotection in the hippocampal CA1, suggesting a common signaling pathway of neuroprotection after IPC and single transient resveratrol application.
In previous studies in the brain, postischemic administration of resveratrol was neuroprotective (Wang et al, 2002). The neuroprotective mechanisms of action by resveratrol were attributed to its intrinsic radical scavenger properties and an effect on blood flow (Andrabi et al, 2004; Wang et al, 2002). In the brain, we are unaware of any previous study that showed a single transient resveratrol pretreatment, as would occur with daily red wine intake, would provide a transitory neuroprotective state, as in IPC. In the heart, resveratrol chronic pretreatment for 14 days preserved myocardial recovery after global ischemia (Dernek et al, 2004). Thus, we conjecture that resveratrol initiates a signal transduction pathway, similar to IPC. In addition, it is important to note that our studies used an in vitro model of cerebral ischemia, which excludes any systemic role in the neuroprotection afforded by resveratrol and suggest an intrinsic neuroprotective pathway.
Both resveratrol and IPC mediated neuroprotection via SIRT1 enzyme activation. Resveratrol promotes the SIRT1 activation by binding to SIRT1 leading to a conformational change near the coumarin group of bound p53 acetylpeptide containing 7-amino-4-methycoumarin (p53-AMC peptide) (Borra et al, 2005). To our knowledge, our observation is the first study to show that IPC promotes the activation of SIRT1. We infer that the signaling pathways elicited by IPC lead to SIRT1 activation, although the precise pathway remains undefined. A possible clue emerges from the fact that any metabolic alteration in NAD + nicotinamide adenine dinucleotide (reduced form) (NADH) ratio activates SIRT1 enzyme (Lin et al, 2004). We and others have established over the years that major changes in NAD+/NADH ratios occur after anoxia/ ischemia (Centeno et al, 1999). For example, after ‘lethal’ anoxic or ischemic insults, a profound hyperoxidation of NADH levels were observed during the initial 30 mins of reperfusion (Centeno et al, 1999). However, when preconditioning is induced, not only NADH did not change during the reperfusion phase but also in turn increased during the ‘test’ anoxic insult (Centeno et al, 1999). At that time, these results were perplexing. However, in light of the results of the present study, these results suggest a possible mechanism by which IPC could activate the SIRT1 pathway. Studies are underway to define the possible link between increases in NADH levels and SIRT1 activity after IPC.
Although both resveratrol and IPC were neuroprotective against ‘test’ ischemia, the time course for the activation of SIRT1 was different between the two paradigms. As expected, resveratrol rapidly and transitorily increased SIRT1 activity, whereas IPC-induced increase of SIRT1 activity was only observed before ‘test’ ischemia. The resveratrol results suggest that the transitory increase in SIRT1 activity initiates a neuroprotective state lasting at least 48 h. Loss of neuroprotection because of inhibition of SIRT1 activation during 48 h of reperfusion after resveratrol treatment confirms the fact that SIRT1 activation mediates the neuroprotective state. Further studies are needed to define the time range whereby resveratrol promotes neuroprotection after a single administration.
These data lead us to propose a novel SIRT1-mediated signal transduction pathway by which IPC/resveratrol protect CA1 hippocampal neurons against cerebral ischemia. However, the SIRT1 site of action in the CA1 region of the hippocampus remains undefined. Further studies will define the CA1 cell types where SIRT1 activity increases. In addition, our study supports the efficacy of resveratrol as a prophylactic agent against cerebral ischemia. The most direct and significant application of these findings is that resveratrol may provide pharmacological access to this protective state, especially during cardiopulmonary bypass surgery, cardiac transplantation, and other types of brain surgeries, where necessary periods of ischemia are provoked in healthy tissue to repair other central nervous system anomalies. Additional studies will be required to define whether daily administration of resveratrol provides similar or more neuroprotection against ischemia as the paradigm presented in the current study.
Footnotes
Acknowledgements
We are thankful to Dr Howitz (BIOMOL international, PA, USA) for his thoughtful suggestions during standardization of SIRT1 assay in our laboratory.