
Abstract
The authors sought to determine whether Zn2+ translocation associated with neuronal cell death occurs after transient global ischemia (TGI) in mice, as has been previously shown in rats, and to determine the effect of mild hypothermia on this reaction. To validate the TGI model, carbon-black injection and laser-Doppler flowmetry were compared in three strains of mice (C57BL/6, SV129, and HSP70 transgenic mice) to assess posterior communicating artery (PcomA) development and cortical perfusion. In C57BL/6 mice, optimal results were obtained when subjected to 20-minute TGI. Brain and rectal temperature measurements were compared to monitor hypothermia. Results of TGI were compared in normothermia (NT; 37°C) and mild hypothermia groups (HT; 33°C) by staining with Zn2+-specific fluorescent dye, N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) and hematoxylin– eosin 72 hours after reperfusion. The Zn2+ translocation observed in hippocampus CA1, CA2, and Hilus 72 hours after 20 minutes of TGI was significantly reduced by mild hypothermia. The number of degenerating neurons in the HT group was significantly less than in the NT group. Mild hypothermia reduced mortality significantly (7.1% in HT, 42.9% in NT). Results suggest that mild hypothermia may reduce presynaptic Zn2+ release in mice, which protects vulnerable hippocampal neurons from ischemic necrosis. Future studies may further elucidate mechanisms of Zn2+-induced ischemic injury.
Several mechanisms such as glutamate release, toxic calcium influx, apoptosis, free radicals, and excitotoxicity are reported to be associated with neuronal cell death after transient global ischemia (TGI). Recently, Zn2+ has been emphasized as a key mediator in neuronal cell death after TGI (Choi and Koh, 1998; Koh et al., 1996; Weiss et al., 2000). Zn2+ is located in presynaptic vesicles in excitatory nerve terminal and colocalized with glutamate (Martinez-Guijarro et al., 1991). After neuronal injury, Zn2+, in addition to glutamate, is released to synaptic clefts and enters postsynaptic neurons through glutamate channels (Choi and Koh, 1998; Wiess et al., 2000). Accumulated Zn2+ in postsynaptic neurons may activate free radical generation or apoptotic cascades, which results in neuronal cell death (Kim et al., 1999; Lobner et al., 2000; Park et al., 2000).
Mild hypothermia is known to show a strong neuro-protective effect against both focal ischemia (Barone et al., 1997; Ginsberg et al., 1992) and global ischemia (Busto et al., 1989a; Chopp et al., 1989). Mechanisms of neuroprotective effect include reduction of brain metabolic rate (Chopp et al., 1989; Lanier, 1995) and glutamate release (Baker et al., 1995; Busto et al., 1989b; Globus et al., 1988, Nakashima and Todd, 1996). Deep hypothermia (29°C) can reduce Zn2+ translocation and cell death after TGI in rats (Johansen et al., 1993). The effect of mild hypothermia on Zn2+ release after TGI in mice has not been previously investigated.
Since genetically altered mice are useful to investigate the role of gene products in cytotoxic reactions during cerebral ischemia, reproducible models for ischemic injury in mice are needed (Chan, 2001). In studies of TGI, several reports describe the use of genetically altered mice (Kelly et al., 2001; Kitagawa et al., 1998a; Murakami et al., 1997; Sheng et al., 2000). The model has not been standardized, however, perhaps because of variability of posterior communicating artery (PcomA) development and collateral cerebral blood flow after carotid artery occlusion in different strains. Furthermore, effects of mild hypothermia and correlation of brain and rectal temperature measurements have not been studied in mice.
The initial goal of this study was to compare PcomA patency in C57BL/6, SV129, and HSP70 transgenic mice for correlation with cortical perfusion during TGI and to establish the TGI model in mice. C57BL/6 mice were used to assess the effect of varying durations of bilateral common carotid artery occlusion (BCCAO) since it is often used as a background strain in developing transgenic mice (Fujii et al., 1997). Neuronal cell death was localized in order to better establish the TGI model in mice. Rectal and brain temperature measurements were compared over the 32°C to 37°C range for assessment of hypothermia in mice. The hypothesis that Zn2+ translocation is induced after TGI in mice and that mild hypothermia reduces presynaptic Zn2+ release and translocation associated with neuronal cell death after TGI was tested. Results of TGI produced by 20-minute BCCAO were compared after 72 hours in normothermic and hypothermic mice.
MATERIALS AND METHODS
Animal protocol
This study was conducted in accordance with animal care guidelines issued by the National Institutes of Health. The Animal Care and Use Committee at the Veterans Affairs Medical Center, San Francisco, approved all protocols. Male adult C57BL/6 and SV129 mice (24 to 28 g) were obtained from Charles River Laboratory (Wilmington, MA, U.S.A.). Dr. W. H. Dillmann (University of California, San Diego) provided transgenic mice overexpressing heat-shock protein 70 (HSP70 Tg, 24 to 26 g), which were used because they were available for other studies in our laboratory. HSP70 Tg mice were bred on a CB6F1 background, which was hybridized by C57BL/6 and BAL B/C. Mice were given free access to food and water before and after surgery. Anesthesia was induced with 2% isoflurane in a closed chamber and maintained with 1.5% isoflurane in 30% O2 and 70% N2O using a face mask. Rectal temperature was monitored and maintained by Homeothermic Blanket Systems (Harvard Apparatus Inc., Holliston, MA, U.S.A.). Temporalis muscle temperature was monitored by a 33-G needle probe and Dural Channel Thermometer (Fischer Scientific, Pittsburgh, PA, U.S.A.).
Because repeated blood withdrawal influences surgical and histological outcome in mice, separate groups of mice were used for evaluating physiological parameters. A PE10 femoral catheter was placed to monitor blood pressure using a Research Grade Blood Pressure System (Harvard Apparatus), and to sample arterial blood gas (pH, P
Carbon-black injection
Carbon black was injected in C57BL/6, SV129, and HSP70 Tg mice (n = 8 each) as previously described (Majid et al., 2000). Mice were anesthetized deeply with pentobarbital (2 mg/kg). Transcardiac injection of carbon black (in equal volume of 20% gelatin dissolved in distilled water) was performed. After approximately 1.5 mL carbon black was infused, mice were decapitated and brains were removed immediately. The brains were immersed in 4% paraformaldehyde in 0.1-mol/L phosphate buffer (pH 7.4) overnight, followed by 20% sucrose at 4°C. Development of PcomA was evaluated by inspection under the surgical microscope (40x). The score was as follows: grade 0 = absent, grade 1 = poor development, and grade 2 = well formed. The arteries were scored bilaterally and the higher grade was selected for each animals.
Laser-Doppler flowmetry
Cerebral cortical perfusion was monitored with laser-Doppler flowmetry (Vasamedics, St. Paul, MN, U.S.A.) in C57BL/6, SV129, and HSP70 Tg mice (n = 8 each). After mice were anesthetized, a 1.9-mm diameter needle probe was placed directly on the skull 1-mm posterior and 2-mm lateral to the bregma. Cortical perfusion values were monitored before and 1 minute after onset of BCCAO. Values 1 minute after BCCAO were expressed as a percentage relative to baseline.
Induction of transient global ischemia
Transient global ischemia was produced by BCCAO. Mice were placed in the supine position and a midline skin incision was made. The common carotid arteries (CCA) were exposed bilaterally and carefully isolated from the adjacent vagus nerve to prepare for clip application. A microvascular clip (B-2; Fine Science Tools, Inc., Foster City, CA, U.S.A.) was temporary applied to each CCA. Mice were observed under anesthesia during occlusion and for 20 minutes after onset of reperfusion. After 20 minutes of occlusion, clips were removed and patency of CCA was confirmed by inspection under the surgical microscope. Rectal and temporalis muscle temperatures were monitored continuously until 20 minutes after reperfusion. After recovery from anesthesia, mice were returned to the cage and observed for 2 hours. Temperature inside the cages was maintained at 25 °C.
To determine the optimum duration of BCCAO, a pilot study using C57BL/6 mice was performed to evaluate mortality and neuronal cell death after 15, 20, 25, and 30 minutes of occlusion (n = 4 each).
Histological assessment
After 72-hour survival, mice were anesthetized with ketamine (80 mg/kg) and xylazine (32 mg/kg) and decapitated. Brains were frozen immediately in dry ice and stored at −80°C. The brains were then sectioned with a cryostat (10-μm thickness).
To visualize intracellular zinc ions, zinc-specific fluorescent dye, N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ; Molecular Probes, Eugene, OR, U.S.A.) was used as previously described (Frederickson et al., 1987). Sections at the level of 2.4-mm posterior to the bregma were immersed in TSQ staining solution for 1 minute followed by saline for 1 minute. Immediately after rinsing in saline, sections were examined under the fluorescence microscope (excitation, 360 nm; emission, 480 nm; 10x) without cover slip while still wet.
To evaluate neuronal cell death after TGI, adjacent coronal sections were stained with hematoxylin-eosin (H&E) after fixation in 70% alcohol overnight. In this study, separate adjacent sections were stained with TSQ and H&E because TSQ degrades cell integrity, interfering with accurate counting of degenerating neurons on H&E-stained sections. After H&E staining, degenerating neurons exhibit intensively eosinophilic cytoplasm, shrunken cell bodies, and triangulated nuclei. Owing to possible occurrence of unilateral lesions or asymmetric density of hippocampal neuronal cell death, the number of degenerating and viable neurons in hippocampal subregions (CA1, 2, 3, and Hilus) only in the most severely damaged hemisphere was counted under a light microscope (200x) by two investigators masked to the study design. Results were averaged and expressed as the percentage of degenerating neurons in each hippocampal subregion.
Brain temperature assessment
Correlation of brain, temporalis muscle, and rectal temperature was examined in C57BL/6 mice (n = 5). Anesthesia was administered, and mice were placed in the prone position on a heating blanket. Rectal temperature was monitored and maintained at 37°C. Through a scalp incision, a small burr hole was made 1.5-mm lateral and 2.5-mm posterior to the bregma. A 33-G needle probe was lowered 2 mm from skull surface, where the probe tip reached the hippocampus. Another 33-G needle probe was placed into right temporalis muscle. After securing the probes, the heating blanket was disconnected from its control unit. Then rectal temperature was gradually reduced to 32°C for 60 to 90 minutes, while room temperature was maintained at 25 °C. Brain and temporalis muscle temperature were recorded for every 0.5°C in reduction of rectal temperature.
Mild hypothermia
Animals were assigned to two temperature groups monitored with rectal and temporalis muscle probes: normothermia (NT, n = 14) and mild hypothermia (HT, n = 14). In the NT group, rectal and temporalis muscle temperatures were maintained with a heating blanket at 37.0°C ± 0.5°C and 36.0°C ± 0.5°C, respectively. In the HT group, rectal and temporalis muscle temperatures were maintained at 33.0°C ± 0.5°C and 32.0°C ± 0.5°C, respectively. Mild hypothermia was induced by disconnecting the heating blanket from a control unit and allowing rectal temperature to decrease spontaneously. When rectal temperature decreased to 33°C, BCCAO was induced. Rectal and temporalis muscle temperatures were further reduced slightly during occlusion, but were maintained within defined ranges without warming. After reperfusion, warming was started by reconnecting the heating blanket to its control unit. Rectal temperature recovered to 37°C within 15 minutes after onset of warming. The mean arterial blood pressure and arterial blood gasses were monitored during and after TGI in separate groups to avoid the effects of blood loss due to sampling on biological outcome.
Statistical analysis
The unpaired Student's t-test was used to determine the statistical significance in physiological variables and the percentage of degenerating neurons in hippocampal subregions in groups. The one-way analysis of variance (ANOVA) followed by post hoc Student-Newman-Keuls test was used to determine the statistical significance in PcomA development score, cortical perfusion values, and the percentage of degenerating neurons in hippocampal subregions. Pearson correlation coefficient followed by a linear regression was used to determine correlation between rectal and brain temperature. Chi-square analysis was used to determine the statistical significance in mortality between groups. Statistical significance was defined as P < 0.05. All values are expressed as mean ± SD. StatView 5.0.1 (SAS Institute Inc., Cary, NC, U.S.A.) was used for all analyses.
RESULTS
Transient global ischemia model in mice
Posterior communicating artery development was assessed using transcardiac carbon-black injection in C57BL/6, SV129, and HSP70 Tg mice (n = 8 each). The PcomA development scores in C57BL/6, SV129, and HSP70 Tg mice were 1.00 ± 0.76, 1.38 ± 0.75, and 1.63 ± 0.52, respectively. Although C57BL/6 mice showed the lowest score, there was no significant difference between the three strains (Fig. 1A). However, PcomA development scores after carbon-black injection are consistent with variability in PcomA patency, which might influence residual blood flow after BCCAO.
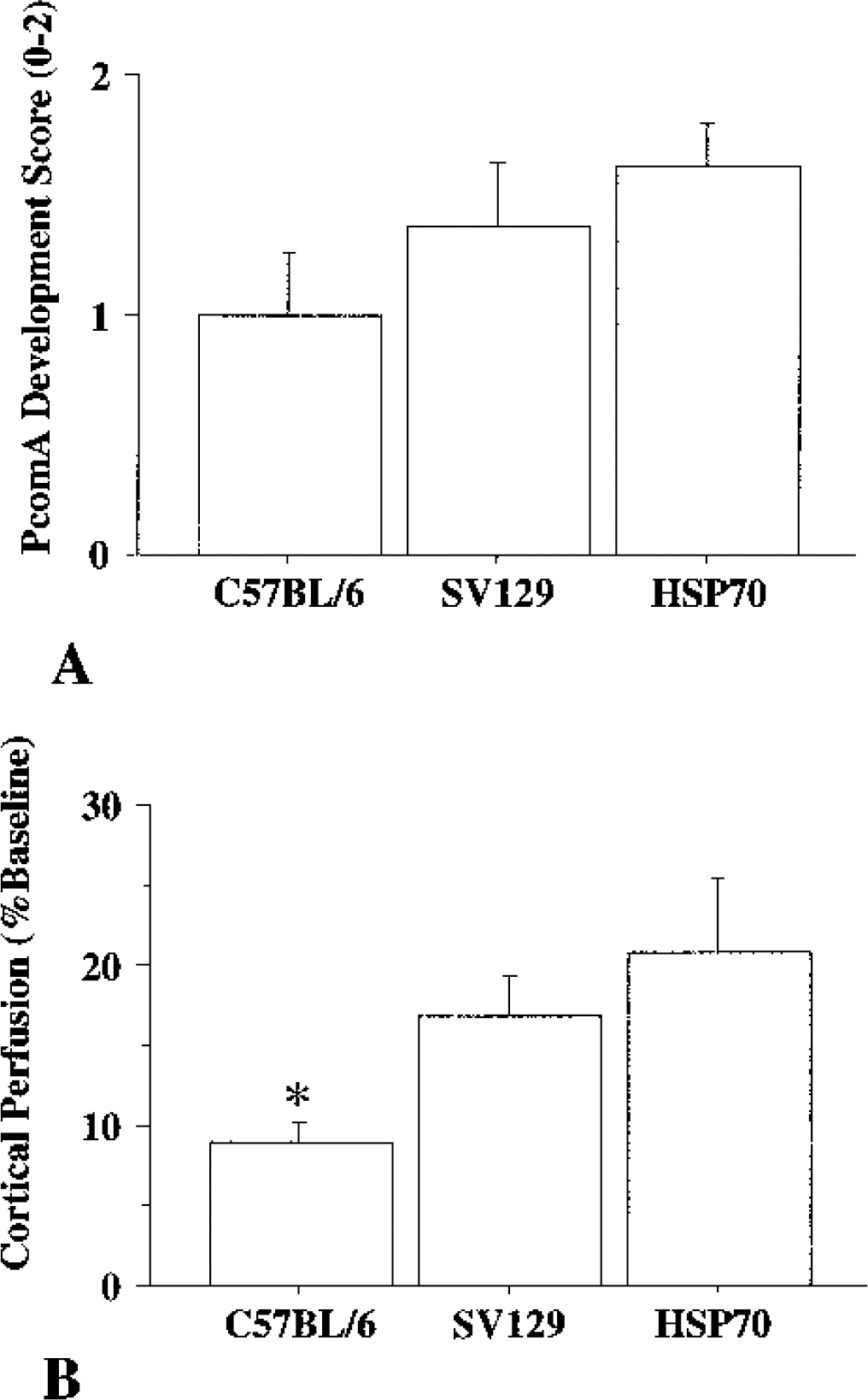
(A) Posterior communicating artery (PcomA) development score after carbon-black injection in C57BL/6, SV129, and heat-shock protein 70 transgenic (HSP70 Tg) mice. There is no significant difference between strains.
Cortical perfusion values 5 minutes after BCCAO in C57BL/6, SV129, and HSP70 Tg mice were, 8.87 ± 3.84, 16.85 ± 7.22, and 20.80 ± 12.91, respectively (Fig. 1B). There is a significant difference between C57BL/6 and HSP70 Tg mice. Because results suggested that C57BL/6 mice were more vulnerable to ischemic neuronal injury, the C57BL/6 strain was used for subsequent experiments.
The mortality rates 72 hours after BCCAO in 15-, 20-, 25-, and 30-minute occlusion models were 25%, 50%, 75%, and 100%, respectively. Although 15-minute occlusion produced the lowest mortality, histological evaluation demonstrated substantial variability in the percentage of degenerating neurons in hippocampus. Therefore, 20-minute occlusion was used in this experiment.
Neuronal cell death was demonstrated with H&E staining 72 hours after 20 minutes of BCCAO in C57BL/6 (n = 13). Laterality of the lesions, which may have been due to variations in collateral circulation of the PcomA in hippocampal subregions, is presented in Fig. 2A. In CA1, CA2, and Hilus, more than 80% of mice showed either bilateral or unilateral lesions. Unilateral lesions, however, were commonly observed. Only 30.8% of mice showed lesions in CA3. The percentage of degenerating neurons in hippocampal subregions was assessed on the most severely injured side for purposes of this study because the number of injured cells was too small to count in the less damaged hemisphere (Fig. 2B). CA2 showed the highest percentage of injury. There was a significant difference between the percentage of degenerating neurons in CA2 and in CA3. Although CA1 and Hilus are known to be vulnerable regions after transient global ischemia in rats and gerbils, CA2 was found to be the most vulnerable region in these mice.
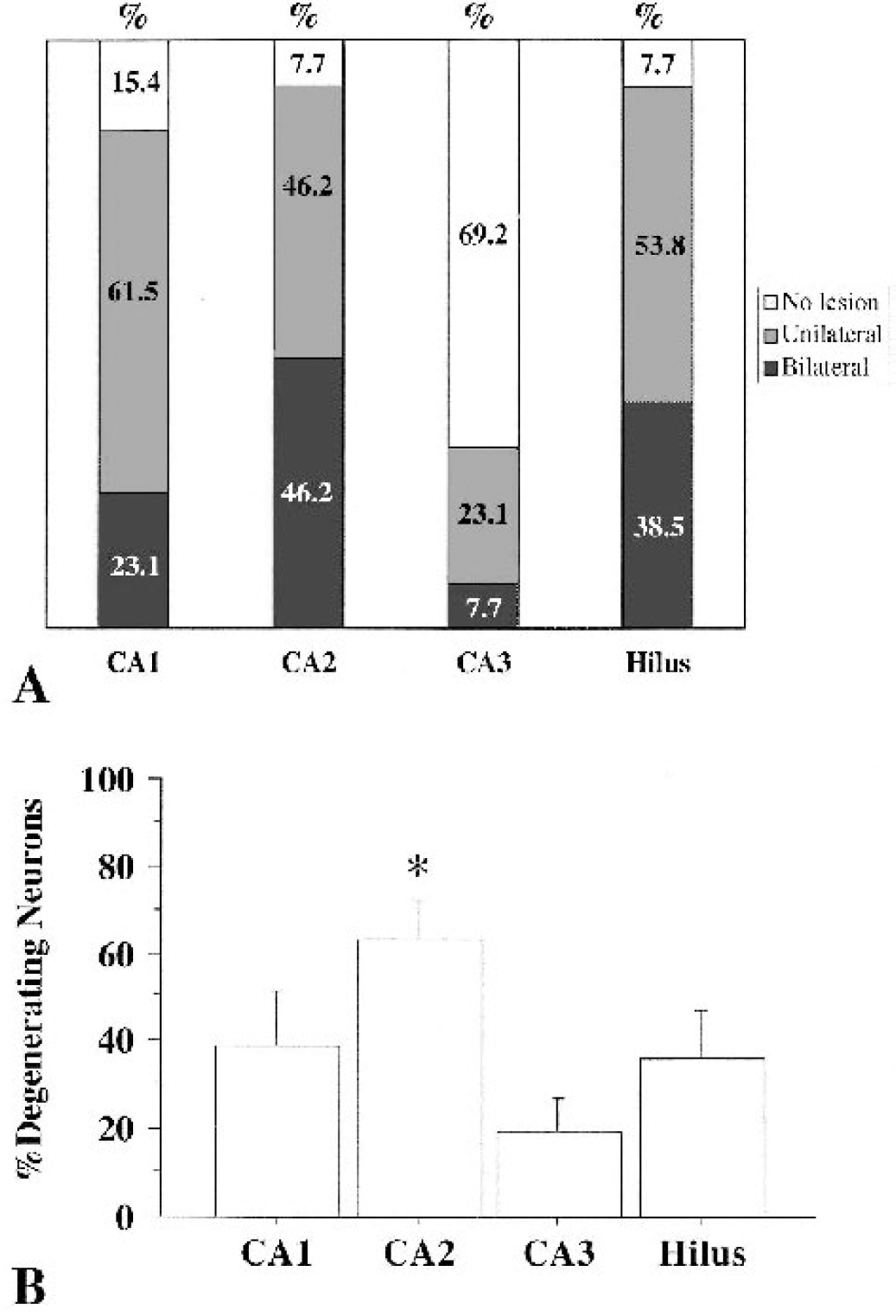
(A) Laterality of lesions in CA1, CA2, CA3, and Hilus 72 hours after 20 minutes of bilateral common carotid artery occlusion (BCCAO) in C57BL/6.
Zinc translocation after transient global ischemia in mice
Zinc translocation was observed 72 hours after 20 minutes of BCCAO (Fig. 3). In normal control mice, TSQ staining was seen in mossy fibers, CA1 to CA4 stratum radiatum and oriens, and dentate gyrus (Fig. 3A). Zn2+ staining was also present in cortical layer I-III and IV, striatum, thalamus, and amygdala. In CA1, a dark band was observed in the pyramidal cell layer in control mice (Fig. 3B). After TGI, dense Zn2+ translocation was seen in the pyramidal layer neurons (Fig. 3C). Similarly, in both CA2 and Hilus, Zn2+-translocated neurons were observed (Figs. 3F and 3I). Ischemic change in TSQ-positive cells was validated with H&E staining on the same sections (Figs. 3D, 3G, and 3J). Zn2+-translocated necrotic neurons were more commonly found in CA2 than in CA1 or Hilus.
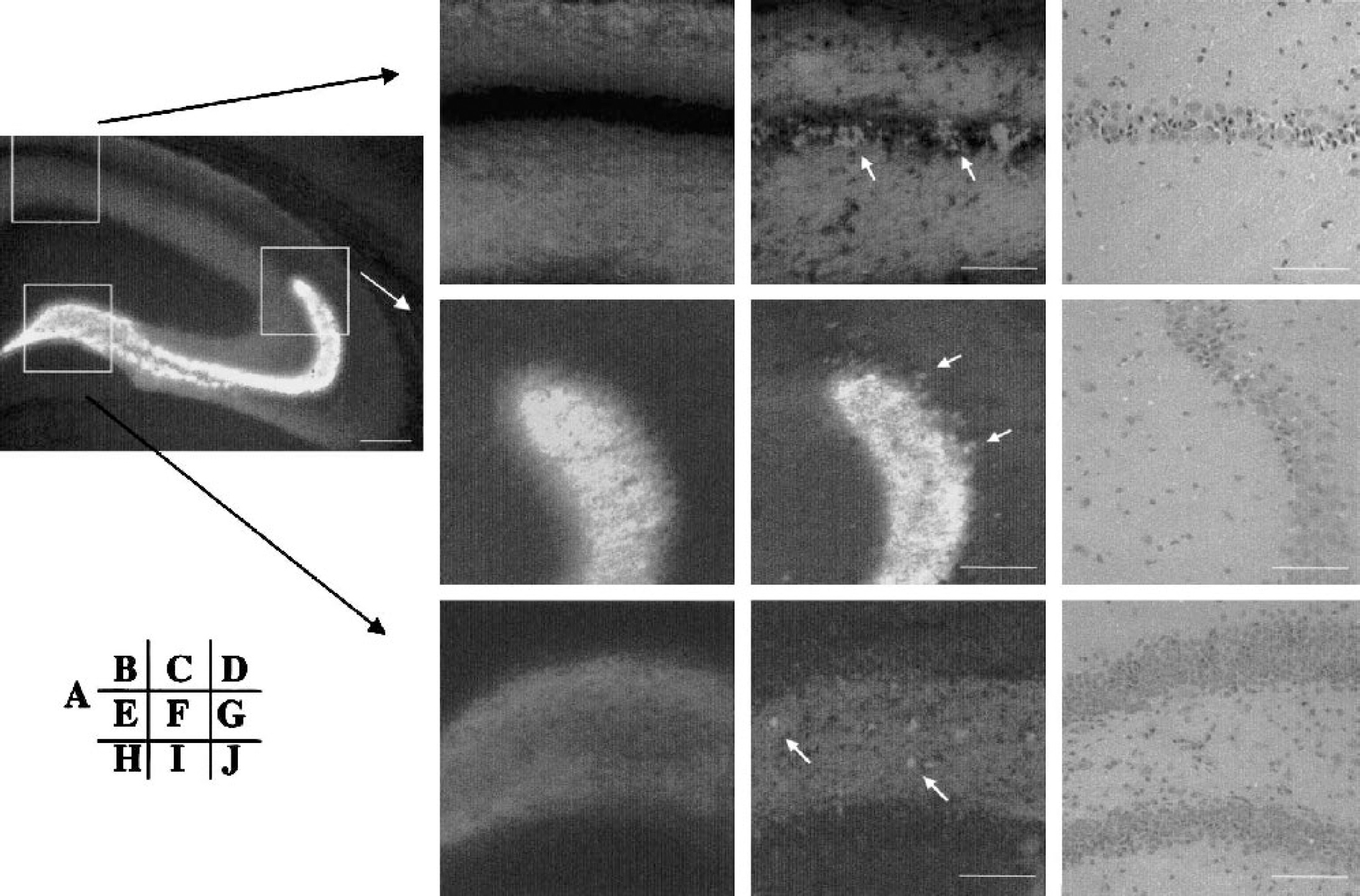
N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) staining in normal control mouse (
Correlation of brain, temporalis muscle, and rectal temperature measurements in mice
Although comparison of brain and temporalis muscle temperature measurements demonstrated equivalent values at 32°C to 37°C, undetected differences could develop after BCCAO because BCCAO reduces the blood supply to temporalis muscle. Therefore, rectal temperature may reflect brain temperature more accurately after BCCAO. A strong correlation between brain and rectal temperature measurements was observed (r = 0.96, P < 0.0001). However, the relation was not entirely linear. When rectal temperature was 36°C to 37°C, brain temperature was 1°C lower. Brain temperature was consistent with rectal temperature when rectal temperature was 33°C to 34°C. When rectal temperature was 32°C, brain temperature was 0.5°C higher.
Reduction of Zn2+ translocation by mild hypothermia
There was no significant difference in physiological variables in the NT and HT groups during and after TGI. In both groups, mean arterial blood pressure increased during occlusion and decreased after reperfusion. Rectal and temporalis muscle temperatures during BCCAO and for 20 minutes after reperfusion are shown in Fig. 4. In both groups, temperatures were maintained within the defined ranged. Temporalis muscle temperature was reduced in both groups during BCCAO. In the HT group, rectal and temporalis muscle temperature increased and recovered to 37.0°C ± 0.5°C within 15 minutes after warming was initiated (Fig. 4).
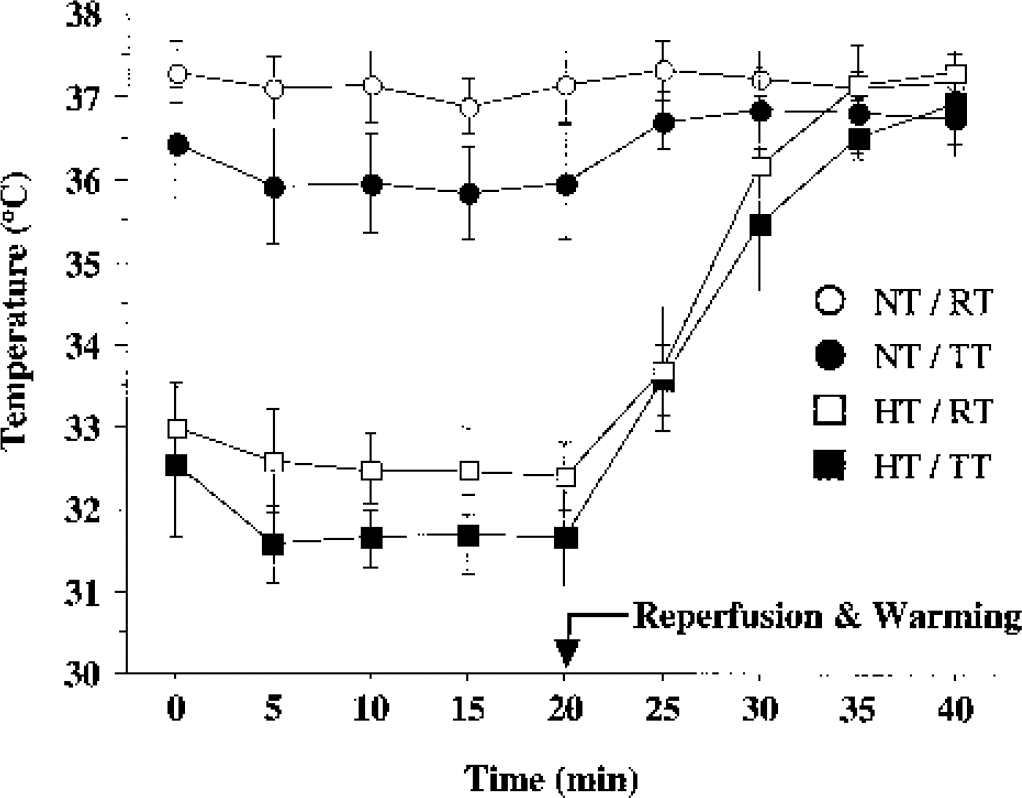
The time course of changes in rectal and temporalis muscle temperatures in NT and HT. Values are mean ± SD; error bars are SD. NT, normothermia group (n = 8); HT, hypothermia group (n = 9); RT, rectal temperature; TT, temporalis muscle temperature.
The mortality after 20-minute BCCAO was significantly different in the NT and HT (42.9% vs. 7.1%, P < 0.05). In the NT group, 6 of 14 mice died, 1 within 24 hours after surgery and 5 within 72 hours after BCCAO. In the HT group, only 1 mouse died within 48 hours after BCCAO.
The TSQ fluorescent and H&E stains in a nonischemic control, as well as in NT and HT ischemic mice, are shown in Fig. 5. In the control mouse, zinc staining is present in stratum radiatum and oriens in CA1. Zinc staining, however, was not observed in the pyramidal layer in CA1 (Fig. 5A). In the NT group, dense zinc translocation was seen in the neuronal cell bodies of the pyramidal layer in CA1 (Fig. 5B). A large number of eosinophilic pyknotic neurons were observed in the pyramidal cell layer on adjacent sections with H&E staining (Fig. 5E). In the HT group, only scattered TSQ-positive and degenerating neurons were observed in pyramidal layer (Figs. 5C and 5F). The frequency of TSQ-positive and degenerating neurons was markedly reduced in the HT group.
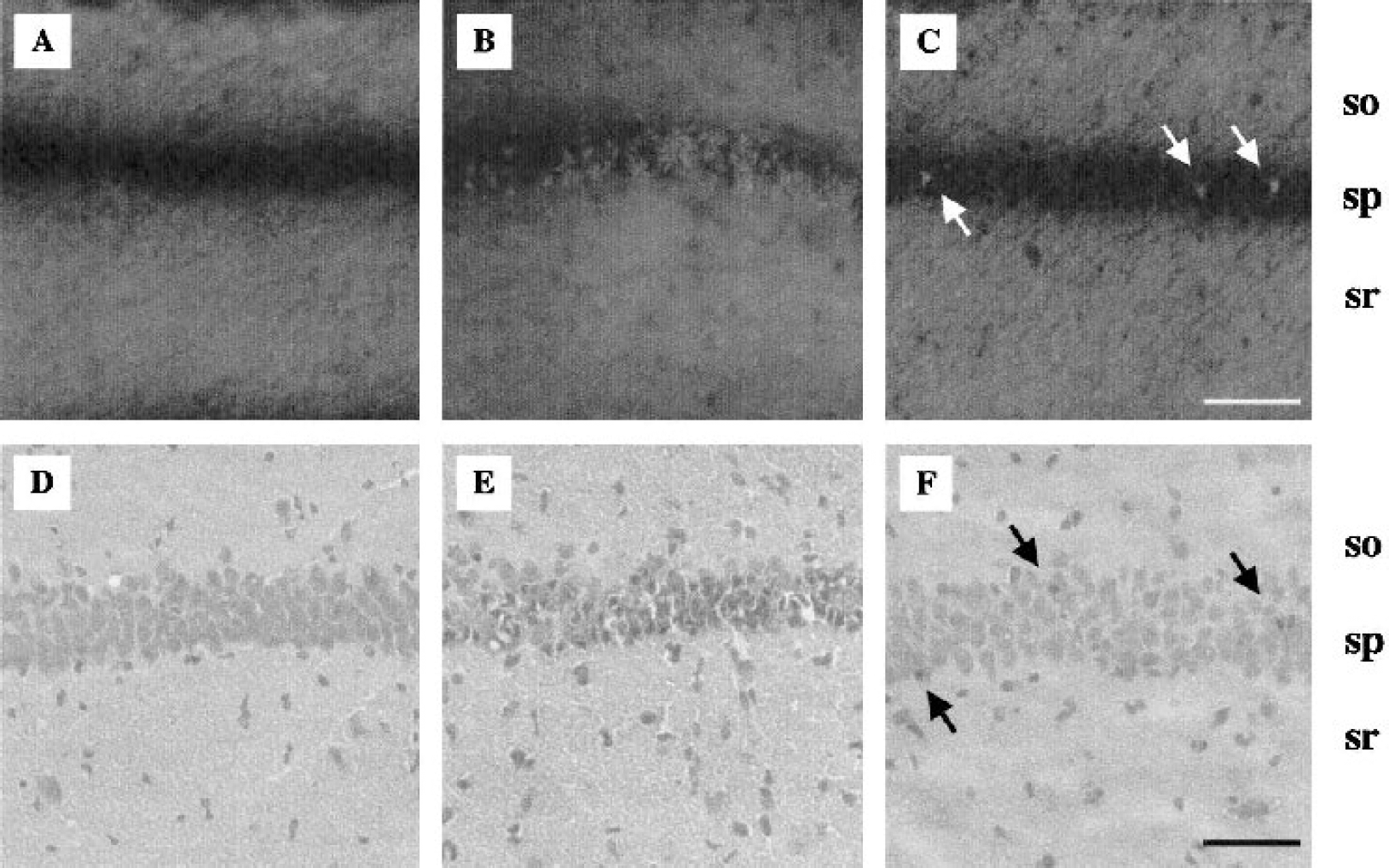
N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) fluorescent (
In the NT group, the percentages of degenerating neurons in CA1, 2, 3, and Hilus were 61.0% ± 44.2%, 73.0% ± 29.5%, 28.2% ± 33.2%, and 39.8% ± 44.9%, respectively. In HT groups, the percentages of degenerating neurons in CA1, 2, 3, and Hilus were 17.9% ± 35.6%, 11.2% ± 27.5%, 4.3% ± 12.9%, and 2.2% ± 0.7%, respectively (Fig. 6). In CA1, CA2, and Hilus, there were significant differences in between the NT and HT groups. In CA3, although the difference was not significant (P = 0.064), there was a trend towards reduction of the percentage of degenerating neurons by mild hypothermia.
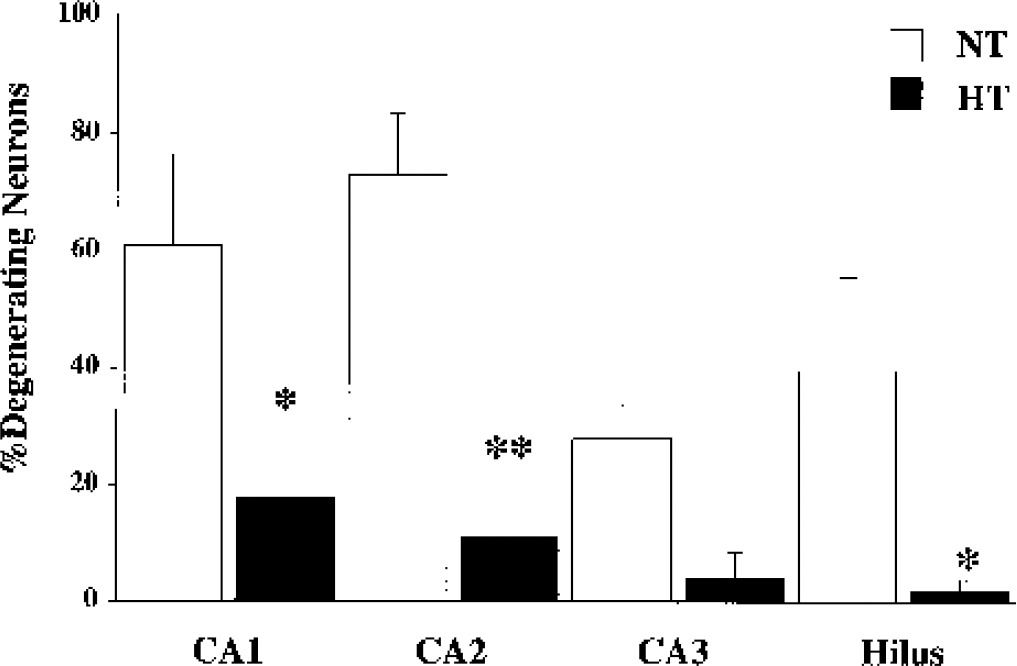
The percentage of degenerating neurons in hippocampal subregions 72 hours after 20 minutes of bilateral common carotid artery occlusion in normothermia (NT) and hypothermia (HT) groups. There are significant differences between NT and HT in CA1, CA2, and Hilus. Although the percentage of degenerating neurons in CA3 was less in HT, the difference was not significant (P = 0.06). *P < 0.05, **P < 0.01. Error bars are SEM.
DISCUSSION
Transient global ischemia model in mice
The results of preliminary experiments in this study establish 20-minute BCCAO in C57BL6 mice as a reproducible method for inducing selective neuronal cell death after TGI in mice. Species dependent differences in extent of reduction of cortical perfusion and severity or location of neuronal cell death after BCCAO are documented using this model.
Recently, genetically altered mice overexpressing BCL2, CuZn-superoxide dismutase (SOD), extracellular SOD, and inducible HSP70 have been used to investigate the mechanisms of neuronal death after TGI (Kelly et al., 2001; Kitagawa et al., 1998a; Murakami et al., 1997; Sheng et al., 2000). Different methods, however, have been used for the induction of TGI. Different strains demonstrate different PcomA development, which may influence collateral CBF and histological outcome. Thus, BCCAO with longer occlusion duration, BCCAO with hypotension, and BCCAO monitored with laser-Doppler flowmetry have been tested (Kelly et al., 2001; Kitagawa et al., 1998a; Sheng et al., 2000). Initially, BCCAO with systemic hypotension was applied in preliminary studies in our laboratory. However, high mortality, technical difficulty, and variability of histological outcome rendered it difficult to standardize this model. To predict the residual blood flow after BCCAO, PcomA development with carbon-black injection and cortical perfusion with laser-Doppler flowmetry were compared in three strains of mice. Consistent with previous reports (Fujii et al., 1997; Kitagawa et al., 1998a; Yang et al., 1997), results suggest that there was a variability of PcomA patency detected among strains with a trend toward smaller vessel size in C57BL6 mice. Laser-Doppler flowmetry after BCCAO was useful to indicate extent of blood flow reduction after BCCAO.
Variability of PcomA development is a problem when genetically altered mice are to be subjected to TGI produced by BCCAO. In this study, HSP70 Tg mice showed the highest PcomA development score and cortical perfusion value. Variability of PcomA development in genetically altered mice depends on the strains used as genetic background. To date, C57BL/6 and SV129 strains have been most commonly used to develop transgenic animals (Fujii et al., 1997). Therefore, it is important to consider the results of PcomA development demonstrated in this study when genetically altered mice are used in TGI experiments. Laser-Doppler flowmetry may be useful to predict histological outcome (Kitagawa et al., 1998b).
Zn2+ neurotoxicity
Among the possible mechanisms of neuronal cell death after transient global ischemia, Zn2+ neurotoxicity has been emphasized (Choi and Koh, 1998; Weiss et al., 2000) recently. In brain, most zinc binds to proteins. Chelatable Zn2+ is located in presynaptic vesicles (Choi and Koh, 1998) and is stained with the zinc-specific fluorescence dye TSQ (Frederickson et al., 1987). In rats, TSQ staining was present in cortex (layer I-III and V), hippocampus (mossy fiber, CA1–4 stratum radiatum and oriens, and dentate gyrus), subiculum, amygdala, thalamus, and striatum (Frederickson et al., 1987). Distribution of Zn2+ staining observed in C57BL/6 control mice in this study was consistent with previous reports in rats.
The mechanism of neurotoxic effects of Zn2+ has been investigated. In cultured cortical neurons, extracellular exposure to Zn2+ has been shown to be neurotoxic (Choi et al., 1988). Exposure to Zn2+ in culture produces free radical generation (Kim et al., 1999; Sensi et al., 1999) and activates apoptotic pathways. Neurons degenerating after Zn2+ exposure were stained by terminal deoxynucleotidyl transferase-mediated nick-end labeling (Kim et al., 1999) and reduced by caspase inhibitors (Lobner et al., 2000; Park et al., 2000).
Zn2+ translocation has been observed in vivo after transient global ischemia, brain trauma, and kainate-induced seizure (Frederickson et al., 1989; Koh et al., 1996; Suh et al., 2000; Tonder et al., 1990). Koh et al. (1996) reported that Zn2+ influx preceded ischemic degeneration after transient global ischemia. With TSQ and subsequent acid fuchsin staining on the same sections 24 hours after TGI, TSQ-positive cells had already appeared in CA1 pyramidal cells, but neuronal degeneration was not observed. Correspondence of TSQ-positive cells and degenerating neurons, however, was observed 72 hours after ischemia. Furthermore, administration of a Zn2+-specific chelator reduced neuronal cell death after TGI (Koh et al., 1996). Kainate-induced seizures in ZnT3 (zinc transporter in synaptic vesicle) knockout mice, however, were recently shown to produce neuronal zinc accumulation, suggesting that vesicles may not be the source of Zn2+ (Lee et al., 2000). The precise source of zinc needs to be further elucidated.
Protective effects of hypothermia
Various mechanisms of neuroprotective effects associated with mild hypothermia have been reported including reduction of metabolic rate (Chopp M 1989), ATP depletion (Welsh et al., 1990), protein synthesis inhibition (Shigeno et al., 1990), neurotransmitter release (Baker et al., 1995; Busto et al., 1989b; Globus et al., 1988; Nakashima and Todd, 1996), and free radical production (Kil et al., 1996). Suppression of glutamate release is considered to play an important role (Baker et al., 1995; Busto et al., 1989; Globus et al., 1988; Nakashima and Todd, 1996). A variety of reactions, however, occur during cerebral ischemia (Sharp et al., 2000), and many additional metabolic events may be influenced by temperature alteration.
Zn2+ is stored with glutamate in synaptic vesicles and is released after ischemia together with glutamate into synaptic clefts. Excessive Zn2+ influx is associated with neuronal death. Results of this study suggest that mild hypothermia may reduce presynaptic Zn2+ release along with a reduction of glutamate release, resulting in protection of vulnerable hippocampal neurons against combined Zn2+- and glutamate-induced neuronal cell death after TGI. However, the mechanisms of both neurotoxicity of Zn2+ and neuroprotection of mild hypothermia remain uncertain. In the present study, the mouse global ischemia model with hypothermia was validated. Although hypothermia effects are nonspecific with respect to mechanism, future studies with genetically altered mice may elucidate these issues.