
Abstract
Estrogen can ameliorate brain damage in experimental models of focal cerebral ischemia. In vitro, estrogen increases levels of apolipoprotein E (apoE), which also has neuroprotective effects in brain injury. The authors tested the hypotheses that physiologically relevant levels of 17β-estradiol are neuroprotective in global cerebral ischemia and that neuroprotection is mediated via apoE. In the first study, subcutaneous implants of 17β-estradiol were tested in female C57Bl/6J mice (ovariectomized and nonovariectomized) and plasma levels measured by radioimmunoassay to validate that physiologically relevant levels could be achieved. In the second study, female C57Bl/6J and apoE-deficient mice were ovariectomized and implanted with 17β-estradiol or placebo pellet. Two weeks later, transient global ischemia was induced by bilateral carotid artery occlusion and the mice killed after 72 hours. Ischemic and normal neurons were counted in the caudate nucleus and CA1 pyramidal cell layer and the percentage of neuronal damage was compared between the treated groups. In C57Bl/6J mice, there was less neuronal damage in the 17β-estradiol-treated group compared with placebo group in the caudate nucleus (15 ± 20% versus 39 ± 27%, P = 0.02) and in the CA1 pyramidal cell layer (1.8 ± 2% versus 10 ± 14%, P = 0.08). In contrast, neuronal damage was not significantly different between the 17β-estradiol and placebo groups in apoE-deficient mice in the caudate nucleus (47 ± 35% versus 53 ± 29%, P = 0.7) or in the CA1 pyramidal cell layer (24 ± 19% versus 24 ± 19%, P = 1.0). The data indicate a neuroprotective role for estrogen in global ischemia, the mechanism of which is apoE-dependent.
Estrogen replacement therapy ameliorates the neuronal dysfunction associated with Alzheimer disease, brain injury, and stroke. The neuroprotective role of estrogen has been well documented in several in vitro and in vivo models of brain injury (for review see Hurn and Macrae, 2000). These studies have demonstrated that 17β-estradiol, the predominant and most biologically active estrogen, markedly attenuates the extent of brain damage. In particular, this effect has been demonstrated in models of focal cerebral ischemia where the extent of tissue damage is markedly ameliorated with estrogen (Simpkins et al., 1997; Rusa et al., 1999).
A number of mechanisms have been proposed for the neuroprotective effects of physiologic levels of estradiol on the brain. These include antioxidant (Behl et al., 1995), antiinflammatory (Iliev et al., 2001), and antiexcitotoxic (Weaver et al., 1997) mechanisms and promotion of neurite outgrowth and synaptogenesis (Woolley and McEwen, 1992; Woolley et al., 1997). Increasing evidence has indicated that the actions of estrogen parallel those of the lipid transport protein, apolipoprotein E (apoE). Apolipoprotein E is also known to be neuroprotective in experimental models of brain injury whereas apoE deficiency exacerbates the extent of brain damage in response to the initial insult (Laskowitz et al., 1997; Horsburgh et al., 1999, 2000a). The mechanisms by which apoE confers protection, similar to estrogen, are suggested to involve antioxidant (Lomnitski et al., 1997) and antiinflammatory (Laskowitz et al., 2001) effects and an effect of apoE on dendritic and neuronal remodeling (Nathan et al., 1994; Fagan et al., 1996; White et al., 2001). It has been well documented that in the periphery estradiol produces marked alterations in lipoprotein profiles. There is also increasing evidence of interactions between apoE and estrogen in the central nervous system (CNS). Administration of estrogen increases the expression of apoE mRNA in mouse brain (Srivastava et al., 1997; Levin-Allerhand et al., 2001). This increase in apoE mRNA in response to increased estrogen levels was demonstrated to occur in microglia and astrocytes (Stone et al., 1997). In addition, there is some evidence that endogenous apoE mediates the effects of estrogen in models of synaptic plasticity. Estrogen promotes sprouting after hippocampal denervation in wild-type mice, but this mechanism is ineffective in apoE-deficient mice both in vitro (Teter et al., 1999) and in vivo (Stone et al., 1998).
In view of the evidence that both estrogen and apoE are neuroprotective in models of ischemia and that estrogen stimulates apoE expression, we hypothesized that the neuroprotective effect of estrogen in cerebral ischemia is mediated via apoE. In this study we determined first, whether 17β-estradiol replacement in ovariectomized mice would give physiologic levels in plasma; second, whether physiologically relevant levels of estradiol are neuroprotective in a mouse model of global ischemia; and third, whether apoE mediates estradiol-induced neuroprotection.
MATERIALS AND METHODS
Mice
Adult female C57Bl/6J mice were obtained from Charles River, U.K. ApoE-deficient mice were derived from the Maeda line and had been backcrossed at least 10 times into C57Bl/6J mice. Homozygous apoE-deficient mice were bred at University of Glasgow for the purpose of these studies. The genotype of the mice was confirmed by polymerase chain reaction (Nicoll et al., 1997). All procedures were carried out under license from the Home Office with the approval of the University Ethical Review Panel and were subject to the Animals (Scientific Procedures) Act 1986.
Surgical preparation and experimental groups
Measurement of 17β-estradiol levels by radioimmunoassay
In the first study, we measured the plasma levels of 17β-estradiol in C57Bl/6J mice using a radioimmunoassay (Diagnostic Products Corporation, Los Angeles, CA, U.S.A.). Mice were initially anesthetized with 3% halothane in 70% N2O/30% O2, the halothane concentration was reduced to 1.5% in all animals, and anesthesia was continued via a face mask, the animals breathing spontaneously. Mice underwent a bilateral ovariectomy via a dorsal incision. Then, a pellet containing 17β-estradiol (0.025 mg or 0.25 mg, n = 8 per group) or the corresponding placebo pellet (n = 8 per group) was implanted subcutaneously at the nape of the neck. The animals then recovered for 2 weeks. An additional group of mice (n = 8) that did not undergo ovariectomy or receive a subcutaneous pellet served as a control group to determine 17β-estradiol levels in the intact mouse. The mice were decapitated, blood collected, spun, and the plasma retained for measurement of 17β-estradiol levels by radioimmunoassay.
In the second study, the mice were ovariectomized and a subcutaneous pellet containing either placebo or 17β-estradiol (0.025 mg/pellet) implanted; two weeks later the mice underwent global ischemia. Both common carotid arteries were isolated and then occluded using microaneurysm clips applied bilaterally for 17 minutes. In both procedures, mice were initially anesthetized with 3% halothane in 70% N2O/30% O2, and after induction the halothane concentration was reduced to 1.5% in all animals and anesthesia was continued via a face mask. The duration of ischemia was based on previous observations made in studies of neuronal damage after different durations of global ischemia in C57Bl/6J mice (Kelly et al., 2001) and in studies in which neuronal damage was measured in apoE-deficient mice compared with wild-type mice after global ischemia (Horsburgh et al., 1999, 2000a). After the period of ischemia, the clips were removed and blood flow through the arteries was confirmed before the incision was sutured. The body temperature was strictly controlled at 37°C during the procedure. The animal was then placed in an incubator (2 hours), after which it was returned to the animal house and allowed to recover for 72 hours. The operator was blind to the genotype and drug treatment of the mice. Animals were killed 72 hours later by perfusion fixation under deep halothane anesthesia. The brain was perfused via the ascending aorta with heparinized physiologic saline followed by 4% paraformaldehyde. The brains were removed and postfixed in 4% paraformaldehyde for 2 hours and then embedded in paraffin. Sections (6-μm) were cut and stained with hematoxylin and eosin. The numbers of morphologically normal neurons and neurons showing the features of ischemic cell change (Graham, 1992) were counted in the hematoxylin- and eosin-stained sections using a 100-mm2 grid at x40 magnification. Neurons were counted in 10 different fields in each region at x40 magnification using defined areas of 25 mm2 on the grid (equivalent to 0.0156 mm2) in the caudate nucleus and 10 mm (equivalent to 0.00625 mm2) in the CA1 pyramidal cell layer. The percentage of ischemic neurons in placebo-treated mice was compared with 17β-estradiol-treated mice using the Mann-Whitney U statistic. For information on neuronal density, the total number of neurons was calculated by adding together the number of ischemic and normal neurons counted in the defined area in the 10 different fields. The total number of neurons, therefore, is expressed in the caudate nucleus/0.156 mm2 and in the CA1 pyramidal cell layer/0.0625 mm2.
RESULTS
Plasma levels of 17β-estradiol
Ovariectomized female mice received subcutaneous pellets containing either 0.025 mg or 0.25 mg 17β-estradiol or a placebo pellet (n = 8 per group). The 17β-estradiol pellets release a constant concentration of 17β-estradiol over 21 days. The 17β-estradiol plasma levels (as determined by radioimmunoassay) were compared with those of female mice that did not receive an ovariectomy or implant (intact mice with normal physiologic 17β-estradiol levels). The plasma levels of estradiol recorded 2 weeks later are given in Table 1. The plasma levels of estradiol were determined to be lower in the ovariectomized mice with placebo compared with the intact (nonovariectomized) mice. The ovariectomized mice with 17β-estradiol implants, however, had markedly greater levels of estradiol compared with the placebo-treated mice. There was approximately a 15-fold difference in estradiol levels between the 0.025-mg and 0.25-mg 17β-estradiol pellets, with the 0.025-mg pellet giving rise to plasma estradiol levels closest to the physiologic range. Consequently, the 0.025-mg pellet was chosen as the most appropriate dose for the subsequent studies on ischemic injury.
Plasma concentrations in intact and in placebo-or 17β-estradiol-treated ovariectomized mice
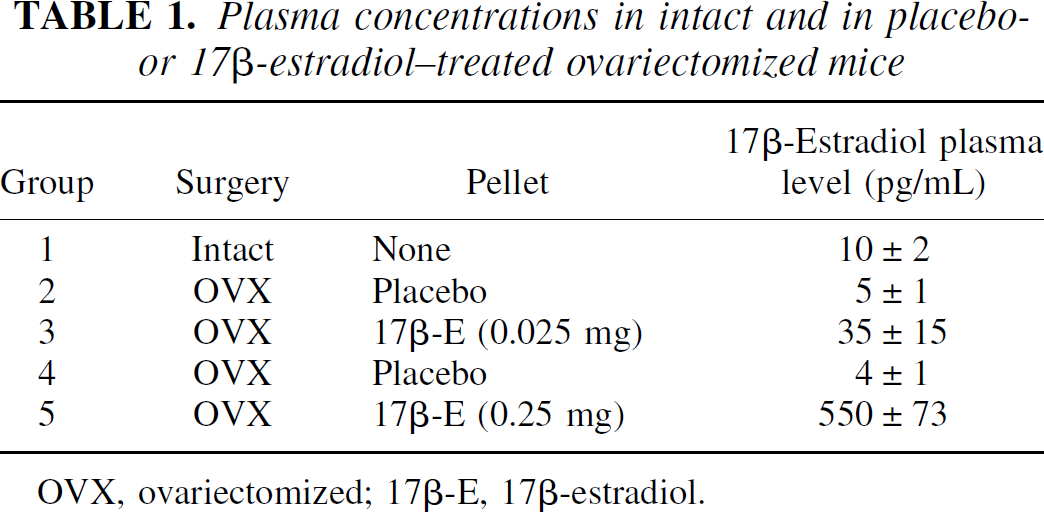
OVX, ovariectomized; 17β-E, 17β-estradiol.
Histopathologic outcome after global ischemia
Transient global cerebral ischemia of 17-minutes duration is associated with selective neuronal damage. The extent of neuronal damage was quantified in two brain regions that are associated with selective neuronal damage: the CA1 pyramidal cell layer in the hippocampus and the caudate nucleus. The extent of neuronal damage was compared between placebo- or 17β-estradiol-treated C57Bl/6J wild-type or apoE-deficient mice (Fig. 1). In wild-type mice, the estrogen-treated mice (n = 11) exhibited a significant reduction in neuronal damage compared with placebo-treated (n = 12) mice in the caudate nucleus (15 ± 20% versus 39 ± 27% ischemic neurons, P = 0.02; Fig. 2). The percentage of ischemic neuronal damage was also lower within the CA1 pyramidal cell layer of estrogen-treated compared with placebo-treated mice but failed to attain statistical significance (1.8 ± 2% versus 10 ± 14%, P = 0.08). In apoE-deficient mice, however, there was no significant difference between estrogen treatment (n = 9) and placebo treatment (n = 9) in the caudate nucleus (47 ± 35% versus 53 ± 29%, P = 0.7) and in the CA1 pyramidal cell layer (24 ± 19% versus 24 ± 19%, P = 1.0; Fig. 3). The neuronal density in the wild-type and apoE-deficient mice was similar in the caudate nucleus (278 ± 24 versus 296 ± 26, total number of neurons/0.156 mm2) and in the CA1 hippocampal region (307 ± 24 versus 313 ± 17, total number of neurons/0.0625 mm2).
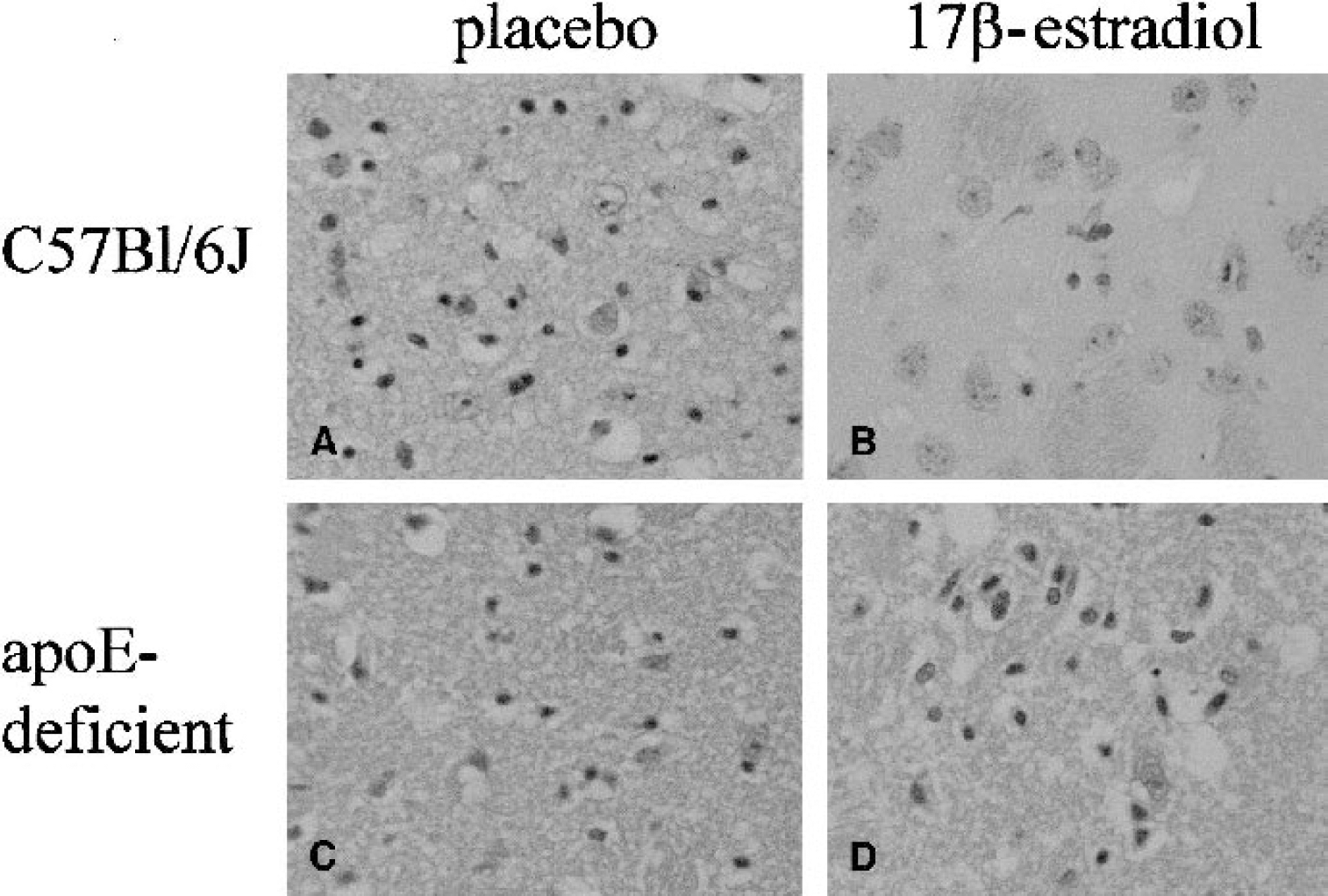
Illustrative examples of the extent of ischemic neuronal damage in hematoxylin- and eosin-stained sections of placebo- (
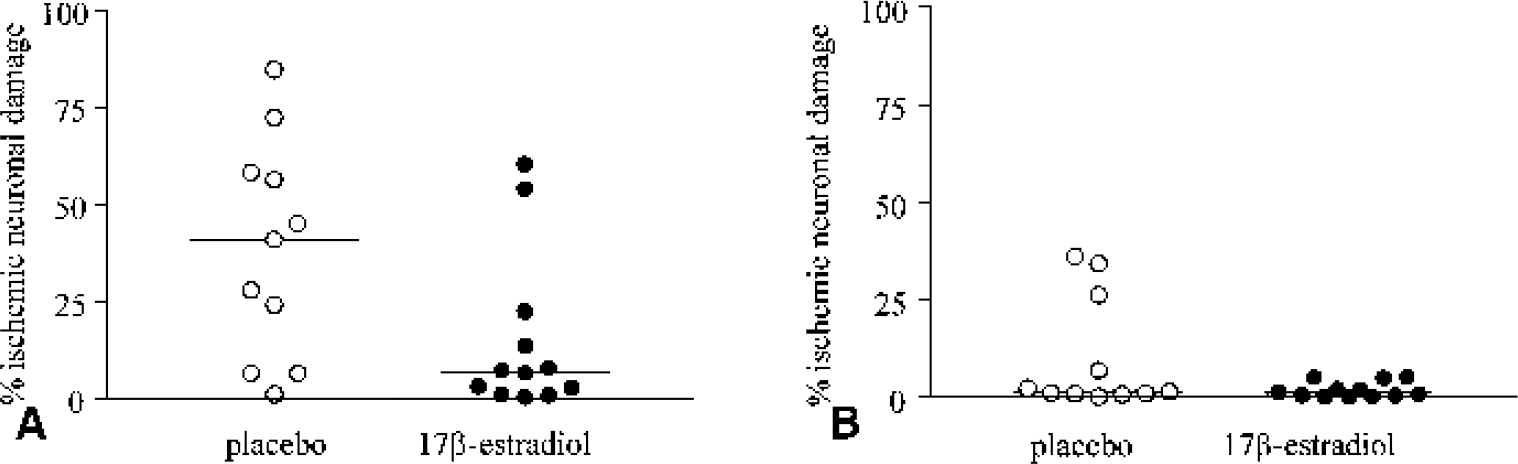
Quantification of ischemic neuronal damage in the caudate nucleus
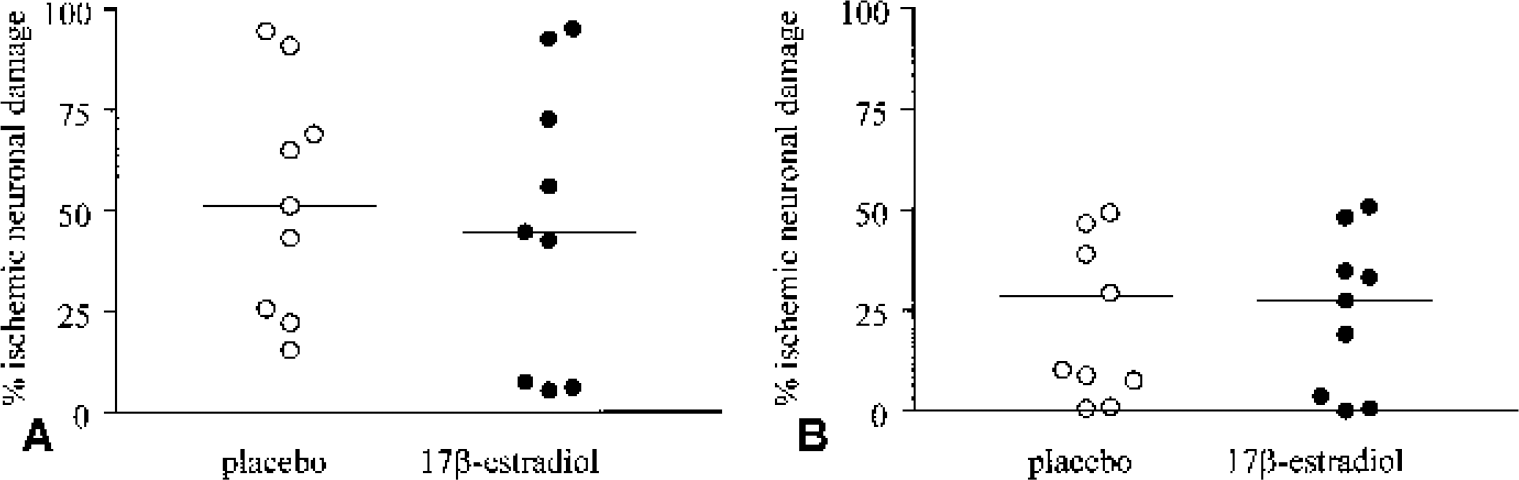
Quantification of ischemic neuronal damage in the caudate nucleus
DISCUSSION
The present study demonstrated a neuroprotective effect of physiologically relevant plasma levels of estradiol in a mouse model of global cerebral ischemia. We propose that this neuroprotective effect of estradiol was dependent on endogenous apoE since there was no neuroprotection afforded by estradiol replacement in the apoE-deficient mice.
Estrogen has demonstrated great potential as a therapeutic agent in experimental stroke. Exogenous 17β-estradiol, with plasma levels in the physiologic range, improves outcome in different models of focal ischemic brain injury, in female ovariectomized rodents and male rats (Alkayed et al., 1998; Dubal et al., 1998, 2001; Fukuda et al., 2000; Rusa et al., 1999; Simpkins et al., 1997; Toung et al., 1998). The present study shows that, in female ovariectomized C57Bl/6J mice, 17β-estradiol within the physiologic range is neuroprotective in a 2-vessel occlusion model of global ischemia. 17β-Estradiol neuroprotection in transient forebrain ischemia has also been shown in gerbils (Chen et al., 1998). In this study, CA1 pyramidal cell loss was reduced by high doses of 17β-estradiol (30 μg intraventricularly [intracerebroventricularly] and 4μg/kg intraperitoneally) whereas low doses (3 or 10 μg intracerebroventricularly) were ineffective (Chen et al., 1998). In another study the effect of 17β-estradiol was demonstrated to be dose-dependent, where 0.25 μg/d intracerebroventricularly ameliorated the loss of CA1 pyramidal neurons and ischemia-induced deficit in an avoidance task (Sudo et al., 1997), but neither the 0.05 μg/d nor 1.25 μg/d intracerebroventricularly was effective. In two rat forebrain ischemia studies, neuroprotection was demonstrated by 17β-estradiol (Pelligrino et al., 1998; Wang et al., 1999) whereas in another study 17β-estradiol actually exacerbated the extent of neuronal damage (Harukuni et al., 2001). The difference in efficacy of estrogen in models of transient global ischemia may arise from the different durations and severity of ischemia, the different brain regions affected or the different estrogen plasma levels achieved. It is recognized that increased duration of ischemia is associated with increased neuronal damage. With 2-vessel occlusion in C57Bl/6 mice, 17-minute occlusion produces extensive neuronal damage in the caudate nucleus and moderate neuronal damage in the CA1 region of the hippocampus whereas 20-minute occlusion produces more extensive damage in these regions (Horsburgh et al., 1999; Kelly et al., 2001). Furthermore, the brain regions affected are different in mouse compared with rat transient cerebral ischemia. For example, there is more extensive damage in the caudate nucleus in the mouse than in the rat (Kelly et al., 2001). In the present study estrogen neuroprotection was demonstrated in the caudate nucleus and there was a trend towards protection in the CA1 region. The effectiveness of estrogen in the CA1 region, however, might have been limited by the low levels of damage within this region. In terms of different estrogen plasma levels, in the present study in mice, the 0.025-mg dose was associated with a 7-fold increase in plasma levels of 17 β-estradiol compared with placebo-treated ovariectomized mice and a 3.5-fold increase compared with intact (nonovariectomized) mice. An equivalent dose in the rat, however, produced minimal alterations in plasma levels of estradiol compared with the placebo-treated group (Harukuni et al., 2001).
In both placebo- and estradiol-treated groups, there was more extensive neuronal damage in the apoE-deficient compared with wild-type controls, and the magnitude of this difference was greatest in the CA1 region as compared with the caudate nucleus. This may be explained by regional differences in mechanisms of injury (e.g., apoE is more important in CA1 than in caudate) or by differences in ischemic severity (effect of apoE gene deletion is less apparent in caudate, for example, because of a very severe ischemia). Although physiologic levels of 17β-estradiol afforded protection in C57Bl/6J mice after global ischemia, the equivalent dose was ineffective in apoE-deficient mice in both the caudate nucleus and CA1 region. It is therefore unlikely that the ineffectiveness of estrogen in apoE deficient mice was due to either region or severity of ischemia. To our knowledge, this is the first report to demonstrate that estradiol neuroprotection is mediated via apoE in a model of global ischemia. In the periphery, estradiol produces marked alterations in lipoprotein profiles. This effect may be different in apoE-deficient mice. Estradiol and apoE have been shown to have protective effects on the vasculature (Bourassa et al., 1996; Hodgin et al., 2001), and endothelial nitric oxide synthase was not essential for this effect of exogenous estradiol (Hodgin et al., 2002). In transient focal ischemia, the mechanism of protection by estrogen may also be mediated by vascular mechanisms, with evidence that blood flow during reperfusion is enhanced by estrogen (Shi et al., 1998). Although we did not determine cerebral blood flow in the present study, there is evidence that blood flow is not altered in apoE-deficient compared with wild-type mice in response to focal ischemia (Bart et al., 1998). It would be of value to determine whether any effects of estradiol on blood flow in the global ischemia model are different in apoE-deficient compared with wild-type mice.
Although physiologic levels of 17β-estradiol afforded protection in C57Bl/6J mice after global ischemia, the equivalent dose was ineffective in apoE-deficient mice. There has been no evidence to date to conclude that estradiol neuroprotection in cerebral ischemia is mediated via apoE. There has been some evidence, however, that endogenous apoE is necessary for 17β-estradiol-mediated effects on neuronal plasticity. Compensatory synaptic sprouting after hippocampal denervation is enhanced by estrogen in wild-type mice but not in apoE-deficient mice. This effect has been demonstrated both in vivo (Stone et al., 1998) and in vitro (Teter et al., 1999). It is suggested that sprouting may be stimulated by estrogen through its upregulation of apoE expression, leading to increased recycling of membrane lipids for use by sprouting neurons. In vivo, the dose of estrogen associated with enhanced neuronal plasticity is similar to the dose presently associated with neuroprotection. It remains to be determined whether the effect of estrogen is on neuronal or on glial apoE. A previous study indicated that estrogen upregulation of apoE occurs in microglia and astrocytes (the predominant source of apoE) (Stone et al., 1997).
In addition to estradiol's actions on the vasculature, a number of additional potential mechanisms have been suggested to underlie the protective actions of estradiol. It is notable that these actions parallel mechanisms by which apoE can modulate neuronal stability. Evidence exists for estradiol-induced attenuation of excitatory amino acid toxicity associated with ischemia. Overactivation of NMDA receptors occurs after ischemia. 17β-Estradiol pretreatment has been reported to markedly reduce ischemia-induced aspartate and glutamate release in the gerbil hippocampal CA1 region (Chen et al., 2001). Estradiol also protects against NMDA-induced neuronal death by directly inhibiting the NMDA receptor (Weaver et al., 1997), and high doses of 17β-estradiol, which reduced CA1 neuronal loss in transient forebrain ischemia, were shown to inhibit release of calcium from the intracellular stores and influx into neurons from the extracellular space (Chen et al., 1998). Therefore, 17β-estradiol may protect neurons from ischemic damage by attenuating excitotoxicity mediated by an increase in extracellular excitatory amino acids and intracellular calcium levels. Similarly, apoE can modulate NMDA receptor activation and calcium influx via the low-density lipoprotein receptor (Herz, 2001).
In addition to an effect on NMDA receptor activation, both 17β-estradiol and apoE have antioxidant activities (Behl et al., 1995; Lomnitski et al., 1997) as well as similar antiinflammatory properties. Estrogens are good scavengers of free radicals and have potent lipid antioxidant activity (Ruiz-Larrea et al., 2000). ApoE has also been shown to influence the extent of oxidative injury, with deficiency of apoE being associated with greater lipid peroxidation and oxidative damage (Horsburgh et al., 2000a; Lomnitski et al., 1997; Ramassamy et al., 2001). These actions have been suggested to underlie the neuroprotective effects of both estrogen and apoE in oxygen radical-induced brain injury (Green and Simpkins, 2000; Horsburgh et al., 2000b; Hurn and Macrae, 2000). Additionally, 17β-estradiol (Vegeto et al., 2001) and apoE (Laskowitz et al., 2001) both reduce microglial activation. 17β-Estradiol (Zhou et al., 1996) and apoE (Ohkubo et al., 2001) both stimulate cAMP-response element-binding (CREB) protein. Indeed, studies have shown increased expression of CREB-dependent genes such as bcl-2 associated with 17β-estradiol (Dubal et al., 1999; Alkayed et al., 2001) and apoE (Ohkubo et al., 2001), implying antiapoptotic mechanisms, and c-fos expression associated with 17β-estradiol (Balog et al., 2001) and apoE (Ohkubo et al., 2001), implying plasticity mechanisms.
17β-Estradiol mediates such mechanisms probably through upregulation of apoE, either at the transcriptional level (Stone et al., 1997) or posttranscriptional level (Srivastava et al., 1997). Increased apoE mRNA induced by 17β-estradiol (Stone et al., 1997) may be mediated by genomic estrogen receptor activation, although at present it is unclear whether an estrogen response element is present on the apoE gene. Posttranscriptional upregulation of apoE cannot be excluded, and this may involve either increased mRNA translation or decreased catabolism of apoE. A study by Srivastava et al. (1997) indicated that estradiol upregulates apoE gene expression by increasing levels of apoE mRNA in the polysomal translating pool and that this effect was mediated by estrogen receptors. A recent study found that supraphysiologic plasma 17β-estradiol levels in female ovariectomized C57Bl/6 mice gave rise to 1.5-, 2.01-, and 2.32-fold increases in apoE protein levels in hippocampus, cortex, and diencephalon, respectively, with no significant change in apoE mRNA levels (Levin-Allerhand et al., 2001). In the same study, upregulation of apoE protein was demonstrated in the cortex of ovariectomized estrogen receptor α (ER α)–knockout mice, and 17α-estradiol (a poor estrogen receptor agonist) induced apoE protein in the cortex of C57Bl/6 mice. These results suggest a non-ER α-receptor-mediated mechanism is implicated in the cortex, which may point to the involvement of other estrogen receptors or non-receptor-mediated mechanisms.
In summary, this study shows that 17β-estradiol is neuroprotective in a mouse model of global cerebral ischemia. In addition, the key finding of this study is that this effect of 17 β-estradiol is mediated via endogenous apoE. Estrogen and apoE may therefore interact in their modulation of recovery from CNS injury.
Footnotes
Acknowledgements
The technical assistance at the Wellcome Surgical Institute is gratefully acknowledged.