
Abstract
Transgenic technology provides a powerful means of studying gene regulation and specific gene function with complex mammalian systems. In this study, the authors exploited the specific and discrete neuronal expression pattern mediated by promoter 1 of the Lmo-1 gene to study the neuroprotective effects of the inducible form of heat shock protein 70kD (hsp70i) in primary hippocampal cultures in a mouse model of global cerebral ischemia. Targeting expression of hsp70i to hippocampal neurons protected these cells significantly from toxic levels of glutamate and oxidative stress (for example, exposure to 10 μmol/L free iron produced a 26% increase in lactate dehydrogenase release from neurons cultured from wild-type mice, but a 7% increase in neurons cultured from hsp70i transgenic mice). Bilateral carotid occlusion (25 minutes) produced significantly less neuronal damage in the caudate nucleus and posterior thalamus in hsp70i transgenic mice than in wild-type littermates (for example, 21% ± 9.3% and 12.5% ± 9.0% neuronal damage in lateral caudate nucleus of wild-type and hsp70i transgenic mice, respectively, P < 0.05). The current study highlights the utility of targeted expression of transgenes of interest in cerebral ischemia and demonstrates that expression of hsp70i alone is sufficient to mediate the protection of primary neurons from denaturing stress and that expression of human hsp70i in vivo plays crucial role in determining the fate of neurons after ischemic challenge.
Keywords
Cells respond to a sublethal heat shock and several other forms of stress by suppressing normal cellular transcription and translation and activating the transcription of heat shock or stress proteins that protect them from damage. The inducible form of heat shock protein 70kD (hsp70i) is particularly important in protecting prokaryotic and nonneuronal eukaryotic cells from heat stress (Parcell and Lindquist, 1993). In vivo studies have shown that heat shock proteins are induced rapidly at the site of tissue injury in mammalian brain after ischemia, epilepsy, trauma, hyperthermia, and exposure to excitotoxins (Uney, et al., 1988, 1993a; Nowak, 1990; Brown, 1994), and these proteins (particularly hsp70i) may play a role in enhancing neuronal survival. In the hippocampus, the expression of hsp70i immunoreactivity identified the neurons destined to survive a transient ischemic insult, with sequential appearance of staining in dentate granule cells and CA3 pyramidal neurons; in contrast, cells destined to die within the vulnerable CA1 neurons showed minimal accumulation of hsp70i (Vass et al., 1989). Rordorf et al. (1991) and Lowenstein et al. (1991) have shown that the induction of heat shock proteins by exposure of cortical and hippocampal cultures to a sublethal elevation in temperature protect these cells against glutamate toxicity. Furthermore, calcium phosphate and adenoviral-mediated transfection of primary neurons with hsp70i significantly increased the survival of neurons and glia exposed to a severe heat stress (Uney et al., 1993b; Beaucamp et al., 1998). These data support the hypothesis that overexpression of hsp70i plays an important role in protecting neurons and glia from the denaturing effects of severe thermal stress and that inducing the expression of specific heat shock proteins may lead to the development of novel treatment strategies for central nervous system (CNS) diseases.
The mechanism of action of hsp70i is not well defined; however, it shares 80% protein homology with hsp70c (the constitutively expressed form of hsp70i), and its physiologic effects are therefore likely to be mediated through similar pathways. hsp70c putatively acts to prevent inappropriate protein interactions (Pelham, 1986), is involved in the transport of precursor proteins across membranes (acting as an unfoldase or chaperone protein), and is involved in protein catabolism as the ATPase responsible for clathrin uncoating (Chappell et al., 1986). These actions suggest that in the central nervous system hsp70i may protect cells against stress by allowing them to stabilize partially denatured proteins, which otherwise may aggregate and/or bind nonspecifically to cellular proteins and interfere with their normal cellular function, and quickly replace and remove proteins irreversibly damaged after stress (Beaucamp et al., 1998).
Transgenic technology provides a powerful means of studying gene regulation and specific gene function with complex mammalian systems. However, it has been difficult to identify promoters that target specific populations/areas of neuronal cells, and many studies have used pan neuronal promoters or promoters that target both glial and neuronal cell populations. Lmo-1, Lmo-2, and Lmo-3 comprises a family of genes encoding LIM-only proteins. Lmo-1 and Lmo-2 (originally rbtn-1 and rbtn-2) were identified adjacent to the breakpoint of a chromosomal translocation, associated with a human T cell tumor (Boehm et al., 1988). Expression of Lmo-1 normally appears to be restricted to the CNS (Greenberg et al., 1990). Transgenic mice expressing the bacterial lac Z gene under the transcriptional control of Lmo-1 promoter 1 has revealed β-gal expression in various regions of the developing CNS (Greenberg et al., 1990). In the embryonic and neonatal brains of Lmo-1–β-gal transgenic mice, β-gal expression is prominent in the hippocampus, neocortex, olfactory bulbs, and septum (Greenberg et al., 1990; Hinks et al., 1997). This pattern of expression persists into adult life, in which Lmo-1 expression is associated with specific subpopulations of cell types in various CNS regions including the hippocampus, caudate putamen, thalamus, amygdala, medial habenula, and cerebral cortex (Greenberg et al., 1990; Hinks et al., 1997). Interestingly, the activity of the Lmo-1 promoter 1 has been shown to be increased after seizure activity and it has been suggested that this may reflect an involvement in cell phenotype regulation (Hinks et al., 1997). This observation suggests that Lmo-1 promoter 1 driven transgene expression will be increased by an excitatory stress, such as that caused by glutamate release during ischemic stress.
In this study, the authors exploited the specific and discrete expression mediated by Lmo-1 promoter 1 to study the neuroprotective effects of hsp70i using transgenic technology. The use of this approach has a number of advantages: (1) transgene expression is turned on at embryonic day 12 and continues to be expressed throughout the life of the animal; (2) expression is driven to discrete populations of neurons and glia (that is, transgene expression occurs in hippocampal neurons, it is not expressed in hippocampal glial cells), and it is therefore possible to investigate the neuroprotective effects of the transgene after expression in neurons per se; (3) transgene expression is driven to the majority of hippocampal neurons and this allows the neuroprotective effects of hsp70i to be assessed in a mouse model of global ischemia in which hippocampal CA1 and CA2 are vulnerable.
MATERIALS AND METHODS
Generation of Lmo promoter 1, HSP70i transgenic mice
Mice were generated as described in Greenberg et al. (1990). Briefly, a 2.3-kb coding fragment of the hsp70i gene (a generous gift from Dr. R. Morimoto) was cloned into the Not 1 site of an Xho I–Nru I fragment of the Lmo-1 gene (containing promoter 1 and approximately 8 kb genomic DNA). After Xho-I restriction digest, an 11-kb fragment was microinjected into fertilized MF1 mouse eggs. hsp70i transgenic mice were identified through Southern blot analysis of tail biopsy material. The restriction enzymes HindIII and DraI were used to digest the tail DNA, because these enzymes cut two sites within the intact transgene. Southern blots then were hybridized with 32−P labeled hsp70i probe and to a radiolabeled sequence specific to the transgene alone. Five independent lines were identified and established for the initial study. Line 205.4 were mated to homozygosity and used in these studies (Fig. 1C). Mice expressing B-galactosidase and luciferase under the control of the Lmo-1 promoter 1 also were made.
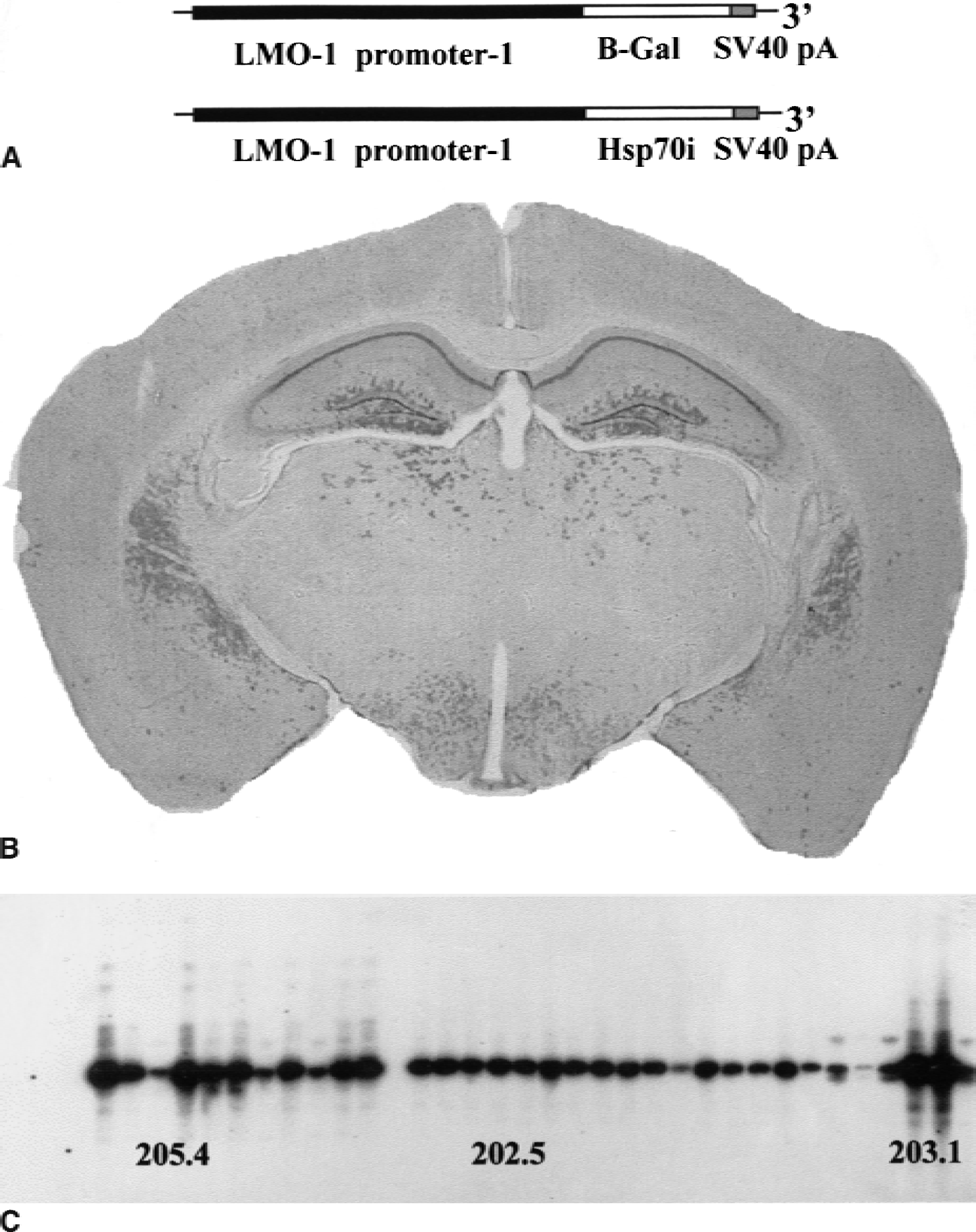
Construction of the inducible form of heat shock protein 70kD (hsp70i) transgenic mice.
RNA extraction
Using sterile conditions at 4°C, the hippocampal formation was dissected bilaterally under an operating microscope and immediately homogenized in 5 mol/L guanidinium isothiocyanate, the cell lysate then was layered on top of a 3 mL 5.7 mol/L CsCl cushion, and the RNA isolated by centrifugation at 100,000 g at 18°C for 18 hours in a Beckman SW-55 rotor (Palo Alto, CA, U.S.A.). After centrifugation, the supernatant was discarded and each RNA pellet was resuspended in 200 μL diethylpyrocarbonate-treated (0.02% v/v) water. RNA then was ethanol precipitated and quantitated by spectrophotometry and stored at −80°C. To confirm that the animals were transgenic for human hsp70i mRNA, reverse transcriptase–polymerase chain reaction (RT-PCR) assays for the human and mouse forms of hsp70i were developed. To achieve this, the mouse and human hsp70i cDNA sequence was analyzed and regions of maximal heterogeneity were identified. Then primers to these sequences were synthesized. Primers to the human hsp70i sequence were 5′-GCGGAGAAGTACAAAGCG-5 and 3′-AGTCCCAGGGTTCCCTCCC-5. Mouse primers were 5′- GCCGAGCGCTACAAGGCC-3′ and 5′-CGGGTCCGCGGCGGCTTT-3. To ensure that the PCR-amplified sequences encoded the hsp70i sequence, the PCR products were transferred by Southern blotting and hybridized against a 32P-labeled probe that recognized a sequence (CATGAAGAGCGCCGTGGAGGA) common to mouse hsp70i and human hsp70i.
Preparation of hippocampal primary cultures
Hippocampi were dissected from 8 to 10 embryonic mouse brains (E15–17) and digested in Hank's balanced salt solution (HBSS) without calcium and magnesium with 1 mg/mL trypsin for 40 minutes at 37°C. Cells were washed 3× in HBSS and tritirated in 500 μL Dulbecco's modified eagles medium (DMEM) supplemented with 5% B-27 supplement (Gibco BRL, Paisley, Scotland), 2% inactivated fetal calf serum (Sigma, St. Louis, MO, U.S.A.), glutamine (100 mmol/L), penicillin (100 U/mL), and streptomycin 100 μg/mL (this medium is DMEM). After trituration, 2 mL DMEM was added and 50 μL of the suspension aliquoted onto 10-mm wells that had previously been coated with polylysine (0.1 mg/mL) and 500 μL DMEM then was added to each well; the final cell concentration was 1 × 106 cells/well. The plates were incubated in 5% CO 2 at 37°C. Cytosine/ Arabinoside (2 μg/mL medium) was added to each well 48 hours after plating.
Cells were cultured for 7 days and then exposed to glutamate (0 to 200 μmol/L) for 6 hours or iron (0 to 100 μmol/L) for 2 hours as described previously by Uney et al. (1993a). Toxicity caused by the excitotoxin, glutamate, and due to free radical production (Uney et al., 1993a, 1993b) by the Fenton reaction (catalyzed by iron) was then estimated by light photomicroscopy, 3-(4,5,-Dimethylthiazoe-2-yl)-2,5diphenyltetrazolium bromide (MTT), and lactate dehydrogenase (LDH) assays.
MTT assay
This assay estimates the metabolic activity of cells by measuring the mitochondrial-dependent conversion of the tetrazolium salt, MTT, to a colored formazan product. At each time point, 20 μL MTT (5 mg/mL in phosphate-buffered saline) was added to each well of primary hippocampal neurons and incubated for 2 hours at 37°C. The medium then was carefully aspirated and 100 μL acidified isopropanol was added to solubilize the colored formazan product deposited in the viable cells. Absorbance was read at 550 nm on a scanning multiwell spectrophotometer after agitating the plates for 5 minutes on a shaker.
Lactate dehydrogenase assay
The LDH assay (Sigma) used was adapted for use in a 96-well plate format. This method is based on a series of linked enzyme reactions, the final reaction being the reduction of a tetrazolium salt to a highly colored insoluble formazan product. At each time point, 10 μL of medium was removed from each well and placed in the base of another 96-well plate. Two hundred microliters of the reconstituted LDH “Cellu-stain” buffer was added to each sample, and the plate was incubated in the dark for 30 minutes at 37°C. Absorbance again was read at 550 nm. The data generated were analyzed using one-way analyses of variance and Student's t-tests.
Global cerebral ischemia
Global cerebral ischemia was induced in the mouse by bilateral carotid artery occlusion (BCCAO). Surgical anesthesia was induced in a Perspex chamber with halothane (3%) added to a mixture of nitrous oxide and oxygen (70:30) for 2.5 minutes. Anesthesia was maintained throughout the procedure with halothane (1.2% to 1.7%) delivered through a facemask. Core body temperature was recorded using a rectal temperature probe and maintained at 37°C using a heating lamp throughout the procedure. Through a small skin incision in the neck, the common carotid arteries were freed from connecting tissues by blunt dissection. Lengths of surgical silk (4–0) were placed around both common carotid arteries. Carotid arteries were occluded for 25 minutes applying tension to these silks and placing an aneurysm clip (50 to 80 g closing pressure) on each vessel. Complete vessel occlusion was observed and the incision site irrigated with sterile saline throughout the course of the occlusion. Sham-operated mice had surgical silks (4–0) placed around both common carotid arteries and the incision site was irrigated with sterile saline. Clips were removed from occluded animals after 25 minutes. Vessel patency was checked and the incision site was sutured using surgical silk (6–0). Each mouse was injected subcutaneously with sterile saline (0.5 mL), and ventilated with 100% oxygen until recovery from anesthesia. The mouse was placed in a thermally controlled incubator (29°C) for 2 hours before being returned to its cage. A 72-hour (3-day) survival period was chosen to investigate ischemic neuronal damage based on observations in a pilot study that indicated that ischemic neuronal damage at 3 and 7 days post-BCCAO were similar.
Assessing ischemic neuronal damage
Hematoxylin and eosin staining was used to identify ischemic neurons. Morphologically abnormal neurons exhibiting features of ischemic cell change, including shrunken cell bodies, triangulated nuclei, and intensely eosinophilic cytoplasm, were determined in eight brain regions. Ischemic neurons and nonischemic neurons in a defined region were counted using a 100-mm2 grid (×400 magnification), and the percentage of ischemic neurons was determined. Ischemic neuronal damage in wild-type and hsp70i transgenic mice were compared after 25-minute BCCAO and 25-minute sham-BCCAO using a one-way analysis of variance followed by an unpaired Student's t-test with a Bonferroni correction factor of 4 (as 4 comparisons were made;Table 3).
Ischemic neuronal damage in transgenic mice overexpressing hsp70i

Ischemic neuronal damage in transgenic mice overexpressing hsp70i and their wild-type littermates after 25-minute bilateral carotid artery occlusion (BCCAO) were compared. Data are expressed as % of necrotic neurons in regions ± SD. Four groups were statistically analyzed with one-way analysis of variance followed by unpaired Student's t-test with Bonferroni correction factor of four. Significant differences in ischemic neuronal damage were observed in three of the comparisons made: *hsp70i transgenic sham-BCCAO versus hsp70i transgenic BCCAO; **wild-type littermate sham-BCCAO versus wild-type littermate BCCAO; and ‡hsp70i transgenic BCCAO versus wild-type littermate BCCAO.
P < 0.001.
P < 0.05.
P < 0.01.
P < 0.01.
P < 0.001.
P < 0.001.
P < 0.05.
Physiologic variables in transgenic mice overexpressing hsp70i and their wild-type littermates
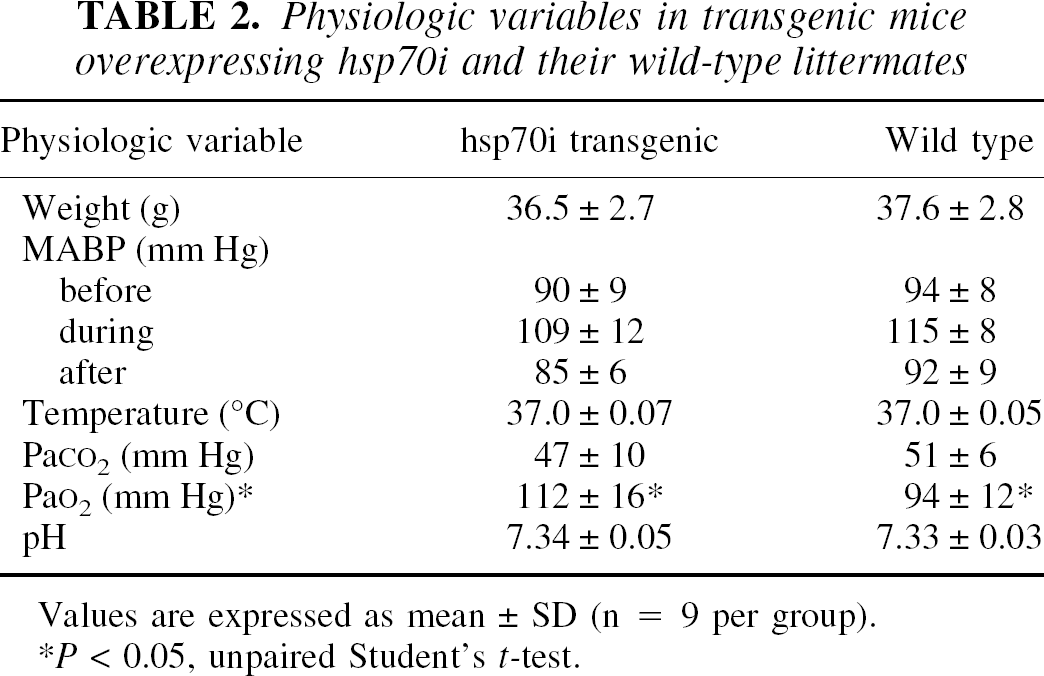
Values are expressed as mean ± SD (n = 9 per group)
P < 0.05, unpaired Student's t-test.
hsp70i immunoreactive neurons in transgenic mice overexpressing hsp70i
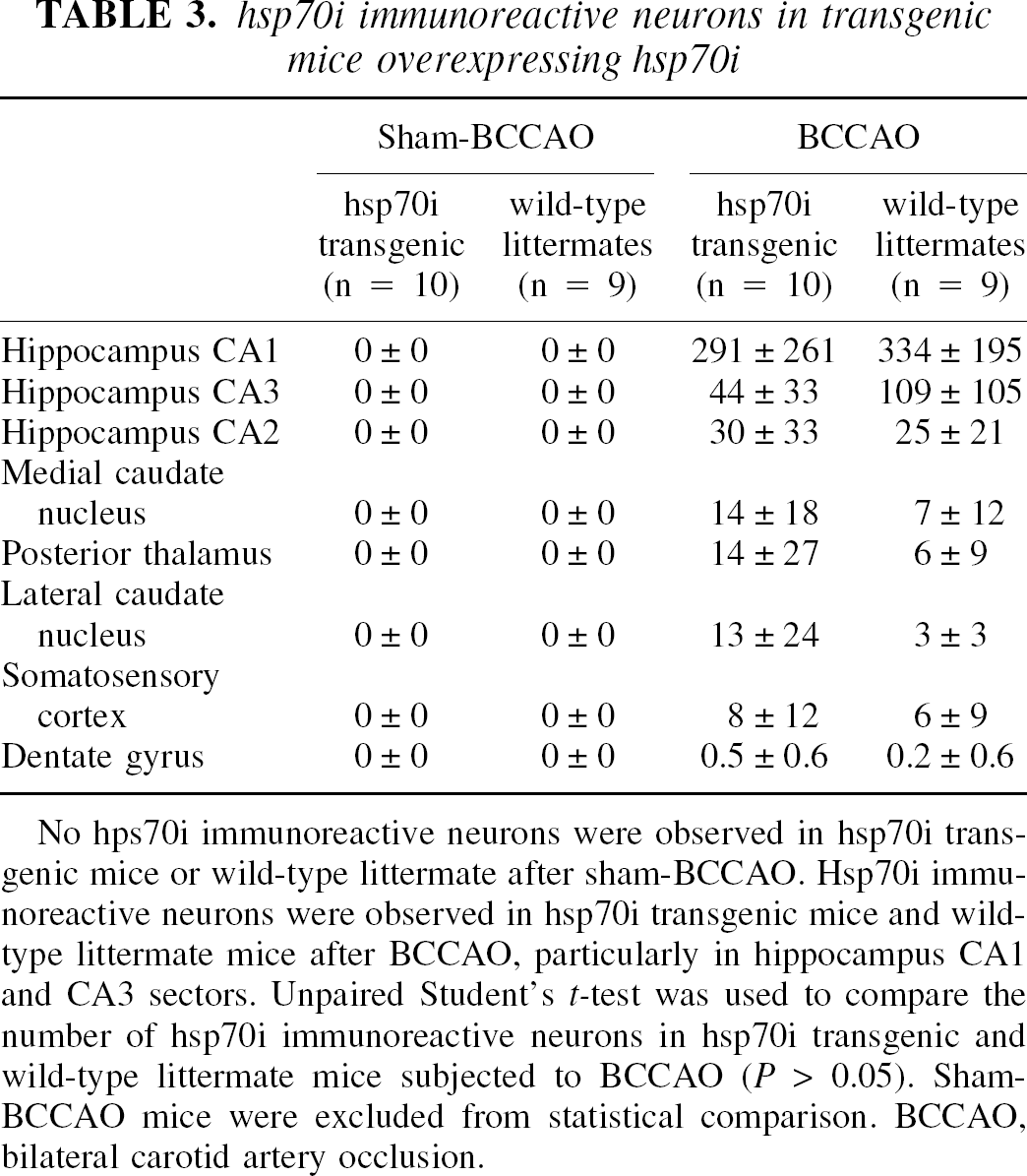
No hps70i immunoreactive neurons were observed in hsp70i transgenic mice or wild-type littermate after sham-BCCAO. Hsp70i immunoreactive neurons were observed in hsp70i transgenic mice and wild-type littermate mice after BCCAO, particularly in hippocampus CA1 and CA3 sectors. Unpaired Student's t-test was used to compare the number of hsp70i immunoreactive neurons in hsp70i transgenic and wild-type littermate mice subjected to BCCAO (P> 0.05). Sham-BCCAO mice were excluded from statistical comparison. BCCAO, bilateral carotid artery occlusion.
Immunocytochemistry
Immunohistochemistry was performed in adjacent sections used to define ischemic neuronal damage. Sections were immunostained with anti-HSP72/73 (1:1000; Calbiochem-Nova, San Diego, CA, U.S.A.) using an avidin biotin method with diaminobenzidine as the chromogen. Antigen retrieval was required for optimal results and the sections were microwaved in citric acid buffer (0.01 mol/L, pH 6) for 2 × 5 minutes. Sections were lightly counterstained with hematoxylin. All immunoreactive neurons were counted in each brain region.
RESULTS
Lmo-1 promoter 1 human hsp70i transgenics
The DNA constructs used to make the Lmo-1 transgenics are shown in Fig. 1A. Figure 1B shows expression of beta-galactosidase under the control of the Lmo-1 promoter; of particular importance to this study is the neuronal specific expression that is seen in the CA1, CA2, and dentate gyrus neurons of the hippocampus and in the caudate nucleus (Greenberg et al., 1990; Hinks et al., 1997). After microinjection of the Lmo-1 promoter 1 hsp70i construct into MF1 oocytes, Southern blotting identified 5 founder mice positive for the human hsp70i gene. Three of these founder mice were then bred to homozygosity and the 205.4 line was used in all experiments reported in this study (Fig. 1C). To confirm that the animals were expressing the human hsp70i transgene, RNA was extracted from control and transgenic hippocampi, and RT-PCR analysis was performed using human hsp70i and mouse hsp70i specific primers (see Materials and Methods for details). Figure 2A shows that mRNA encoding mouse hsp70 was present in control and transgenic mice; however, human hsp70i mRNA was present only in transgenic animals (Fig. 2C). To confirm that both amplified sequences did indeed code for hsp70i mRNA species, Southern blot analysis was performed. The RT-PCR amplified mouse and human DNA was transferred to nylon membranes and hybridized to a 32P-labeled oligonucleotide probe specific to a sequence common to both mouse and human hsp70i (Fig. 2B and 2D). Both the control and transgenic samples amplified using the mouse specific primers hybridized with the hsp70i probe. The DNA samples derived from transgenic animals and amplified with the human hsp70i specific probes also hybridized with the hsp70I-specific probe.
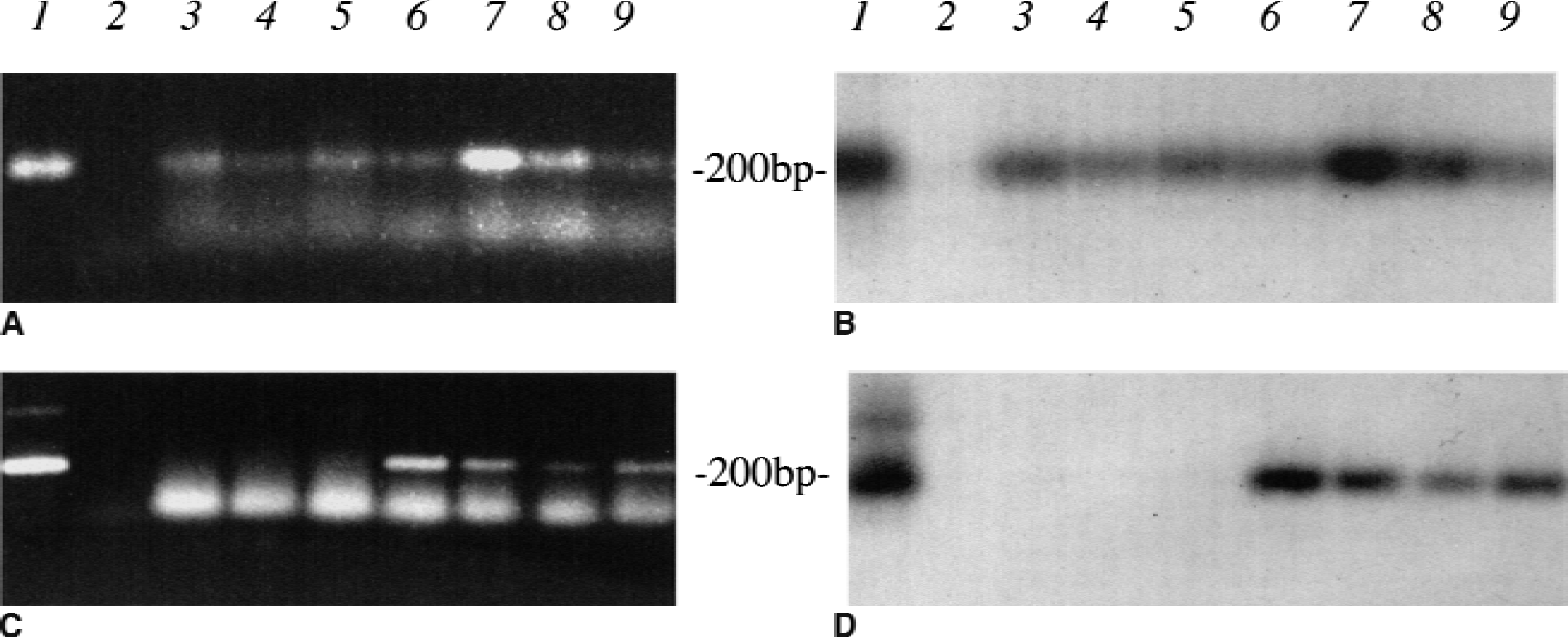
hsp70i mRNA expression in Lmo-1 promoter 1 hsp70i transgenic mice. RNA was extracted from the hippocampi of control (lanes 3 to 5) and hsp70i transgenic mice (lanes 6 to 9), and reverse transcriptase–polymerase chain reaction (RT-PCR) analysis was performed using primers specific for a 200-bp region of mouse hsp70i and human hsp70i (plasmid positive controls in lane 1). When the mouse specific primers were used, endogenous mouse hsp70i was found in the hsp70i transgenics and its controls
In vitro protection assays
To investigate the neuroprotective effect of overexpressing human hsp70i in neuronal cells, primary cultures of hippocampal neurons were prepared from hsp70i transgenics and MF1 controls (derived from the original founder littermates) and were exposed to glutamate (50 to 200 μmol/L) and iron (10 to 100 μmol/L) to catalyze free radical formation. Exposure to free iron affected a concentration-dependent reduction in MTT activity in neurons cultured from wild-type mice; in contrast, free iron produced minimal alterations in MTT activity in neuronal cultures prepared from hsp70i transgenic mice (Fig. 3). Exposure to free iron affected increases in LDH release in neurons cultured from wild-type mice. Increases in LDH release with iron (25 and 50 μmol/L) were significantly attenuated in neuronal cultures prepared from hsp70i transgenic mice (Fig. 3).
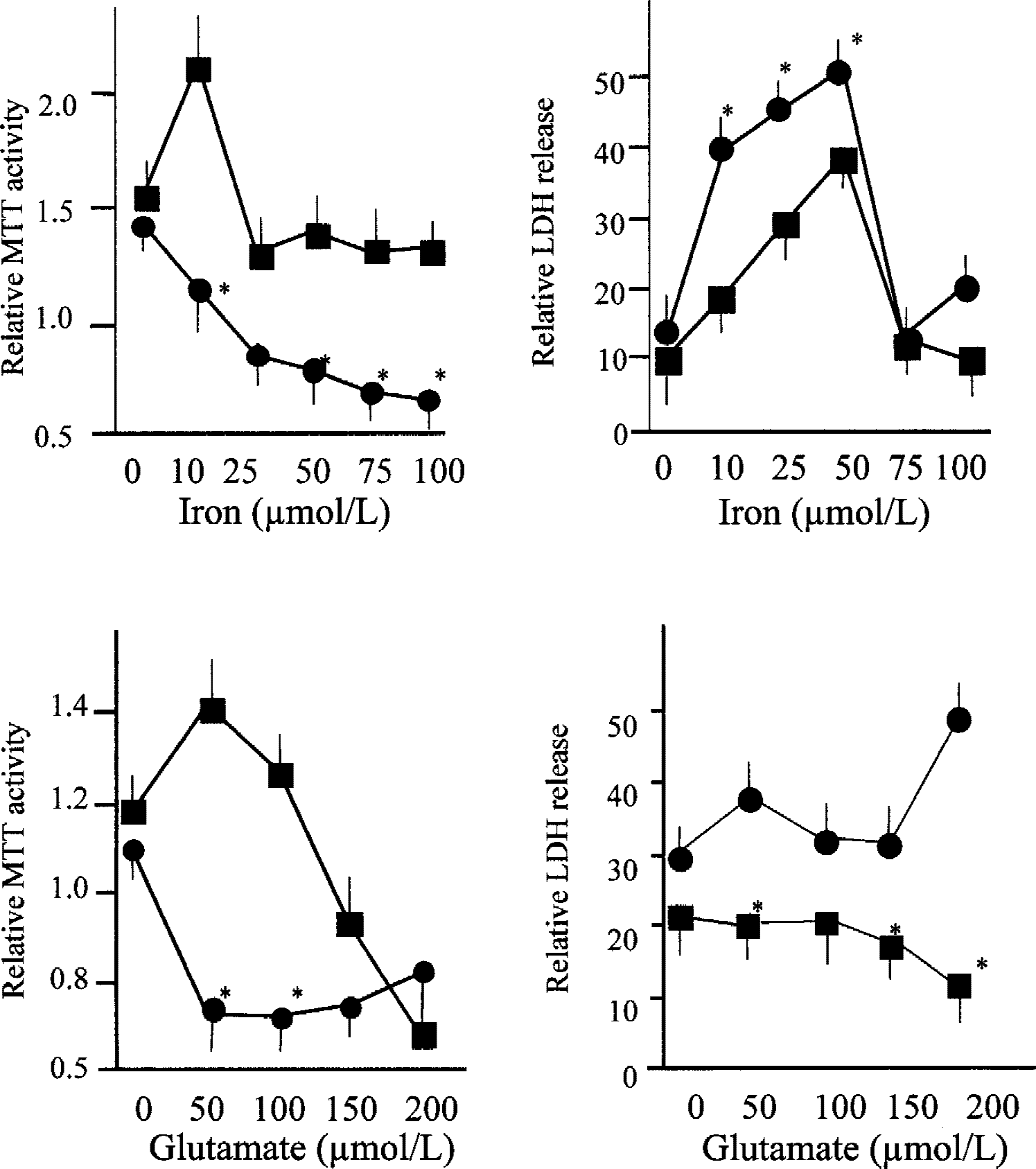
Effect of toxic concentration of glutamate and free iron on hippocampal cultures derived from hsp70i transgenic mice and nontransgenic control littermates. To estimate cell toxicity, MTT (n = 9) and lactate hydrogenase (LDH) assays (n = 9) were performed on hippocampal cultures derived from hsp70i transgenics (filled squares) and control littermates (filled circles) after exposure to iron and glutamate. *Significant difference was found when this value was compared with its equivalent control, P < 0.05 by Student's t-test.
Exposure to glutamate (at 50 μmol/L and greater) affected a marked reduction in MTT activity in neurons cultured from wild-type mice. In cultures prepared from hsp70i transgenic mice, glutamate (50 and 100 μmol/L) minimally altered MTT activity, though reductions were observed with the highest concentrations examined (150 and 200 μmol/L) (Fig. 3). Lower levels of LDH release were seen in neuronal cultures prepared from hsp70i transgenic mice compared with cultures from wild-type animals across the range of glutamate concentrations examined (Fig. 3). The lower level of neuronal damage in hippocampal cultures derived from the hsp70i transgenic mice when exposed to glutamate and free iron was confirmed by photomicroscopy (Fig. 4). When hippocampal cultures derived from the hsp70i transgenic cells and those derived from Lmo-1 promoter 1 luciferase transgenic animals also were exposed to glutamate, the hsp70I-derived cultures were again found to be protected (data not shown). These results indicate that the neuroprotective effects of hsp70i are not caused by any insertional effects.
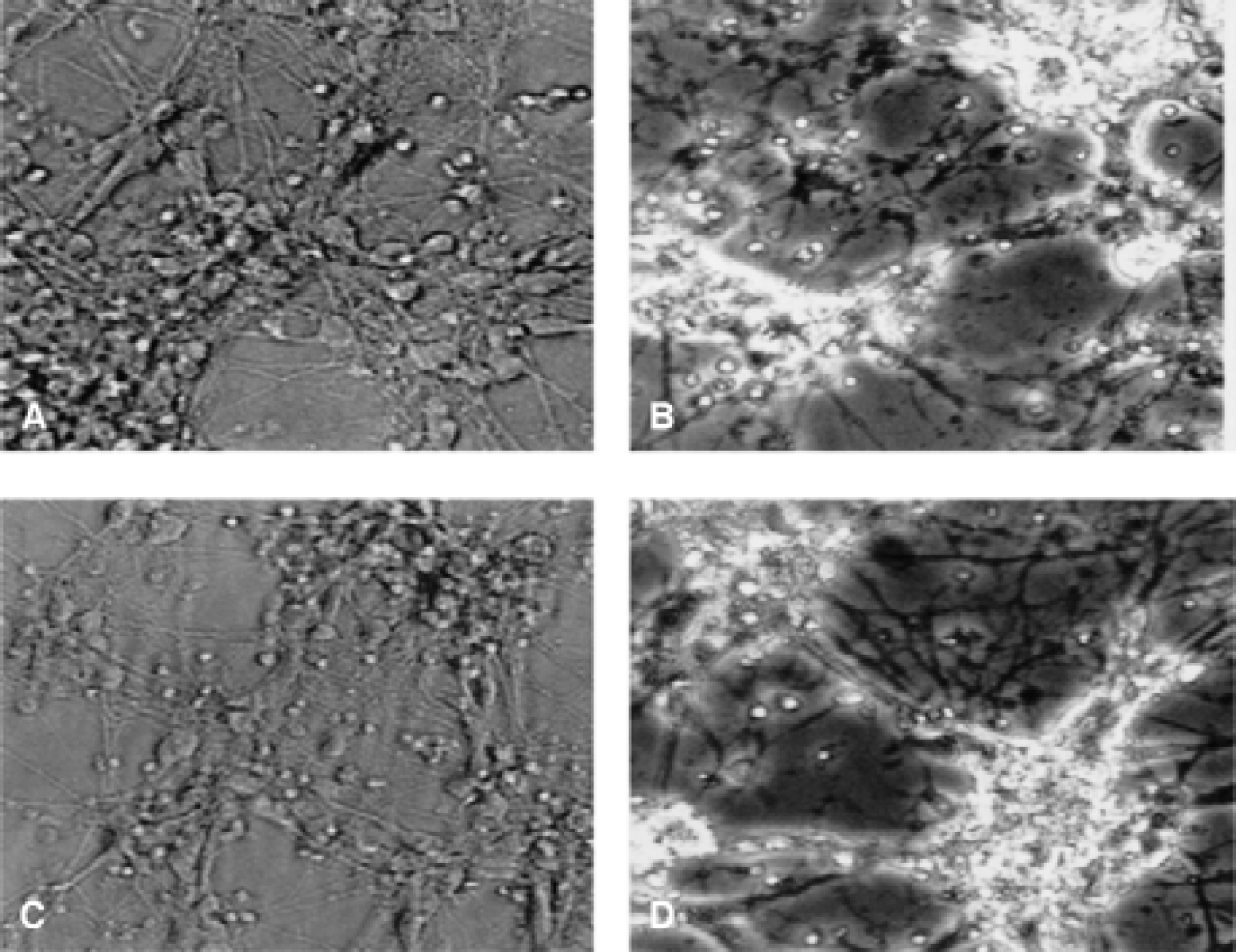
Photomicrographs of primary hippocampal derived from Lmo-1 promoter 1 hsp70i transgenic
Global ischemia in hsp70i transgenic mice
In wild-type littermate mice, 25-minute bilateral carotid occlusion significantly increased neuronal damage in the lateral and medial caudate nucleus, CA1, CA2, and CA3 sectors of the hippocampus, posterior thalamus, and somatosensory cortex compared with sham-occluded mice (Table 1). In hsp70i transgenic mice, 25-minute bilateral carotid occlusion produced significantly lower levels in the lateral and medial caudate nucleus and posterior thalamus from that seen after carotid occlusion of similar duration in wild-type littermates (Table 1). In a separate group of animals in which femoral arteries had been cannulated, no significant differences were observed between hsp70i transgenic animals and wildtype littermates for various cardiovascular and respiratory parameters during or after carotid occlusion (Table 2).
hsp70i immunoreactivity in hsp70i transgenic mice after BCCAO
No hsp70i immunoreactive positive neurons were detected in sham-operated hsp70i transgenic mice (Fig. 5A) or in wild-type control mice (Fig. 5C). After 25-minute BCCAO there was a marked increase in hsp70i immunoreactive neurons in wild-type and hsp70i transgenic mice, most notably in the CA1 and CA3 sectors of the hippocampus (Table 3, Fig. 5). These results show that expression of endogenous and transgenic hsp70i protein before BCCAO is less than the level detectable by this assay. No significant differences in the number of hsp70i immunoreactive neurons could be demonstrated in any region between transgenic and wild-type animals after BCCAO, although the staining appeared more intense in transgenic animals. This may reflect the considerable interanimal variability and the detection of endogenous murine and transgenic human hsp70i by the antibody. Also, in these experiments, hsp70i expression levels may be influenced by the ischemic stress, because hsp70i mRNA have been shown to be stabilized after a stress (Theodorakis and Morimoti, 1987). Furthermore, the activity of the Lmo-1 promoter 1 is itself induced by increased neuronal activity (Hinks et al., 1997). The authors have substantiated this finding using quantitative RT-PCR analysis (data not shown) and have demonstrated that transgenic hsp70i mRNA levels are increased threefold in the hippocampus after the injection of glutamate; however, endogenous hippocampal hsp70i mRNA levels were elevated to even greater levels after exposure to glutamate.
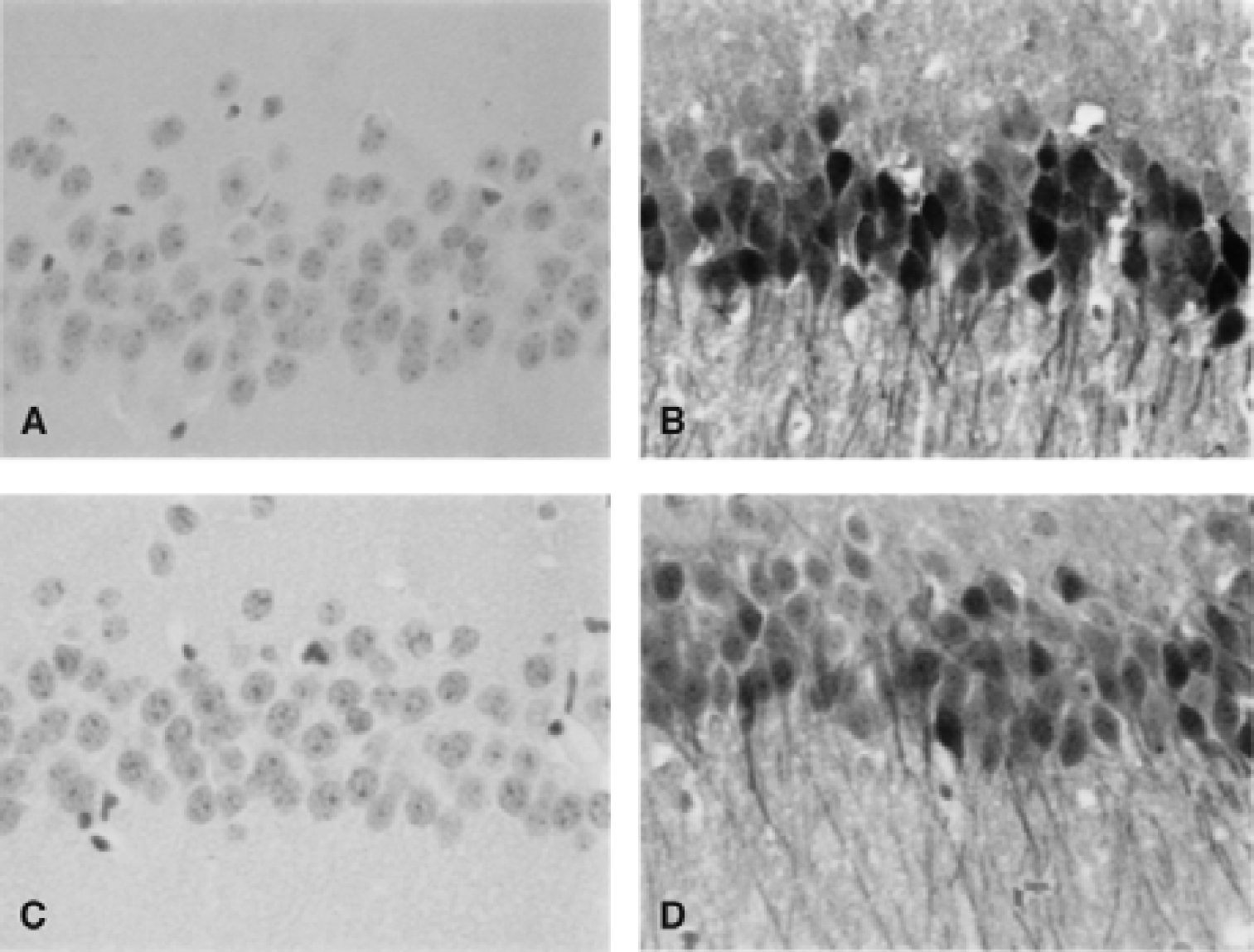
hsp70i immunoreactivity measured in the hippocampus of sham-operated hsp70i transgenic mice
Circle of Willis anatomy in MF1 mice
Vascular anomalies of the circle of Willis have been directly linked with the degree of ischemic neuronal damage after BCCAO in mice with the most frequent anomaly being posterior communicating artery hypoplasticity. The frequency of posterior communicating artery hypoplasticity was similar in the hsp70i transgenic mice and their littermate controls (Table 4).
Posterior communicating arteries in transgenic overexpressing hsp70i
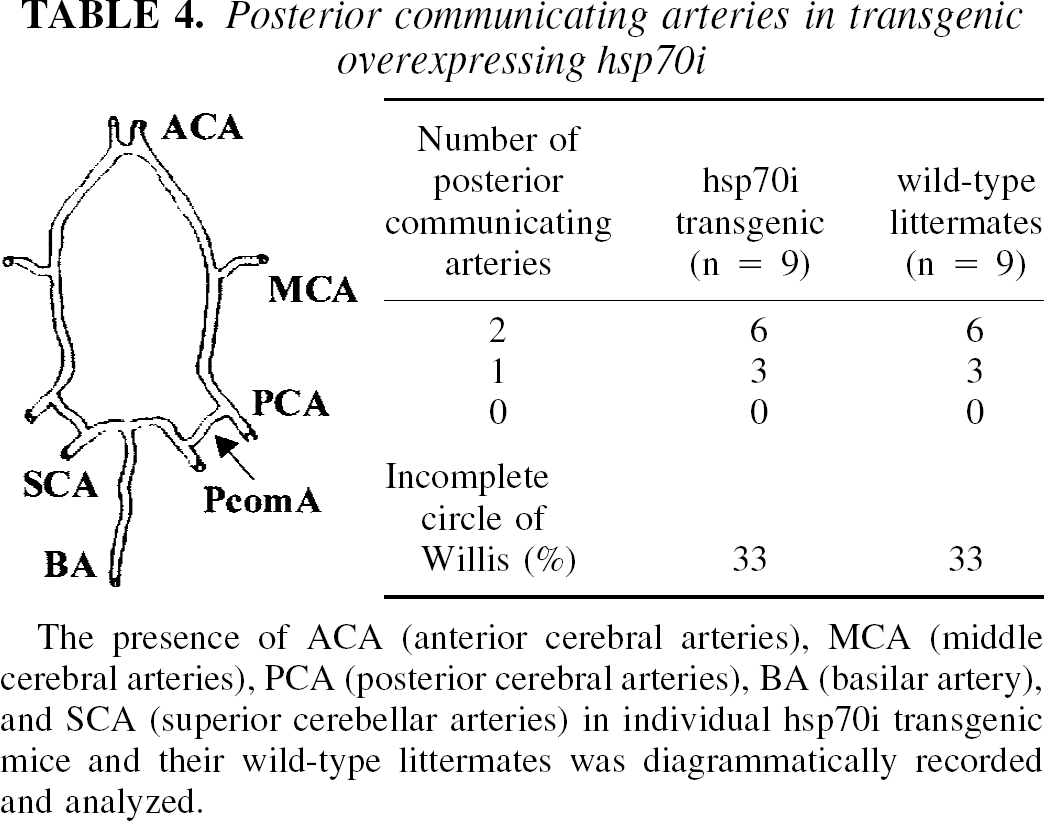
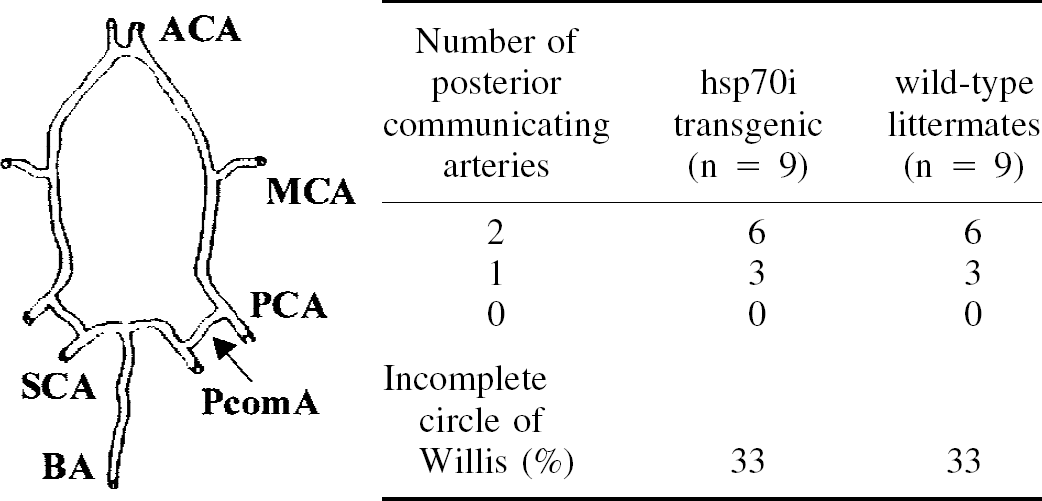
The presence of ACA (anterior cerebral arteries), MCA (middle cerebral arteries), PCA (posterior cerebral arteries), BA (basilar artery), and SCA (superior cerebellar arteries) in individual hsp70i transgenic mice and their wild-type littermates was diagrammatically recorded and analyzed.
DISCUSSION
Previous studies have shown that promoter 1 of the Lmo-1 gene drives expression to structurally distinct populations of neurons and glia within the CNS (Greenberg et al., 1990; Hinks et al., 1997), and the authors have used the Lmo-1 promoter 1 to drive expression in the hippocampus and caudate nucleus to investigate the neuroprotective effect of overexpressing hsp70i. The in vitro results obtained in this study showed that targeting expression of hsp70i to hippocampal neurons protected these cells from toxic levels of glutamate and oxidative insult. Rordorf et al. (1991) and Lowenstein et al. (1991) showed that exposure of primary neuronal cultures to a sublethal hyperthermia induced heat shock protein expression (including hsp70i expression) and protected these cells against a subsequent stress. The current findings show that overexpression of hsp70i alone is sufficient to protect neurons from neurotoxic insults, although this does not exclude the possibility that overexpressed human hsp70i acts in conjunction with critical co-chaperones (for example, hsp40s) that are present at high concentration in cultured cells (see below).
The protective effects of overexpression of hsp70i in vivo were defined in a model of global ischemia. In C57bl/6 mice, brief (10 minutes) bilateral carotid artery occlusion per se produces widespread ischemic neuronal damage, the distribution and amount of which correlates with the duration of occlusion. The susceptibility of the C57bl/6 mice to damage after carotid occlusion has been attributed to the high frequency (near 100%) of abnormalities within the circle of Willis in this strain. In the MF1 strain of mice, which were used to generate the hsp70i transgenic lines, bilateral carotid artery occlusion of longer duration (25 minutes) is required to produce moderate neuronal damage (20% of the population) in the caudate nucleus with lower levels of damage in the hippocampus and cortex (Kelly et al., 1999). Significantly reduced damage could be demonstrated in hsp70i transgenic mice in the caudate nucleus, the region in which the most marked and most consistent (coefficient of variations of 45%) neuronal damage was observed in the wild-type control group. The authors' inability to clearly demonstrate significant attenuation of neuronal damage in the hippocampus CA1 in hsp70i transgene animals (the region in which the highest level of transgene expression is expected) may reflect the low frequency of damage in the region and its high variability (a coefficient variation of 85%).
Alternatively, expression of the human hsp70i transgenic protein may be relatively low and/or the marked expression of murine hsp70i in the CA1 region in both the wild-type and transgenic animals may mask the effects of hsp70i transgene expression. However, when discussing Lmo-1–driven hsp70i transgene expression levels after an ischemic stress it is important to consider that Lmo-1 promoter 1 activity is increased after seizure activity (Hinks et al., 1997) and that hsp70i mRNA stability is increased after stress (Theodorakis and Morimoto, 1987). The authors have corroborated the former findings using quantitative RT-PCR to measure levels of hippocampal murine hsp70 mRNA and human hsp70 mRNA before and after the in vivo injection of an excitotoxic dose of glutamate (data not shown). These data showed that expression of hsp70i mRNA driven by promoter 1 of Lmo-1 was increased threefold after excitotoxicity; however, the induction of murine hsp70i mRNA was greater and reached higher levels than that of transgenic hsp70i after excitotoxicity. In the caudate nucleus and posterior thalamus, where only small increases in murine hsp70i are observed after ischemia, there was evidence that the number of neurons expressing hsp70i was 100% greater in transgenic mice (although these changes failed to achieve accepted levels of significance). Therefore, the impact of transgenic hsp70i on neuronal survival after ischemia may be greater in regions with low endogenous hsp70i expression. In contrast, in regions such as hippocampus CA1 with marked endogenous hsp70i expression (and low ischemic neuronal pathology), transgenic hsp70i had no significant effect. Overall, the in vitro and in vivo results of this study strongly suggest that the overexpression of human hsp70i played a crucial role in determining the fate of neurons after the authors' ischemic challenge. This conclusion is also supported by previous studies in which hsp70 has been constitutively overexpressed using transgenic (Plumier et al., 1997; Rajdev et al., 2000) and viral strategies (Yenari et al., 1998). Using the beta-actin promoter to constitutively overexpress human hsp70i in both neurons and glia, Plumier et al. (1997) showed that hippocampal neurons were not resistant to severe ischemic injury but were protected against delayed ischemic death. Using a similar strategy, Rajdev et al. (2000) constitutively overexpressed rat hsp70i (under the control of a beta-actin promoter and human cytomegalovirus enhancer) and showed that transgenic animals were markedly protected against ischemic infarction in a model of permanent focal cerebral ischemia.
The physiologic function of the hsp70 proteins has been less well defined than their constitutive homologues (hsc70, grp78, bip); however, because of their high degree of sequence homology with the hsc70s, they are thought to act as molecular chaperones. HSP70 proteins have been shown to be monomeric proteins consisting of three domains. An N-terminal domain is the most highly conserved and functions as an ATPase, whereas a C-terminal domain is less highly conserved and is of unknown function. The third domain lies between the N- and C-terminal domains and plays a pivotal role in peptide binding, transport of proteins through intracellular membranes, and regulation of the heat shock response (James et al., 1997; Parsell and Lindquist, 1993). Hsc70 proteins have been shown to transport precursor proteins across membranes, act as an unfoldase or chaperone protein, and be the ATPase responsible for clathrin uncoating (Chappell et al., 1986). A number of recent studies, conducted with recombinant hsp70i protein and with virally expressed hsp70I, have shown that hsp70i alone can mediate the refolding of denatured luciferase (Beaucamp et al., 1998). Together, these findings support the hypothesis that hsp70i interacts with cytosolic proteins and that in the CNS, hsp70i may protect cells against stress by allowing them to stabilize partially denatured proteins, which otherwise may aggregate and/or bind nonspecifically to cellular proteins and interfere with their normal cellular function, and to quickly replace and remove proteins irreversibly damaged after stress. Some hsc70 proteins may be directed to different protein substrates by interacting with proteins known collectively as DnaJs or hsp40s (Silver and Way, 1993). Several eukaryotic DnaJ homologues have now been isolated suggesting that DnaJ proteins may participate in at least some of the reactions catalyzed by eukaryotic hsp70 family members (Cyr et al., 1992; Hattori et al., 1993; Kobayashi et al., 2000). Cheetham et al. (1992) isolated a human alternatively spliced DnaJ homologue (hsj1a and hsj1b) and showed that the isoforms were stress inducible and located exclusively in the brain. Another mammalian DnaJ homologue isolated from HeLa cells, hsp40, has been shown to be highly stress inducible, to accumulate in the nucleus and nucleolus during stress, and to colocalize with hsc73 in heat-shocked HeLa cells. These results were interpreted as suggesting that hsp40 and hsc70/hsp70i may act together to prevent nascent proteins from becoming abnormally folded and may also prevent protein aggregates from accumulating (Hattori et al., 1993). The overexpression of HSDJ (a hsp40/DnaJ chaperone protein) prevented the accumulation of ataxin-1 in a transgenic mouse model of spinocerebellar ataxia type 1 (SCA1). Whereas Kobayashi et al. (2000) have shown that combinations of hsp70 and hsp40 were the most effective at reducing aggregate formation and providing cellular protection. These findings suggest that increasing the expression of DnaJ-like proteins in vivo may also facilitate the neuroprotective effects of hsp70i. Indeed, it may be that hsp70i acts to some degree with constitutively expressed DnaJ proteins in vivo to mediate its protective effects. It has also been suggested that hsp70i may prevent cellular damage caused by an inhibitory interaction with proapoptotic molecules. In the u937 cancer cell line, hsp70i has been shown to prevent apoptotic cell death by inhibiting the stress-activated protein kinase pathway (Gabai et al., 1998; Yaglom et al., 1999) and by interacting downstream of caspase 3. In a cell-free system, hsp70i prevents cytochrome c/dATP-mediated caspase activation by preventing the formation of a functional apoptosome complex (Beere et al., 2000; Saleh et al., 2000). Whether hsp70i could be protective in neurons by a similar action is not known, although it has been shown that overexpressing hsp70i in DRG cultures does not prevent them from undergoing apoptosis after NGF withdrawal (Maihos et al., 1994). Regardless of the cellular mechanisms that are ultimately involved in the action of hsp70i, the current study highlights the utility of targeting expression of transgenes of interest in cerebral ischemia. Using the approach, neuroprotective effects of hsp70i have been demonstrated in cell cultures in vitro and after global ischemia in vivo.