
Abstract
Cerebral ischemia triggers acute inflammation, which exacerbates primary brain damage. Activation of the innate immune system is an important component of this inflammatory response. Inflammation occurs through the action of proinflammatory cytokines, such as TNF, IL-1β and IL-6, that alter blood flow and increase vascular permeability, thus leading to secondary ischemia and accumulation of immune cells in the brain. Production of these cytokines is initiated by signaling through Toll-like receptors (TLRs) that recognize host-derived molecules released from injured tissues and cells. Recently, great strides have been made in understanding the regulation of the innate immune system, particularly the signaling mechanisms of TLRs. Negative feedback inhibitors of TLRs and inflammatory cytokines have now been identified and characterized. It is also evident that lipid rafts exist in membranes and play a role in receptor-mediated inflammatory signaling events. In the present review, using this newly available large body of knowledge, we take a fresh look at studies of ischemic tolerance. Based on this analysis, we recognize a striking similarity between ischemic tolerance and endotoxin tolerance, an immune suppressive state characterized by hyporesponsiveness to lipopolysaccharide (LPS). In view of this analogy, and considering recent discoveries related to molecular mechanisms of endotoxin tolerance, we postulate that inhibition of TLR and proinflammatory cytokine signaling contributes critically to ischemic tolerance in the brain and other organs. Ischemic tolerance is a protective mechanism induced by a variety of preconditioning stimuli. Tolerance can be established with two temporal profiles: (i) a rapid form in which the trigger induces tolerance to ischemia within minutes and (ii) a delayed form in which development of protection takes several hours or days and requires de-novo protein synthesis. The rapid form of tolerance is achieved by direct interference with membrane fluidity, causing disruption of lipid rafts leading to inhibition of TLR/cytokine signaling pathways. In the delayed form of tolerance, the preconditioning stimulus first triggers the TLR/cytokine inflammatory pathways, leading not only to inflammation but also to simultaneous upregulation of feedback inhibitors of inflammation. These inhibitors, which include signaling inhibitors, decoy receptors, and anti-inflammatory cytokines, reduce the inflammatory response to a subsequent episode of ischemia. This novel interpretation of the molecular mechanism of ischemic tolerance highlights new avenues for future investigation into the prevention and treatment of stroke and related diseases.
Keywords
The important thing in science is not so much to obtain new facts as to discover new ways of thinking about them.
(Sir William Bragg)
Pre-exposing the brain to a subthreshold level of pathologic stimulus, also termed preconditioning, markedly diminishes neuronal vulnerability to a subsequent ischemic insult (Dirnagl et al. 2003;Kirino 2002). The nature of the induced protection and the underlying molecular mechanisms of preconditioning remain poorly understood. Indeed, even the molecular cascades responsible for ischemic brain injury have not been fully identified despite intense investigation (Lo et al. 2003). Stimuli used to precondition the brain and induce neuroprotection include transient global and focal ischemia, cortical spreading depression (CSD), seizures, hypothermia, hyperthermia, and administration of lipopolysaccharide (LPS), proinflammatory cytokines, anesthetics, ethanol, or polyunsaturated fatty acids (PUFA) (see Table 1). Depending on the specific preconditioning stimulus, a state of neuronal protection can be established within minutes (rapid tolerance) or after a delay of several hours to days (delayed tolerance). Interestingly, preconditioning treatments associated with delayed tolerance induce proinflammatory cytokines, while those associated with rapid tolerance tend to suppress production of the very same cytokines (Table 1). It is important to recognize that ischemic tolerance is not unique to the brain. Other organs can be protected from ischemic damage using many of the same preconditioning stimuli through mechanisms with features similar to those in brain. An obvious question emerges: How can these diverse stimuli all induce tolerance to ischemia?
Interrelation between ischemic tolerance and inflammatory cytokine regulation in the brain
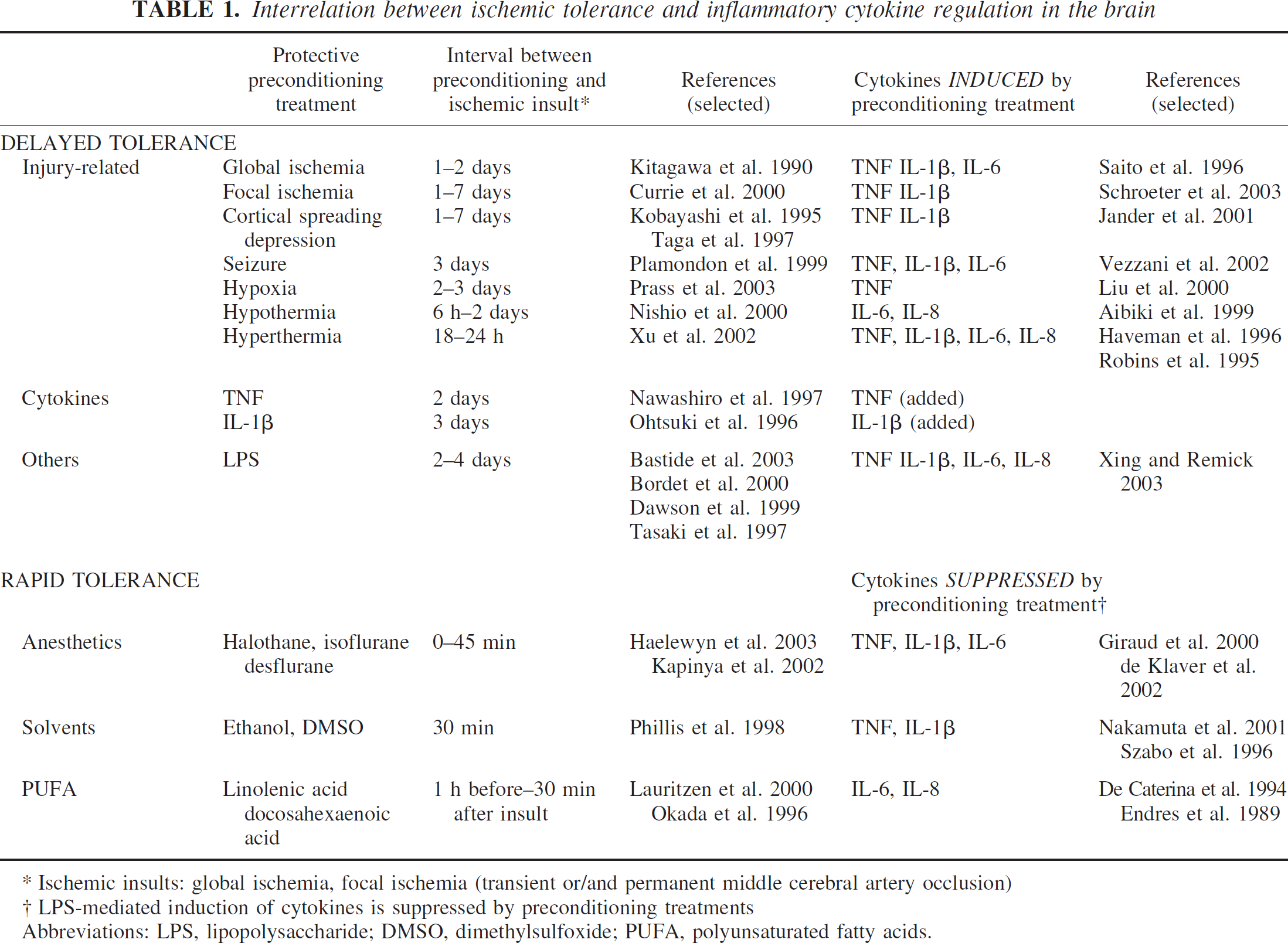
Abbreviations: LPS, lipopolysaccharide; DMSO, dimethylsulfoxide; PUFA, polyunsaturated fatty acids.
Ischemic insults: global ischemia, focal ischemia (transient or/and permanent middle cerebral artery occlusion)
LPS-mediated induction of cytokines is suppressed by preconditioning treatments
A variety of hypotheses have been proposed to answer this question, but no satisfactory explanation with a unifying concept has been provided. Numerous studies have demonstrated that inflammatory mediators play a central role in protective preconditioning (Table 1). Indeed, administration of inflammatory cytokines alone is sufficient to induce tolerance to ischemia (Nawashiro et al. 1997;Ohtsuki et al. 1996). Paradoxically, the same cytokines have also been shown to exacerbate ischemic brain damage (Barone and Feuerstein 1999). This apparent discrepancy, as well as the lack of clear understanding of the basic molecular mechanisms of inflammation, has hindered the formulation of a rational explanation for ischemic tolerance that includes a role for inflammatory cytokines.
Recently, significant advances in the field of innate immunity have furthered our understanding of inflammatory mechanisms. Toll-like receptors (TLRs) recognize and respond to molecules related to microbial infection or host tissue injury (Medzhitov 2001). It has been shown that signaling through TLRs results in inflammation mediated by selected cytokine and chemokine production, such as tumor necrosis factor (TNF), interleukin (IL)-1β, IL-6, and IL-8. Special microdomains, termed lipid rafts, have been identified in the cell membranes, and their importance in receptor-mediated signaling events elucidated. Importantly, an increasing number of feedback inhibitors that negatively regulate TLR and inflammatory cytokine signaling have been identified. The role of these inhibitors is to extinguish or modulate inflammation, thereby protecting the host from an excessive inflammatory response. Investigation of these feedback inhibitors has provided valuable insight into the molecular mechanisms of endotoxin tolerance (Fan and Cook 2004) that may also operate in ischemic tolerance.
Excellent reviews have recently evaluated the literature on ischemic tolerance (Dirnagl et al. 2003;Kirino 2002). Our objective here is to highlight the important characteristics of the positive signaling and negative feedback pathways of TLR and cytokine systems in cerebral ischemia and preconditioning. To this end, we will summarize evidence demonstrating that cerebral preconditioning down-regulates proinflammatory TLR and cytokine signaling, either directly (rapid tolerance) or indirectly (delayed tolerance), thus reducing the acute inflammatory response that exacerbates ischemic brain injury.
DELAYED TOLERANCE: A PARALLEL BETWEEN ISCHEMIC AND ENDOTOXIN TOLERANCE
Brief ischemia and other noxious stresses induce delayed tolerance to ischemia
Numerous studies have demonstrated that preconditioning the brain with a variety of potentially harmful stimuli induces a profound tolerance to a subsequent episode of ischemia (Table 1). A brief ischemic insult can also induce ischemic tolerance in the spinal cord (Zvara et al. 1999) and other organs, such as the heart (Murry et al. 1986), liver (Tsuyama et al. 2000) or kidney (Toosy et al. 1999). The exact molecular mechanisms underlying delayed ischemic tolerance are not well understood, but requirements for de novo protein synthesis (Currie et al. 2000;Nishio et al. 2000), activation of the proinflammatory transcription factor nuclear factor kappa B (NF-κB) (Blondeau et al. 2001;Digicaylioglu and Lipton 2001), and induction of inflammatory cytokines such as TNF, IL-1β, IL-6 and IL-8 have been demonstrated in most of the experimental models (Table 1). These inflammatory cytokines are believed to play a critical role in establishing tolerance because (i) inhibiting their action or production eliminates protection (Cardenas et al. 2002;Ohtsuki et al. 1996) and (ii) administration of TNF or IL-1β alone can directly induce tolerance to cerebral ischemia (Table 1).
LPS induces ischemic tolerance
Pretreatment with LPS, a cell-wall component of gram-negative bacteria, has been shown to induce tolerance to ischemia in a number of experimental models (Table 1). Systemic administration of LPS established a state of tolerance that protected the brain from lethal ischemic insults (Tasaki et al. 1997). Similar protection was observed for other organs including the heart (Eising et al. 1996;Zacharowski et al. 1999), kidney (Heemann et al. 2000), liver (Colletti et al. 1994), and pancreas (Obermaier et al. 2003). Importantly, the tolerance-inducing effect of LPS has the same temporal pattern and involved the same cytokines (TNF, IL-1β, IL-6, and IL-8) characteristic of other preconditioning treatments. LPS-induced protection from ischemic brain damage was abolished when anti-inflammatory agents or protein synthesis inhibitors were co-administered with LPS (Bordet et al. 2000), or when the function of inflammatory cytokines was blocked (Heemann et al. 2000;Tasaki et al. 1997). These results demonstrate an absolute requirement for de novo protein synthesis and a critical role for inflammatory pathways in ischemic tolerance induced by LPS. LPS preconditioning is even better known for its ability to establish tolerance against a subsequent challenge with LPS, a phenomenon termed ‘endotoxin tolerance’. Given these parallels, therefore, an improved understanding of ischemic tolerance may be gained by a closer examination of the mechanisms underlying endotoxin tolerance.
Understanding endotoxin tolerance
Endotoxin tolerance (also called LPS tolerance) was discovered decades ago after recognition that pre-exposure to a low dose of LPS markedly reduced mortality in animals rechallenged with a lethal dose of LPS (Cavaillon et al. 2003;Fan and Cook 2004). In the classic endotoxin tolerance paradigm, the first exposure to LPS activates Toll-like receptor 4 (TLR4), present on monocytes and macrophages. Stimulation of TLR4 triggers a signaling cascade (discussed later in detail) that leads to activation of several transcription factors, including NF-κB, and results in production of inflammatory mediators, including TNF and IL-1β. The signaling events initiated by LPS and these cytokines induce a hyporesponsive state to a subsequent LPS challenge. This classic model uses LPS, a ligand of TLR4, both as the trigger of tolerance and the subsequent challenge. However, endotoxin tolerance can also be induced by ligand-mediated activation of other TLRs or by sole treatment with proinflammatory cytokines (e.g. TNF, IL-1β) through a process called cross-tolerization (Lehner et al. 2001). Studies have demonstrated that noxious stresses, such as surgical procedures, can also establish endotoxin tolerance (Joosten et al. 2003;Kawasaki et al. 2001;Lemaire et al. 1998). Furthermore, an ischemia-reperfusion injury model in a rabbit liver has recently demonstrated that preconditioning with ischemia induces tolerance to subsequent LPS challenge (Langdale et al. 2003). A systemic immune response (e.g., secretion of inflammatory cytokines) has been shown to precede the establishment of the endotoxin tolerance state (Joosten et al. 2003). These findings reveal many common characteristics between ischemic tolerance and endotoxin tolerance. Both ischemic and LPS tolerance are immune suppressed states that can be induced by preconditioning with LPS or ischemia, resulting in production of the same inflammatory cytokines (e.g. TNF, IL-1β, IL-6 and IL-8).
Ischemic tolerance vs. endotoxin tolerance. Is there a difference?
The striking similarities between inducers and mediators of ischemic and endotoxin tolerance suggest that ischemic tolerance and endotoxin tolerance might have identical molecular mechanisms. Endotoxin tolerance has been investigated extensively both in vitro and in animal models. Increasing evidence demonstrates that a variety of molecules (TLRs, their corresponding ligands, cytokines, feedback inhibitors of TLRs and cytokine-signaling, and decoy receptors) play central roles in the molecular mechanism of endotoxin tolerance (Fan and Cook 2004;West and Heagy 2002). To determine whether the same molecules are also involved in ischemic tolerance in the brain, we need to address the following two questions. First, which are the molecules analogous to LPS that activate TLRs during cerebral preconditioning? Second, does cerebral preconditioning induce feedback inhibitors of TLR and cytokine signaling similar to those observed for endotoxin tolerance?
TLR during inflammation
The innate immune system is the first line of defense against invading pathogens (Medzhitov 2001). This system utilizes receptors, which include TLRs, capable of recognizing conserved pathogen-associated molecular patterns (PAMPs) and orchestrating an immune response. TLRs are basic signaling receptors with extra-cellular leucine-rich repeats and a cytoplasmic signaling domain. There are at least 10 distinct TLR family members in humans, and corresponding ligands for most of them have been identified. These ligands include bacterial and viral constituents, such as unmethylated CpG DNA, double-stranded RNA (dsRNA), LPS, and flagellin (Dunne and O’Neill 2003). Generally, TLRs bind to accessory proteins that are specifically associated with PAMPs. The identities of the accessory proteins for most PAMPs are unknown, with the exception of LPS, which stimulates the most studied receptor, TLR4. Optimal signal transduction through TLR4 requires binding of LPS to serum proteins, such as albumin, LPS-binding protein, and MD2, as well as cell surface-associated molecules, such as mindin and the GPI-(glycosylphosphatidylinositol) linked CD14 (Gioannini et al. 2002;He et al. 2004). Because the interacting cytoplasmic protein regions of TLR are highly homologous to the intracellular domain of IL-1 receptor type I (IL-1RI), they have been called Toll/IL-1 receptor (TIR) domains. During intracellular signal transduction, the TIR domain of the TLR associates with an intracellular adaptor protein MyD88 (Fig. 1). A phosphorylation cascade involving IL-1 receptor-associated kinase (IRAK) and TNF receptor-associated factor 6 (TRAF6) ensues, and this signaling activates transcriptional pathways, including NF-κB, c-Jun N-terminal kinase (JNK), stress-activated protein kinase p38, and extracellular signal-regulated kinase (ERK) (Fig. 1). These signals culminate in a proinflammatory response, producing inflammatory cytokines, releasing nitric oxide, and upregulating cell adhesion molecules and the procoagulant tissue factor (Beutler 2004). It is important to note that several TLR mediators, such as MyD88, TRAF6, and IRAK, also participate in the nerve growth factor (NGF)-induced signaling pathway, which is initiated by stimulation of the p75 neurotrophin receptor, leading to NF-κB activation (Mamidipudi et al. 2002). TNF-initiated signal transduction also involves mediators of the TLR signaling cascade (Janssens and Beyaert 2003;O’Neill 2000). TLRs are found primarily on cells of the immune system (macrophages, dendritic cells, B-cells, T-cells) (Visintin et al. 2001), but are also present on epithelial and endothelial cells (Miettinen et al. 2001).
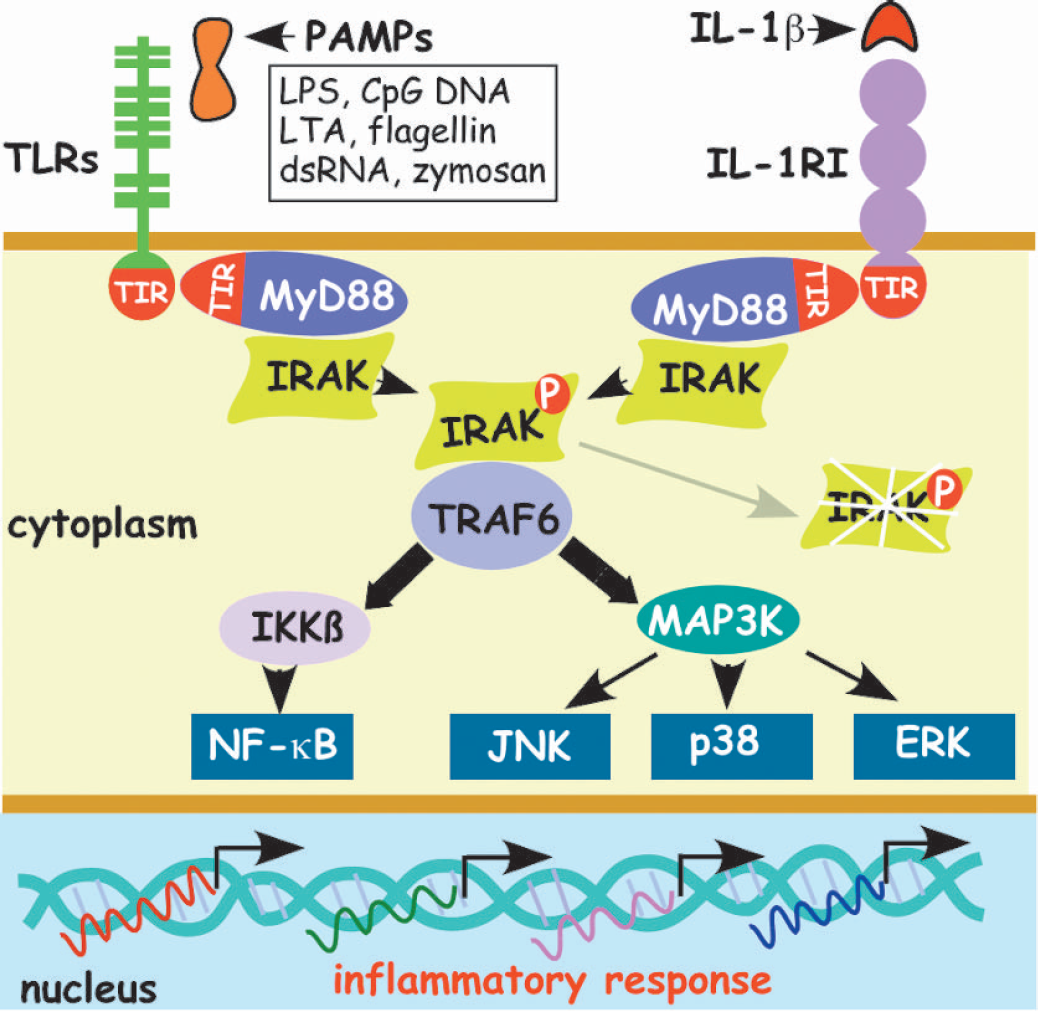
Activation of Toll-like receptors and type I IL-1 receptor induces inflammation in immune cells via shared signaling cascades. Toll-like receptors (TLRs) expressed on professional immune cells (monocytes, macrophages, dendritic cells and microglia) recognize and respond to microbial infection. TLRs are activated by products containing pathogen-associated molecular patterns (PAMPs), derived from bacteria (lipopolysaccharide (LPS), lipotheichoic acid (LTA), DNA with non-methylated cytosine-guanine motifs (CpG DNA), flagellin), from yeast (zymosan), or from viruses (double-stranded RNA (dsRNA)). All TLR family members and the type I interleukin-1 receptor (IL-1RI) have a unique intracellular TIR (Toll/IL-1 receptor) signaling domain. In response to activation by the corresponding ligands, TIR domains interact with the TIR domain of the signaling adaptor MyD88 (myeloid differentiation factor 88), which transduces the signal to a family of IL-1 receptor-associated kinases (IRAKs). Phosphorylation of IRAK, a serine-threonine kinase, by other IRAK family members induces cascades of signaling through TRAF6 (tumor necrosis factor receptor-associated factor 6). TRAF6 transduces the signal to IKKβ (IkappaB kinaseβ) and to MAP3K (mitogen-activated protein kinase kinase kinase). This signaling results in transcriptional responses, mediated primarily by nuclear factor-κB (NF-κB), extracellular-signal regulated kinase (ERK) and stress-activated protein kinases, such as c-Jun N-terminal kinase (JNK) and p38, leads to expression of proinflammatory cytokines. For simplicity not all adaptors and details of signaling are included in the figure.
Toll-like receptors in the CNS
The CNS exhibits a well-organized innate immune reaction in response to systemic or cerebral infection and injury (Nguyen et al. 2002;Rivest 2003). A variety of TLRs have been identified on the cells of the human and murine brain. TLR4 mRNA is constitutively expressed in the circumventricular organs and regions with a rich vascular plexus (Laflamme and Rivest 2001); these same regions express TLR2 mRNA in response to stimulation (Laflamme et al. 2003;Laflamme et al. 2001). Microglia express mRNA for all of the TLRs, while astrocytes primarily express mRNAs for TLR2, TLR3, and TLR4 (Bsibsi et al. 2002;Esen et al. 2004). Recently, murine astrocytes were shown to express TLR2, TLR4, TLR5, and TLR9 mRNAs, and to respond with inflammatory cytokine secretion when stimulated with ligands for these receptors (Bowman et al. 2003). Microglia, which express TLR9, release TNF and IL-12 when stimulated with non-methylated CpG DNA (Dalpke et al. 2002). Recent studies have demonstrated that TLR4 is critical for LPS-induced injury in the CNS and have identified microglia as the major cellular mediator of injury (Lehnardt et al. 2002;Lehnardt et al. 2003). Chronic glial activation, neurodegeneration and IL-1β induction in a rat brain has been demonstrated after administration of dsRNA, the ligand of TLR3 (Melton et al. 2003). Other cells in the brain, such as endothelial and smooth muscle cells of the blood vessels, also express TLRs and can respond to stimulation (Cristofaro and Opal 2003;Miettinen et al. 2001). Thus, TLRs are well positioned in the CNS to mediate the induction of tolerance to ischemia.
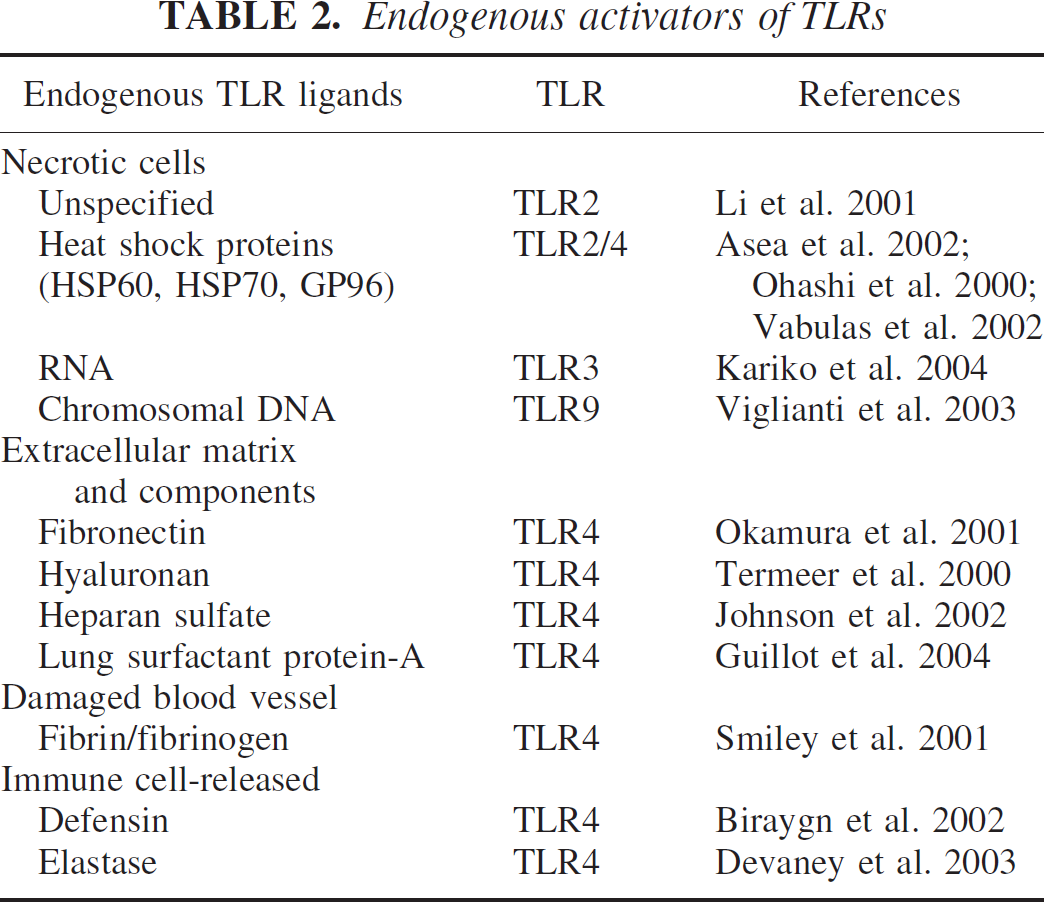
Molecules derived from damaged cells and tissue activate Toll-like receptors
We next consider how ischemia, either as a preconditioning stimulus or as the subsequent severe insult, activates TLR. It has been long recognized that injured tissue and necrotic cells release endogenous adjuvants, which induce inflammatory mediators (Shi and Rock 2002). Recently, several of these endogenous activators were identified as ligands for TLRs (see Fig. 2) (Beg 2002;Frantz et al. 1999;Paterson et al. 2003). Molecules released from damaged blood vessels or extracellular matrix have also been identified as endogenous activators of TLRs (Table 2). Release of TLR ligands from damaged cells and tissues, activate TLRs in the same way that LPS activates TLR4. Robust activation of TLRs induces a high level of inflammatory cytokine secretion, thus augmenting cellular injury. Many models of delayed ischemic preconditioning, while grossly sublethal, are nevertheless associated with a minor degree of cell, blood vessel, or extracellular matrix damage, leading to release of small amounts of TLR ligands. These endogenous ligands will activate TLR-signaling and lead to a low level of cytokine release associated with mild inflammation in a manner analogous to that observed in LPS-induced preconditioning.
Endogenous activators of TLRs
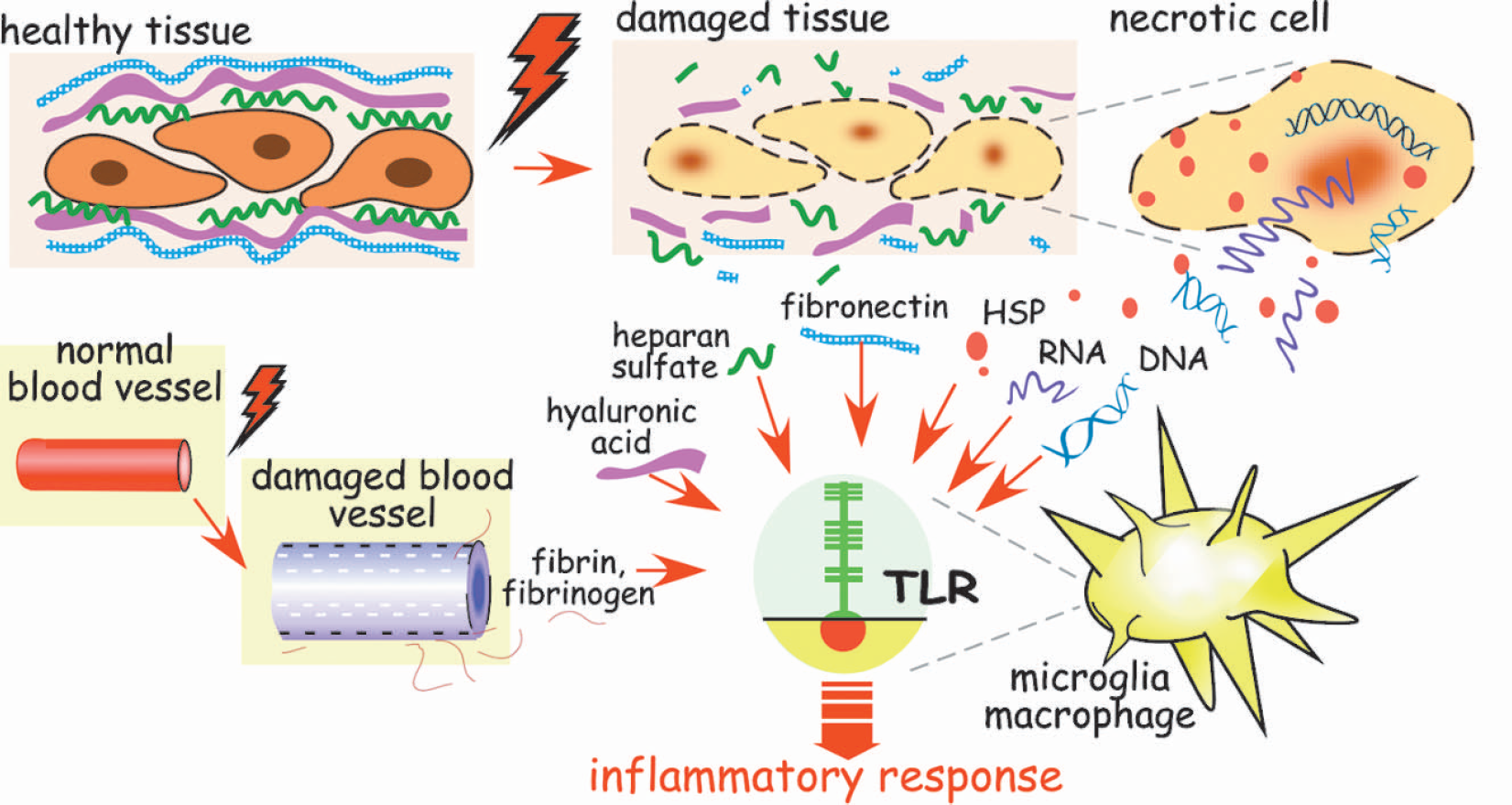
Molecules derived from injured tissue, blood vessels, and necrotic cells activate Toll-like receptors and induce inflammation. Following tissue and cell injury, endogenous ligands of Toll-like receptors (TLRs) are generated and/or released. Fragments of extra-cellular matrices (hyaluronic acid, fibronectin, heparan sulfate), fibrin and fibrinogen, and heat shock proteins (HSP) released from damaged tissue, blood vessels, and necrotic cells, respectively, activate TLR4, the LPS receptor. RNA and chromatin-associated DNA released from necrotic cells activate TLR3 and TLR9, respectively. When endogenous ligands activate TLRs the resulting immune response is similar to that induced by microbial products.
Regulating inflammation
The inflammatory response to tissue injury is essential for the elimination of damaged cells and restoration of normal function. It is important, however, that the intensity of the immune reaction be appropriately tuned to the severity of the injury in order to minimize overstimulation of the innate immune system and exacerbation of tissue damage. There are several regulatory mechanisms that limit the inflammatory response. These negative regulators are induced by inflammatory signaling mechanisms and function to dampen the inflammatory response to subsequent challenge. We postulate that during preconditioning (e.g., exposure to LPS, TNF, IL-1β or to molecules derived from injury-related preconditioning) signals are generated that induce a mild inflammatory response and the production of inflammation inhibitors (Fig. 3A). These inhibitors then limit the inflammatory response to a subsequent episode of severe ischemia (Fig. 3B). Because inflammation exacerbates and extends ischemic brain damage, limiting the inflammatory response will diminish the overall extent of ischemic injury. The feedback inhibitors can be divided into two major groups. First, extracellular inhibitors act to limit dissemination of inflammation. Anti-inflammatory cytokines and decoy receptors of proinflammatory cytokines belong to this group of negative regulators. Molecules of the second group of feedback inhibitors function intracellularly to limit inflammatory signaling. Endogenous inhibitors of the TLR and inflammatory cytokine signaling pathways are members of this group of negative regulators. Expression of these anti-inflammatory molecules requires time for their transcription and translation, thus accounting for “delayed” induction of tolerance. The anti-inflammatory inhibitors induced after preconditioning are then present and positioned to function during a subsequent episode of ischemia.
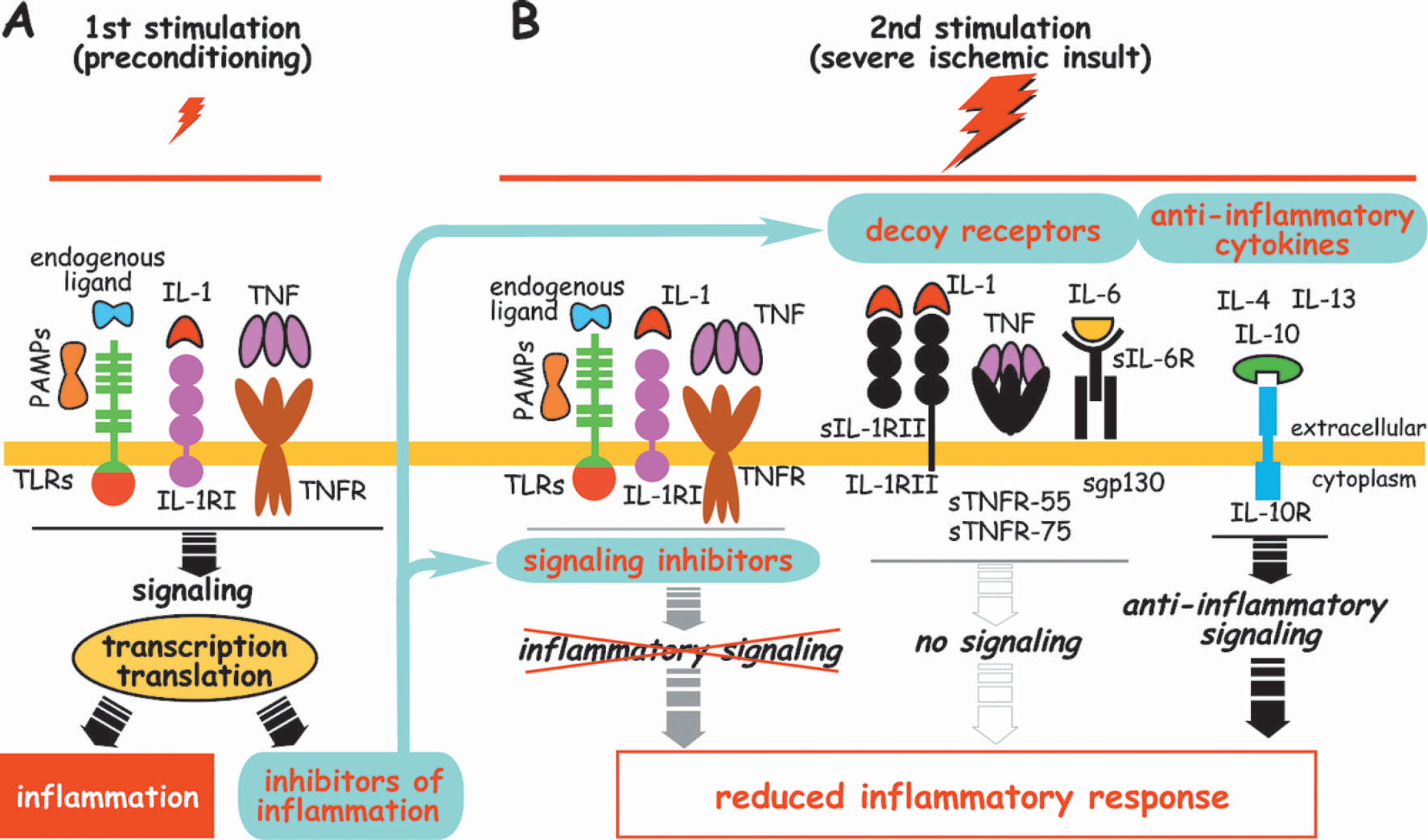
Feedback inhibitors of inflammation reduce the inflammatory response to a second (severe ischemic) insult. Panel A highlights activation of Toll-like receptors (TLRs) and receptors of inflammatory cytokines during preconditioning. Activation of TLRs, either by products with pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), or by injured tissue-derived and cell-derived endogenous TLR ligands (fibronectin, fibrin, heat schock proteins, DNA, RNA, etc.), results in transcriptional and translational induction of proinflammatory molecules (e.g. inflammatory cytokines, tissue factor, co-stimulatory markers) on cells of the immune system. Similarly, treatment with inflammatory cytokines such as tumor necrosis factor (TNF) and interleukin-1 (IL-1) also leads to inflammation through pathways that share several components with TLR signaling. In addition to the inflammatory response, activation of TLRs and cytokine receptors also leads to transcriptional and translational induction of inhibitors of inflammation. The inhibitors, induced by the first (preconditioning) stimulation, include intracellular signaling inhibitors, decoy receptors, decoy ligands, and anti-inflammatory cytokines. Panel B. Signaling inhibitors of TLR and cytokine signal transduction (detailed in Fig. 4) block inflammatory cytokine-induced and TLR ligand-induced intracellular signaling at multiple stages, thus reducing the inflammatory response. Decoy receptors, such as membrane-anchored and soluble type II receptor of IL-1 (IL-1RII and sIL-1RII), and other soluble cytokine receptors, such as sTNFR-55 and sTNFR-75 receptors of TNF, and sIL-6R and sgp130 receptors of IL-6 (all depicted in black), bind functional inflammatory cytokines. However, decoy receptors do not transduce signal; thus, they dampen the cytokine effect and limit inflammation. Anti-inflammatory cytokines inhibit production of proinflammatory cytokines, reduce synthesis of cytokine receptors and TLRs, and inhibit activation of these receptors. Preconditioning also induces IL-1Ra (IL-1 receptor antagonist), the decoy ligand of functional IL-1RI (not shown). IL-1Ra functions as a competitive inhibitor of IL-1 thereby limiting inflammation. Preconditioning-induced feedback inhibitors of inflammation suppress inflammatory responses intracellularly and extracellularly to a second stimulation (e.g. severe ischemic insult). Because inflammation exacerbates ischemic injury, inhibition of ischemia-induced inflammation provides protection.
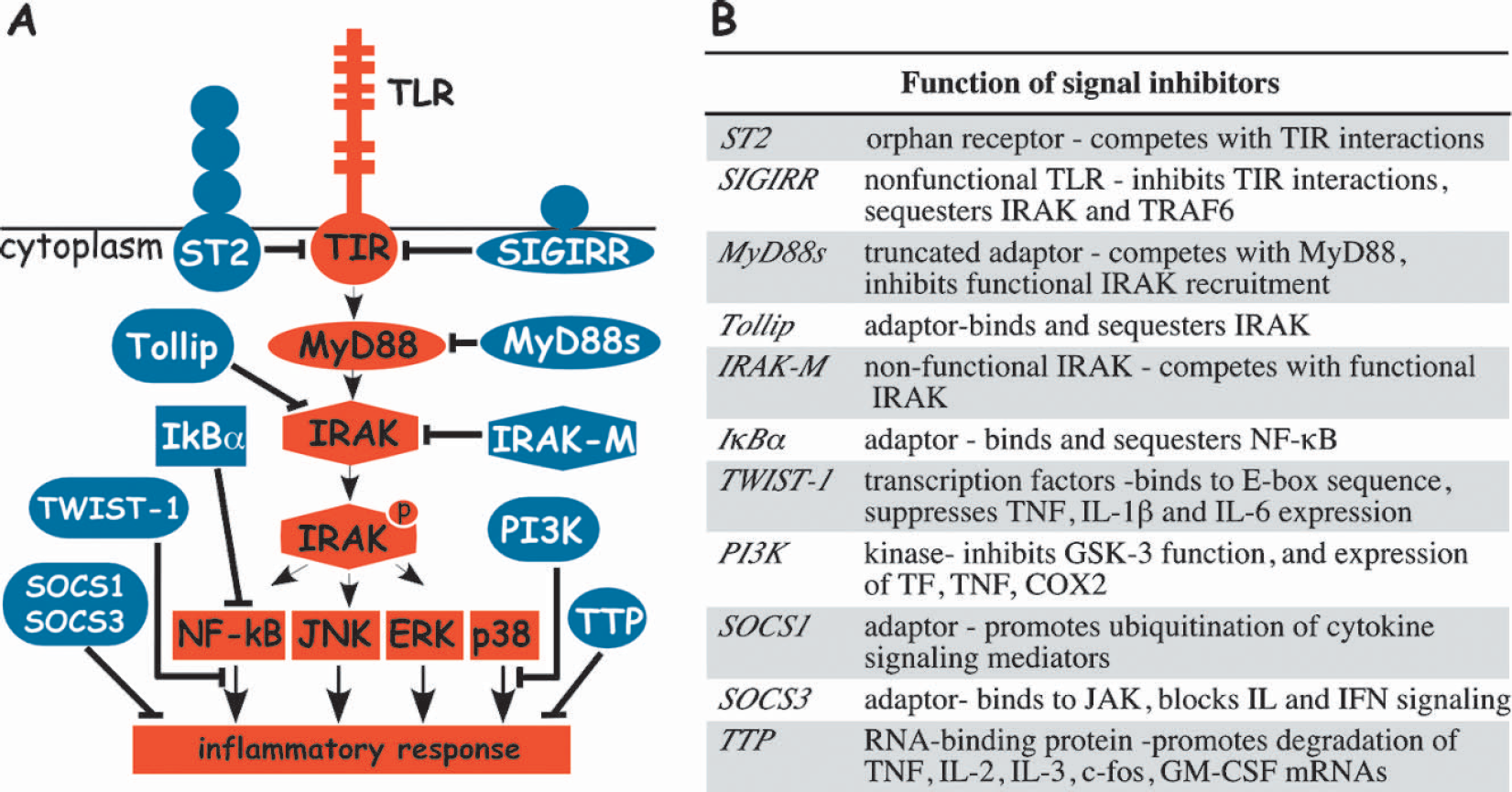
Feedback inhibitors of Toll-like receptor signaling block induction of inflammation. (A) Activation of Toll-like receptor either by microbial or endogenous ligand results in an inflammatory response by delivering a signal from the intracellular TIR (Toll/IL-1 receptor) domain of TLR through a series of intracellular mediators (illustrated as red-filled symbols). These mediators include signaling adaptor MyD88 (myeloid differentiation factor 88), IRAK (IL-1 receptor-associated kinase), NF-κB (nuclear factor-κB), ERK (extracellular-signal regulated kinase), JNK (c-Jun N-terminal kinase) and p38. Feedback inhibitors of TLR signaling (illustrated as blue-filled symbols) can block signal transduction and induction of inflammation at the various sites in the signaling pathway indicated. The inhibitors of TLR-mediated signaling cascades are: ST2, SIGIRR (single immunoglobulin IL-1R-related molecule), sMyD88 (short myeloid differentiation factor 88), Tollip (Toll-interacting protein), IRAK-M (IL-1 receptor-associated kinase M), TWIST-1, PI3K (phosphatidylinositol 3-kinase), TTP (tristetraprolin), IκBα (IkappaBalpha), SOCS1 and 3 (suppressor of cytokine signaling 1 and 3). Other TIR adaptor molecules have been identified, but their role in feedback inhibition is poorly studied and they are not presented in the figure for simplicity. (B) The important characteristics of TLR signal inhibitors are listed. Additional abbreviations: TRAF6, tumor necrosis factor receptor-associated factor 6; GSK-3, glycogen synthase kinase-3; TF, tissue factor; COX2, cyclooxygenase 2; JAK, Janus tyrosine kinase; IL, interleukin; IFN, interferon; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Extracellular feedback inhibitors of TLR and cytokine signaling
Anti-inflammatory cytokines (IL-4, IL-10, IL-13) are negative regulators of inflammatory immune responses (Yoshimura et al. 2003) (Fig. 3B). IL-10, secreted as a feedback inhibitor during inflammation initiated by LPS or tissue damage (Ogata et al. 2000), is synthesized in the CNS, and its receptor is present on microglia, astrocytes, and oligodendrocytes (Strle et al. 2001). The most important anti-inflammatory effects of IL-10 are (i) inhibition of production of all inflammatory cytokines, including IL-1β, TNF, IL-6, IL-8, IFN-γ, and IL-12 (Moore et al. 2001) and (ii) downregulation of expression of TLR4 (Muzio et al. 2000). Studies have demonstrated that IL-10 is an important component of endotoxin tolerance (Ogata et al. 2000) and protects the brain from inflammatory damage induced by LPS (Lynch et al. 2004) or ischemia (Spera et al. 1998).
Once inflammatory cytokines are released into the circulation, their effect can be blocked by decoy receptors, which are induced by inflammatory cytokines (Mantovani et al. 2004) (Fig. 3B). The prototypic decoy receptor is IL-1RII, which is synthesized by cells exposed to IL-1β. IL-1RII is a non-signaling receptor, which exists in membrane-associated and soluble forms. Both forms of the receptor bind and reduce the bioavailability of IL-1α and IL-1β. Functional receptors, such as TNFR and gp130 (the signaling receptor of the IL-6 and IL-6R complex), can also act as decoy receptors. Soluble decoy receptors are generated by shedding membrane-bound receptors or by translation of alternatively spliced mRNAs lacking coding sequences for transmembrane domains (Cui et al. 2003;Muller-Newen et al. 1998). Decoy receptors such as sTNFR-55, sTNFR-75, IL-1RII, sIL-6R, and sgp130 bind, inactivate, and promote clearance of their corresponding ligands (TNF, IL-1α, IL-1β, and IL-6), thereby limiting immune activation. In particular, induction of soluble decoy receptors, sIL-6R, and sgp130 limit bioavailability of IL-6, which has been implicated in the pathology of stroke (Acalovschi et al. 2003). In addition to decoy receptors, inflammation is dampened by binding of decoy ligands such as IL-1 receptor antagonist (IL-1Ra) to functional signaling receptors. Consistent with the anti-inflammatory roles of decoys, local and systemic administration of IL-1Ra has been shown to protect the brain from ischemic injury (Garcia et al. 1995;Relton et al. 1996).
Intracellular inhibitors of TLR and inflammatory cytokine signaling
In addition to the extracellular negative regulators described previously, activation of inflammation also induces intracellular signaling inhibitors. Feedback inhibitors of the TLR pathway are important intracellular components of endotoxin tolerance. Signal inhibitors of TLRs and the mechanisms by which they inhibit signal transduction are illustrated in Fig. 4. Because all TLRs share a common signaling pathway, activation of any individual TLR will induce endogenous inhibitors that reduce signaling through other TLRs (Table 3). This reduced capacity to signal and produce cytokines has been termed “cross-tolerance” (Lehner et al. 2001). Since many of the signaling mediators of TLR and cytokines are shared, most of the signaling inhibitors are also induced by cytokines (Table 3). Importantly, certain feedback inhibitors of TLRs can also block cytokine signal transduction.
Induction and activity of signaling inhibitors of cytokines and TLRs
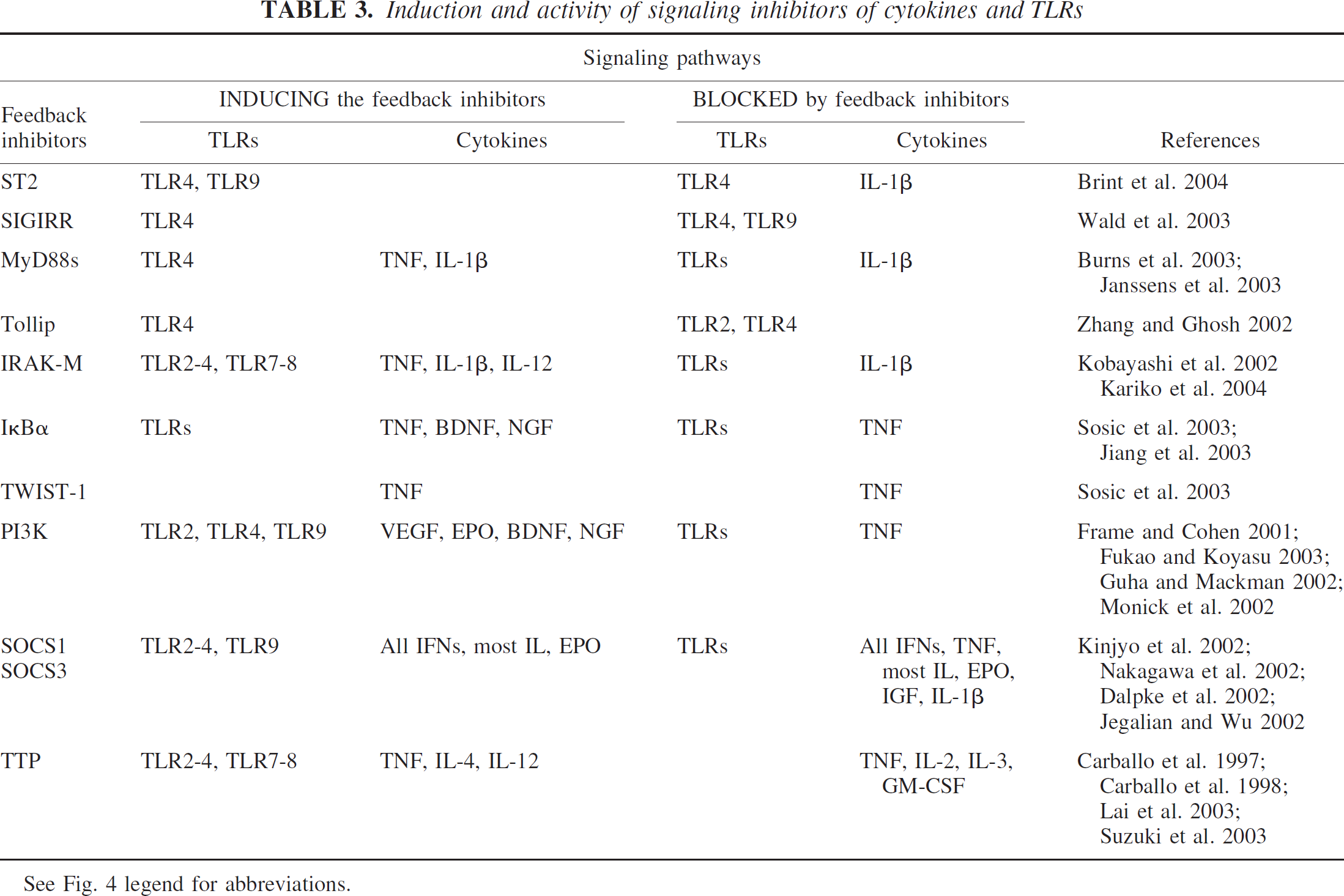
See Fig. 4 legend for abbreviations.
The feedback inhibitors are normally not present in cells, but are transcriptionally induced in response to activation of TLRs or cytokine receptors. Establishment of tolerance through feedback inhibitors, therefore, requires de novo protein synthesis. The inhibitors act on every level of TLR signal transduction to block the inflammatory response (Fig. 4). The functional importance of feedback inhibitors has been demonstrated in null mouse mutants. Mice lacking individual feedback inhibitors, such as the nonfunctional receptor SIGIRR (single immunoglobulin IL-1R-related molecule) (Wald et al. 2003), nonfunctional kinase IRAK-M (IL-1 receptor-associated kinase M) (Kobayashi et al. 2002), adaptor SOCS1 (suppressor of cytokine signaling 1) (Kinjyo et al. 2002), RNA-binding TTP (tristetraprolin) (Carballo and Blackshear 2001), transcription factor TWIST-1 (Sosic et al. 2003), and orphan receptor ST2 (Brint et al. 2004) (see Fig. 4 for details on the inhibitors), suffer from cachexia and inflammation. These mutants also exhibit elevated expression of proinflammatory cytokines in response to LPS and a lack of endotoxin tolerance when rechallenged with LPS. Additional feedback inhibitors of TLR and cytokine signaling, such as the adaptor Tollip (Toll-interacting protein) (Zhang and Ghosh 2002), the inhibitor kappaBalpha (IκBα) (Sosic et al. 2003), the truncated adaptor sMyD88 (Burns et al. 2003), and PI3K (phosphatidylinositol 3-kinase) (Fukao and Koyasu 2003) have also been identified (Fig. 4). Although most of the feedback inhibitors have only recently been discovered, there is already compelling evidence for a role for TTP, SOCSs, and PI3K in ischemic tolerance (see following).
TTP (Tristetraprolin) is a proline-rich protein, that binds to AU-rich elements present in the 3′-untranslated regions of mRNAs encoding proinflammatory cytokines, such as TNF, IL-2, IL-6, and GM-CSF (Carballo et al. 1998;Lai et al. 2003;Stoecklin et al. 2001;Worthington et al. 2002). TTP recruits a deadenylase, which selectively degrades the poly(A)-tail of the bound mRNAs, leading to a decreased half-life of the mRNA and decreased production of proinflammatory cytokines (Lai et al. 2003). TTP also suppresses LPS-induced transcription of TNF and the chemokine IL-8 (Zhu et al. 2001). Resting macrophages do not express TTP, but in response to LPS or TNF stimulation TTP mRNA was detected within 15 min (Table 3) (Carballo et al. 1998). A recent study identified TTP as a key mediator of cardio-protective preconditioning in a canine model of ischemia-reperfusion (Zubakov et al. 2003).
SOCS (suppressor of cytokine signaling) is a family of regulatory proteins that suppresses signal transduction of a variety of cytokines and TLRs, thereby inhibiting inflammation (Kubo et al. 2003). SOCSs are feedback inhibitors, blocking most of the same signaling pathways that lead to their induction (Table 3). Many of the cytokines, including all interferons, IL-6, and erythropoietin (EPO), signal through Janus kinases (JAKs). SOCSs inhibit activity of these cytokines by binding and blocking JAKs directly, but SOCSs also suppress TNF-signaling and TLR in a JAK-independent manner (Kinjyo et al. 2002) (Heeg and Dalpke 2003;Nakagawa et al. 2002). SOCSs are induced by cytokines or activation of TLRs. EPO, for example, induces SOCS1 and SOCS3 production (Jegalian and Wu 2002). Interestingly, because SOCSs suppress immune activation, they may also mediate EPO-induced anti-inflammatory and neuroprotective effects (Prass et al. 2003;Villa et al. 2003). A direct role for SOCS3 in neuroprotection has recently been demonstrated (Rao et al. 2002). SOCS3, which is normally expressed in the brain at low levels, was shown to be induced after ischemia. Interference with SOCS3 synthesis was reported to exacerbate ischemic brain damage, suggesting that SOCS3 plays a role in endogenous neuroprotection (Rao et al. 2002).
Phosphatidylinositol 3-kinase (PI3K) is an endogenous suppressor of inflammatory cytokine production (Table 3) (Fukao and Koyasu 2003). Stimulation of TLRs activates PI3K, leading to the inhibition of expression of many inflammatory genes through diverse mechanisms (Frame and Cohen 2001;Guha and Mackman 2002). Recent studies have demonstrated that PI3K contributes importantly to ischemic preconditioning in the brain and heart (Hashiguchi et al. 2004;Kis et al. 2003).
In summary, the mechanisms by which ischemia and LPS induce self-tolerance and cross-tolerance exhibit many parallels. It has already been demonstrated that LPS induces both tolerance and damage by activating TLR4. LPS tolerance is established by prior stimulation of any TLR, through cross-tolerance. ST-2, SIGIRR, MyD88s, Tollip, IRAK-M, SOCS1, TTP, PI3K, TWIST-1, and IκBα are intracellular inhibitors of the TLR and cytokine signaling cascades. These feedback inhibitors are induced in response to preconditioning stimuli, which activate TLR and/or cytokine signaling through the release of endogenous ligands. The feedback inhibitors then reduce the inflammatory response to a subsequent episode of ischemia, thus diminishing the overall extent of cell injury.
A model for delayed tolerance
A schematic model for preconditioning, ischemic injury, and delayed ischemic tolerance is presented in Fig. 5. We postulate that preconditioning stimuli and severe ischemia induce similar mechanisms. Both preconditioning and ischemia promote a period of inflammation, followed by a period of suppression of innate immunity. The intensity of the inflammation and suppression of innate immunity is proportional to the severity of the insult. Preconditioning causes inflammation that is followed by immune suppression that are mild in degree and short in duration. Severe ischemia causes inflammation and immune suppression that are more intense in degree and more prolonged in duration. Inflammation is mediated primarily by the action of inflammatory cytokines, while suppression of innate immunity is mediated by feedback inhibitors of inflammation. Because the initial insult and the subsequent inflammation both contribute to the induction of the feedback inhibitors, inflammation diminishes gradually while suppression increases. Brain damage from ischemia results from primary mechanisms of cell death and from secondary mechanisms of inflammation and infection. Excessive inflammation and the subsequent extreme suppression of immune responses are both detrimental. It was recognized decades ago (Howard and Simmons 1974) that inflammation is a frequent cause of acute death in injured patients, while infection is a more common cause of death at later times. Indeed, using a mouse stroke model, preventive antibacterial treatment significantly improved neurological outcome (Meisel et al. 2004). It seems reasonable, therefore, that limiting the extent of the immune responses caused by severe ischemia could be beneficial both in preventing local damage and subsequent susceptibility to infection. Both of the harmful immune responses (extreme inflammation and immune suppression) would be reduced if severe ischemia were to occur during suppression of innate immunity induced by prior preconditioning (Fig. 5, lower panel, ischemic tolerance). This interpretation of delayed ischemic preconditioning contrasts with previous hypotheses, which sought to identify unique proteins that were induced exclusively by preconditioning to selectively promote neuronal survival.
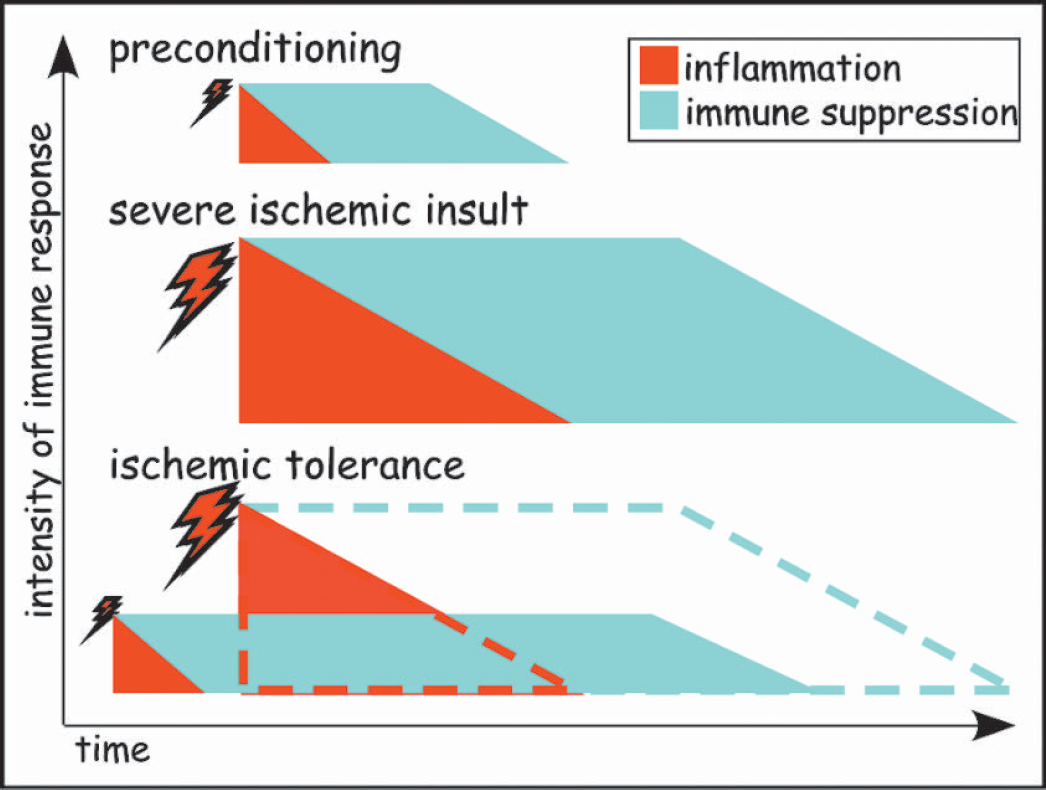
A model for delayed ischemic tolerance. Preconditioning stimuli as well as severe ischemic insults induce inflammation followed by suppression of the innate immune system. The induced immune responses, both inflammation and immune suppression, are proportional to the intensity of the initial insult or treatment. Consequently, immune responses initiated by preconditioning are limited in time and extent. By contrast, a severe ischemic insult induces excessive inflammation, followed by robust immune suppression. Inflammation is mediated primarily by the action of cytokines such as TNF, IL-1, and IL-6. The immune suppression is mediated by feedback inhibitors of inflammation, which include decoy receptors and inhibitors of TLRs and cytokine signaling cascades. Because the initial preconditioning stimulus and the subsequent inflammation both contribute to induction of the feedback inhibitors, inflammation wanes gradually and immune suppression increases. Excessive immune responses are detrimental because severe inflammation promotes coagulation and aggravates ischemia, while massive immune suppression permits infection to spread unchecked. During ischemic tolerance, a severe ischemic insult occurs during an immune suppressed state, both the inflammation and subsequent immune suppression are diminished. The predicted extent of inflammation and immune suppression caused by ischemia without preconditioning is indicated by the red and green dashed lines, respectively. Because severe inflammation and immune suppression exacerbates ischemic damage, limiting these reactions is protective and provides ischemic tolerance.
RAPID TOLERANCE: DIRECT INHIBITION OF TLR AND CYTOKINE SIGNALING
Several pharmacological agents, including anesthetics, ethanol, and polyunsaturated fatty acids (PUFA), provide cerebral protection against ischemia within minutes following their administration (Table 1). Some of the agents are effective even when administered after the ischemic insult, increasing their therapeutic potential. The molecular mechanism of “rapid” ischemic tolerance is not fully understood. Roles for an adenosine receptor and ATP-sensitive potassium channel have been demonstrated in adenosine-induced and ATP-induced rapid tolerance (Nakamura et al. 2002;Perez-Pinzon and Born 1999). Previous studies indicate that anesthetics, ethanol, and PUFA not only provide protection against ischemia but also against inflammation, via induction of rapid endotoxin tolerance (Table 1). We believe that the molecular mechanisms operating in rapid endotoxin tolerance also operate in rapid ischemic tolerance. To describe the molecular mechanism of rapid ischemic tolerance induced by anesthetics, ethanol, and PUFA, we will review the literature on endotoxin tolerance induced by these agents.
Rapid endotoxin tolerance induced by anesthetics, alcohol, and PUFA
It has been recognized for three decades that anesthetics suppress immunity both in vivo and in isolated immune cells (Howard and Simmons 1974). This suppression of host defenses against pathogens contributes to infectious complications following general anesthesia. Investigation of inflammatory responses in anesthetized rats revealed that halothane (Giraud et al. 2000), isoflurane (Plachinta et al. 2003), and ketamine (Taniguchi et al. 2001) all rapidly induce endotoxin tolerance, measured as a reduction in the level of TNF and IL-6 secretion following an in vivo LPS challenge. Endotoxin tolerance can also be induced in vitro. Production of TNF and IL-1β by LPS-treated human monocytes was inhibited when volatile anesthetics were administered concomitantly (Mitsuhata et al. 1995;Sato et al. 1995). Studies have shed light on the mechanism of this limited proinflammatory cytokine release by demonstrating that activation of NF-κB was significantly inhibited by isoflurane (De Rossi et al. 2004). However, this finding fails to identify the direct target of anesthetics and does not explain the molecular detail by which anesthetics inhibit an LPS challenge. The recognition that simultaneous administration of anesthetics and LPS were effective in reducing cytokine release indicates that the inhibitory mechanism has a rapid onset. Even a brief exposure to anesthetics causes a long-lasting inhibition of the effects not only of LPS but also of cytokines. A brief exposure to isoflurane, for example, was shown to block the inflammatory action of TNF, IL-1β, and IFNγ for 2 to 3 days (de Klaver et al. 2002), and thiopental inhibits the effects of TNF (Loop et al. 2003). The ability of the anesthetics to block the action of distinctly different ligands indicates that the inhibitory effects of anesthetics on inflammation are not mediated by a specific receptor, but rather are likely due to a non-specific suppression of receptor signaling.
Ethanol is another drug with potent immunosuppressive activities. Ethanol inhibits immune cell activation and proinflammatory cytokine production in response to microbial challenge (Nelson and Kolls 2002). Ethanol treatment inhibits a range of inflammatory mechanisms induced by endotoxin (Li and Chen 2003) or dsRNA (Hebert and Pruett 2003), ligands of TLR4 and TLR3, or by inflammatory cytokines (e.g., TNF, IL-1β) (Szabo et al. 1996). Evidence indicates that ethanol, similarly to anesthetics, interferes with early signaling events (Szabo et al. 1996).
PUFA, belonging either to the omega-3 or omega-6 family, inhibits LPS-induced and cytokine-induced inflammation by suppressing the secretion of inflammatory mediators (TNF, IL-1β, IL-6, IL-8, etc.) both in patients and in cultured cells (De Caterina et al. 1994;Endres et al. 1989;Furse et al. 2002). Treatment with docosahexaenoic acid (DHA), the most unsaturated fatty acid present in the mammalian membrane, inhibited NF-κB activation in response to TLR2-, TLR3-, TLR4-, TLR5-and TLR9-mediated stimulation by their corresponding ligands (Lee et al. 2003;Lee et al. 2004).
In summary, these findings demonstrate that anesthetics, ethanol, and PUFA rapidly inhibit inflammation induced by TLR ligands and proinflammatory cytokines. Inhibition of the inflammatory response is a potential mechanism by which anesthetics, ethanol, and PUFA exert their neuroprotective effect (Table 1) because endogenous TLR ligands and inflammatory cytokines generated during ischemia are likely the major initiators of inflammation.
How do anesthetics, ethanol, and PUFA block inflammation?
All of these reagents (anesthetics, ethanol, and PUFA) have a common property: the ability to increase membrane fluidity (Goldstein 1984;Goldstein 1986;Jump 2002). Lipid rafts are highly structured membrane microdomains that are critical components of receptor-mediated signaling mechanisms. We hypothesize that anesthetics, ethanol, and PUFA disrupt these lipid rafts by increasing membrane fluidity, thus causing the suppression of inflammatory signaling. As discussed below, compelling evidence from the literature support this possibility as a mechanism for rapid tolerance.
Lipid raft and signaling
Lipid rafts, depicted in Fig. 6A, are dynamic microdomains of the cell membrane enriched in sphingolipids with long saturated fatty acyl chains and cholesterol (Pike 2003). Dispersed in the more fluid phase of the membrane, lipid rafts are rigid structures providing a framework into which selected proteins segregate, interact, and participate in signal transduction (Simons and Toomre 2000). The function of lipid rafts is most studied in immune cells, but the existence of rafts on neurons has also been shown (Hering et al. 2003). Lipid rafts play an important role in T-cell receptor activation (Drevot et al. 2002) and in inflammatory signal transduction initiated by LPS (Pfeiffer et al. 2001) and TNF (Legler et al. 2003). According to a current model, signaling receptors and accessory proteins are sequestered in lipid rafts (Fig. 6B). Stimulation with a ligand facilitates the interaction between relevant receptors and accessory proteins by bringing them in close proximity, which is accomplished by clustering their lipid rafts (Fig. 6B, left side). These interactions culminate in transmitting the signal between proteins within the raft and ultimately from the extracellular domain to the cytoplasmic domain of the signaling receptor. Here, we postulate that anesthetics, ethanol, and PUFA, due to their effect on membrane fluidity, cause rapid disruption of the highly structured lipid rafts (Fig. 6B, right side). Alterations in the lipid rafts then interfere with the clustering of receptors induced by TLR ligands or inflammatory cytokines, both of which are released during ischemia. This interference would then lead to inhibition of signal transduction, suppression of inflammation, and ultimately neuroprotection (Rapid tolerance, Table 1). Consistent with this interpretation, cells exposed to PUFA were shown to change the composition of their lipid rafts, causing displacement of selected proteins from the raft and suppression of immune function (Zeyda et al. 2002). Interestingly, depletion of cholesterol has also been reported to disrupt lipid rafts (Kirsch et al. 2003). This may be relevant to the mechanisms by which the cholesterol-lowering statins exert their long-term immunosuppressive effect (Li and Chen 2003) and prevent stroke in patients (Hankey 2002).
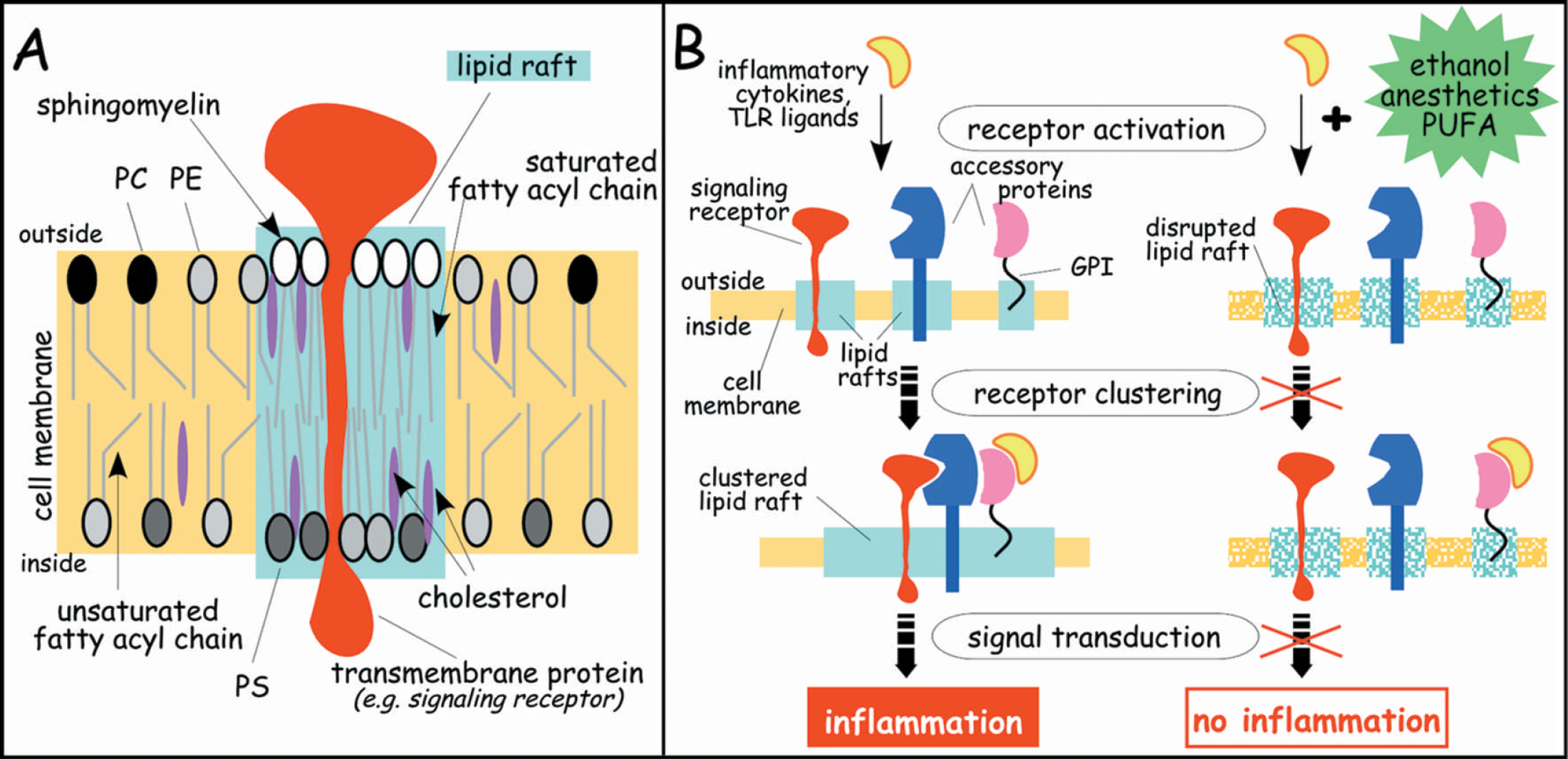
Inducers of rapid tolerance disrupt integrity of lipid rafts of the cell membranes. (A) The plasma membrane of cells is composed of a phospholipid bilayer with patches of highly organized microdomains, termed lipid rafts (green). The raft is a relatively rigid platform consisting primarily of cholesterol and sphingomyelin with long saturated fatty acyl chains. Lipid rafts are enriched in proteins, generally with transmembrane domains. The surrounding membrane domain (yellow) is more fluid, consisting of phospholipids with unsaturated fatty acyl chains and less cholesterol. PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine. (B) Schematic depiction of a potential mechanism for lipid raft-dependent signal transduction. Accessory proteins (transmembrane proteins, glycosylphosphatidylinositol (GPI)-anchored proteins, and signaling receptors), each of which is located in separate lipid rafts, associate in response to receptor activation by exposure to inflammatory cytokines or TLR ligands. This association, facilitated by the clustering of lipid rafts, quickly brings the proteins into close proximity, thereby accelerating signal transduction leading to inflammation. Exposing the cell membrane to reagents that increase membrane fluidity (e.g. ethanol, anesthetics, or PUFA) disrupts lipid rafts and interferes with their clustering, thereby blocking signal transduction and inhibiting inflammation.
CONCLUSION
In the present article, we have advanced the hypothesis that induction of tolerance to ischemia after preconditioning depends on inhibition of TLR and cytokine signaling pathways that suppresses the inflammatory response to ischemia. “Delayed tolerance” occurs when preconditioning induces the expression of endogenous feedback inhibitors of TLR and cytokine signaling. “Rapid tolerance” occurs when there is direct inhibition of TLR and cytokine signaling by structural modulation of plasma membrane lipid rafts by volatile anesthetics, ethanol, and PUFA. Table 4 compares the fundamental features of delayed versus rapid tolerance.
Basic characteristics of delayed and rapid tolerance
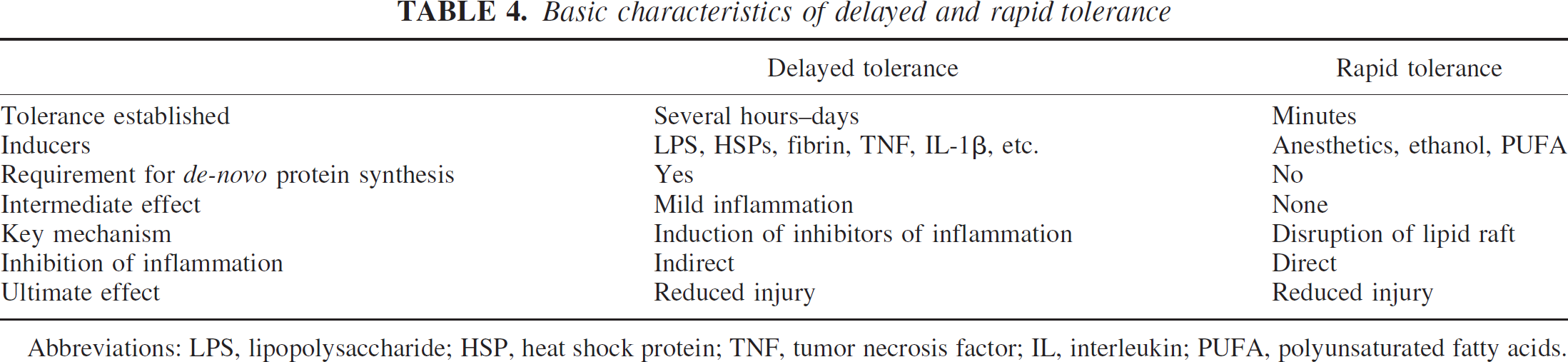
Abbreviations: LPS, lipopolysaccharide; HSP, heat shock protein; TNF, tumor necrosis factor; IL, interleukin; PUFA, polyunsaturated fatty acids.
The incentive to propose this hypothesis was prompted by several important scientific discoveries made in recent years. First, TLRs were identified as the receptors that mediate the innate immune response to microbes. Second, tissue injury releases endogenous ligands of TLRs that activate TLR and cytokine signaling, initiating inflammation. Third, TLR and cytokine signaling trigger counter-regulatory pathways, which increase the expression of signaling inhibitors, decoy receptors, and anti-inflammatory cytokines. Fourth, advances in the understanding of the molecular mechanism of endotoxin tolerance, which closely parallels ischemic tolerance, have served as a template for further investigation of cerebral preconditioning. Fifth, advances in the understanding of the molecular structure and function of lipid rafts and their role in signal transduction led to the concept of direct inhibition of TLR and cytokine signaling in models of “rapid tolerance.” Finally, the hypothesis advanced should apply to all organs because the necessary elements (TLRs, cytokines, signaling pathways) have been identified in other organ systems. Thus, we believe there is strong preliminary evidence allowing us to advance this hypothesis demonstrating the importance of TLR-mediated inflammation and its suppression in ischemic brain injury.
What is the upshot of this hypothesis with respect to therapeutic targets for clinical stroke and other forms of cerebral ischemia? The detrimental effects of inflammation on ischemic brain injury have long been appreciated. In preclinical trials, mainly in small animals, agents that reduce inflammation protect the brain against ischemia. However, in clinical trials, anti-inflammatory therapy has not been effective. Possibly, the therapeutic window for suppression of inflammatory pathways is too limited for efficacious stroke therapy in humans. One of the consequences of a better understanding of TLR and cytokine signaling and suppression will be the design of new agents more specifically targeted to these pathways. Finally, recent clinical evidence indicates that preconditioning may occur naturally in humans after transient ischemic attacks and mild strokes (Wegener et al. 2004). Thus, it is tempting to speculate whether it may be possible to precondition the human brain prior to such procedures as heart surgery, carotid endarterectomy, or invasive brain surgery, and thus increase the tolerance to ischemia. Models presented here indicate that inducing tolerance would require prior activation of TLR or cytokine signaling pathways. Alternatively, agents that directly inhibit TLR and cytokine signaling might be applied. Further investigation will determine the feasibility of these approaches.