
Abstract
Brain ischemia triggers an inflammatory reaction that progresses for days to weeks and seems to have a role in secondary progression of injury. Inflammation induces a complex pattern of signaling molecules with partly contradictory actions, and the responses may be different in the immature and adult brain. The authors characterized the global inflammatory gene expression in the developing brain as a first step toward understanding the protective and deleterious effects of inflammation after hypoxia-ischemia. Oligonucleotide arrays were used to investigate inflammatory genes in cortex, hippocampus, thalamus, and striatum at 2, 8, 24, and 72 hours after hypoxia-ischemia, which was induced in 9-day-old mice by left carotid artery ligation followed by hypoxia. After hypoxia-ischemia, 148 inflammatory genes were differentially expressed. More than 97% of the genes were upregulated and 93% had not previously been reported after hypoxia-ischemia in the immature brain. The results indicate that microglia/macrophages, T-and B-cells, NK-cells, mast cells, dendritic cells, and polymorphonuclear leukocytes may participate in the response to hypoxia-ischemia. In addition, novel cytokines/chemokines, complement-related, interferon-regulated, components of the TIR/nuclear factor-κB pathway, and a number of immunomodulatory genes were induced. Several of these genes may be of pathophysiologic significance after neonatal hypoxia-ischemia.
Inflammation is a complex process involving hundreds of signaling molecules that can arise in any tissue in response to, for example, trauma, infections, and ischemia. The initial proinflammatory phase, which may be tissue damaging, involves the recruitment and activation of diverse immune cells and their interactions with parenchymal cells. Later on in the inflammatory process, a crucial commitment converts the proinflammatory phase into an anti-inflammatory phase, promoting tissue repair and recovery (Nathan, 2002). The molecular phenotype of the immune response in CNS diseases is largely uncharacterized, but brain ischemia triggers an inflammatory reaction that progresses from days to weeks and is believed to contribute to the secondary progression of ischemic injury. Several different mediators participate in the inflammatory reaction, such as chemokines and cytokines, which are induced after ischemia in the adult and neonatal brain (Barone and Feuerstein, 1999; Bona et al., 1999; Cowell et al., 2002; Hagberg et al., 1996; Hedtjarn et al., 2002; Ivacko et al., 1997; Szaflarski et al., 1995). In addition, other inflammatory proteins, such as adhesion molecules (Frijns and Kappelle, 2002), and the activity of inducible nitric oxide synthase (Forster et al., 1999) and cyclooxygenase-2 (Nogawa et al., 1997), are also increased after ischemia.
The involvement of inflammatory mediators in the progression of ischemic injury has been shown by several studies in which neuroprotection was achieved by intervening with their functions. In the adult brain, inhibition of interleukin-1 (Loddick and Rothwell, 1996; Relton and Rothwell, 1992), tumor necrosis factor (TNF)-α (Yang et al., 1998), adhesion molecules (Connolly et al., 1997; Connolly et al., 1996; Zhang et al., 1994), and depletion of leukocytes (Heinel et al., 1994) have proven to be protective strategies after ischemia. In the neonatal brain, mice deficient in caspase-1 (Liu et al., 1999) or interleukin-18 (Hedtjarn et al., 2002) showed reduced injury after hypoxia-ischemia (HI), and inhibition of interleukin-1 by administration of interleukin-1ra decreased CNS vulnerability to HI (Hagberg et al., 1996; Martin et al., 1994).
Despite these findings, no anti-inflammatory drug has been shown to improve outcome after brain ischemia in humans (Brott and Bogousslavsky, 2000). To find efficient neuroprotective strategies based on modulation of the inflammatory response, it is important to gain a better understanding of the underlying molecular mechanisms. Inflammation is a double-edged sword in the sense that it confers both protective and deleterious effects. It is therefore necessary to characterize the molecular signals that sequentially regulate the various components of the pro- and anti-inflammatory responses, as well as the mediators responsible for repair and restitution.
Microarray analyses are useful in studying the expression of many genes in one sample, but are especially useful for studying genes involved in inflammation because the protein expression of most of the mediators involved in immune-inflammatory processes is preceded by de novo transcription. Several microarray analyses have been performed to study the gene expression after ischemic brain injury (Kim et al., 2002; Lu et al., 2003; Schmidt-Kastner et al., 2002; Tang et al., 2002), but none has focused on the inflammatory response, and in the various studies only a limited number of novel genes have been reported.
In this study, microarray analyses were performed in those regions of the gray matter where injury develops after neonatal HI (cortex, hippocampus, thalamus, and striatum), at 2, 8, 24, and 72 hours after HI, to better understand the complex nature of the inflammatory response that arises in the immature brain after HI.
In total, 148 genes belonging to the immune-inflammatory system were differentially expressed after HI. In addition to the inflammatory genes, 343 genes belonging to other functional categories were differentially expressed after neonatal HI, and are presented in our companion article in this issue. Of the inflammatory genes, 70% were previously unreported after ischemia and 93% had not been reported after HI in the immature brain.
MATERIALS AND METHODS
The methods used for neonatal HI, RNA preparation, GeneChip analysis, significance analysis of microarrays, and database searching is described in detail in our companion article.
Real-time polymerase chain reaction
The microarray data for nine genes were confirmed by real-time polymerase chain reaction (PCR), using a Light Cycler (Roche) as described in our companion article. The following primer pairs (from CyberGene AB, Huddinge, Sweden), annealing temperatures, and elongation times were used: CD44: forward: 5′-AATTCCGAGGATTCATCCCA-3′, reverse: 5′-CGCTGCTGACATCGTCATC-3′, 57°C, 12 seconds; glycoprotein 49A (gp49A): forward: 5′-TGTTCTGGATGCTGTTGCTC-3′, reverse: 5′-TGTTCAGCTCTGCATTGTCC-3′, 57°C, 13 seconds; GAPDH: forward: 5′-CATCACCATCTTCCAGGAGCG-3′, reverse: 5′-GAGGGGCCATCCACAGTCTTC-3′, 58°C, 15 seconds; lysozyme M: forward: 5′-CTGCTTATAGGAGACCAGT-3′, reverse: 5′-ACATCCTCTCAAGGGTT-3′, 55°C, 11 seconds; large multifunctional protease 7: forward: 5′-CGGGACAGATGTTTTCCACT-3′, reverse: 5′-CGGAACTCTCCACTTTCACC-3′, 57°C, 9 seconds; MCP-5: forward: 5′-AGCTTTCATTTCGAAGTCTTTG-3′, reverse: 5′-CTCCTTATCCAGTATGGTCC-3′, 56°C, 10 seconds; osteopontin: forward: 5′-ACACTTTCACTCCAATCGTCC-3′, reverse: 5′-TGCCCTTTCCGTTGTTGTCC, 58°C, 10 seconds; MIP-1 gamma: forward: 5′-TCACAACCACGGACCTACAA-3′, reverse: CTCCAGACCTTGCCCATTTA, 57°C, 12 seconds, oncostatin receptor: forward: 5′-GATGTACCCACTAAGCCGCC-3′, reverse: 5′-GAGGACCGTTGAGGTCAAGC-3′, 56°C, 17 seconds; SOCS-3: forward: 5′-TGGAGGGTTCTGCTTTGTCT-3′, reverse: 5′-GCCAATGTCTTCCCAGTGTT-3′, 57°C, 9 seconds.
Immunohistochemistry
Pups were deeply anesthetized by intraperitoneal injection of 150 μL Pentothal (50 mg/mL) and perfused intracardially with 0.9% NaCl followed by 5% buffered formaldehyde (Histofix, Histolab, Göteborg, Sweden) at 3, 10, 24, and 72 hours after HI (n = 5 at each time point). Control pups were killed at postnatal day (PND) 9, 10, and 12 (n = 3), and brains were prepared and immunohistochemistry performed as described elsewhere (Hedtjarn et al., 2002). The following primary antibodies, dilutions, and incubation times were used: rabbit anti–mouse osteopontin (O-17, IBL, Gunma, Japan; 1:400 in phosphate-buffered saline [PBS], overnight at 4°C), rabbit anti–human lysozyme (A48, Biomeda (Foster City, CA, U.S.A.); 1:1,000 in PBS, overnight at 4°C), and goat anti-GP49 (N-20; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.; 1:400 in PBS, overnight at 4°C). Before immunohistochemical staining, sections were deparaffinized and boiled in citric acid buffer (0.01 mol/L, pH 6.0, 10 minutes). For lysozyme and GP49 staining, sections were also treated with proteinase K (10 μg/mL in PBS, 7 minutes; Roche-Boehringer Mannheim, Indianapolis, IN, U.S.A.). For double-labeling experiments, Alexa-Fluor 488 or 594 streptavidin (4 μg/mL in PBS; Molecular Probes, Eugene, OR, U.S.A.) was used after incubation with biotinylated secondary antibody, or secondary antibodies directly conjugated to Alexa-Fluor 488 or 594 (5 μg/mL in PBS) were used. Microglia were detected using FITC-labeled Isolectin B4 (10 μg/mL in PBS, 1 hour, L-2895; Sigma, St. Louis, MO, U.S.A.). Preabsorption of the different antibodies with a 100x excess of the corresponding proteins/peptides (recombinant osteopontin 441-OP/CF from R&D Systems, Inc., Minneapolis, MN, U.S.A. gp49 (N-20) blocking peptide, from Santa Cruz Biotechnology; Lysozyme L-8402, from Sigma) was performed to control for the specificity of the different antibodies. The preabsorption resulted in omitted immunohistochemical stainings for the different antibodies used.
RESULTS
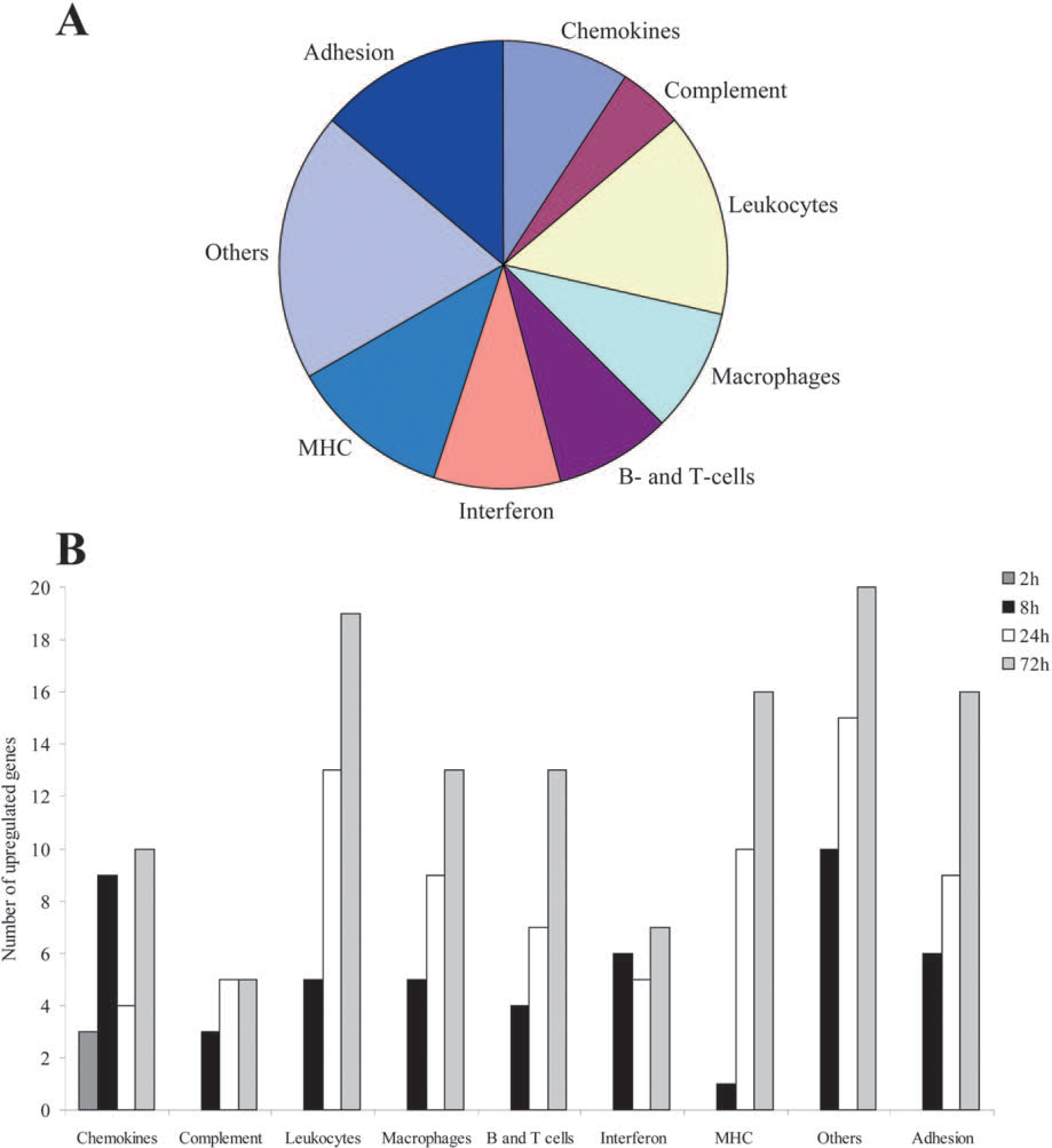
Hypoxia-ischemia in the immature brain induced the expression of a significant number of genes related to the immune-inflammatory response. In total, 148 genes and expressed sequence tags (ESTs) were differentially expressed 2 to 72 hours after HI, using the previously described criteria (false discovery rate < 10%, fold change at least 1.5, and significant change when comparing the ipsilateral hemisphere with the contralateral hemisphere and with the appropriate control). Of the differentially expressed inflammatory genes and ESTs, as many as 144 were upregulated and only four were downregulated. At 2 hours after HI only three genes were upregulated, but at 8 hours after the injury the number of significantly upregulated genes had increased to 49. At 24 hours after HI 77 genes were upregulated, and at 72 hours after HI as many as 119 genes were upregulated. Of the downregulated genes, one was found at 24 hours and three at 72 hours after HI. By searching PubMed and investigating all previous articles concerning changes in gene expression after ischemia, it was estimated that 103 (70%) of the 148 regulated genes had not previously been reported after cerebral ischemia, and 137 (93%) of the genes had not previously been reported after ischemia in the immature brain. The percentage of upregulated genes in different functional categories, and the total number of upregulated genes at each time point is shown in Figure 1.
(
Real-time polymerase chain reaction
The expression of the nine genes chosen for confirmation by PCR—MCP-5, osteopontin, lysozyme M, lmp7, oncostatin receptor, MIP-1 gamma, SOCS-3, CD44, and glycoprotein 49A—was significantly increased even when using this method (Fig. 2), indicating that there is a good agreement between the microarray analysis and real-time PCR. Not only were all genes upregulated, but the “fold change,” when comparing ipsilateral with contralateral hemisphere, was similar between PCR and microarray in that the genes with the highest fold change on the microarray analysis also had the highest fold change with PCR. However, the levels of expression were sometimes increased to an even higher extent using PCR (e.g., osteopontin and gp49A), indicating that real-time PCR is even more sensitive than microarray analysis.
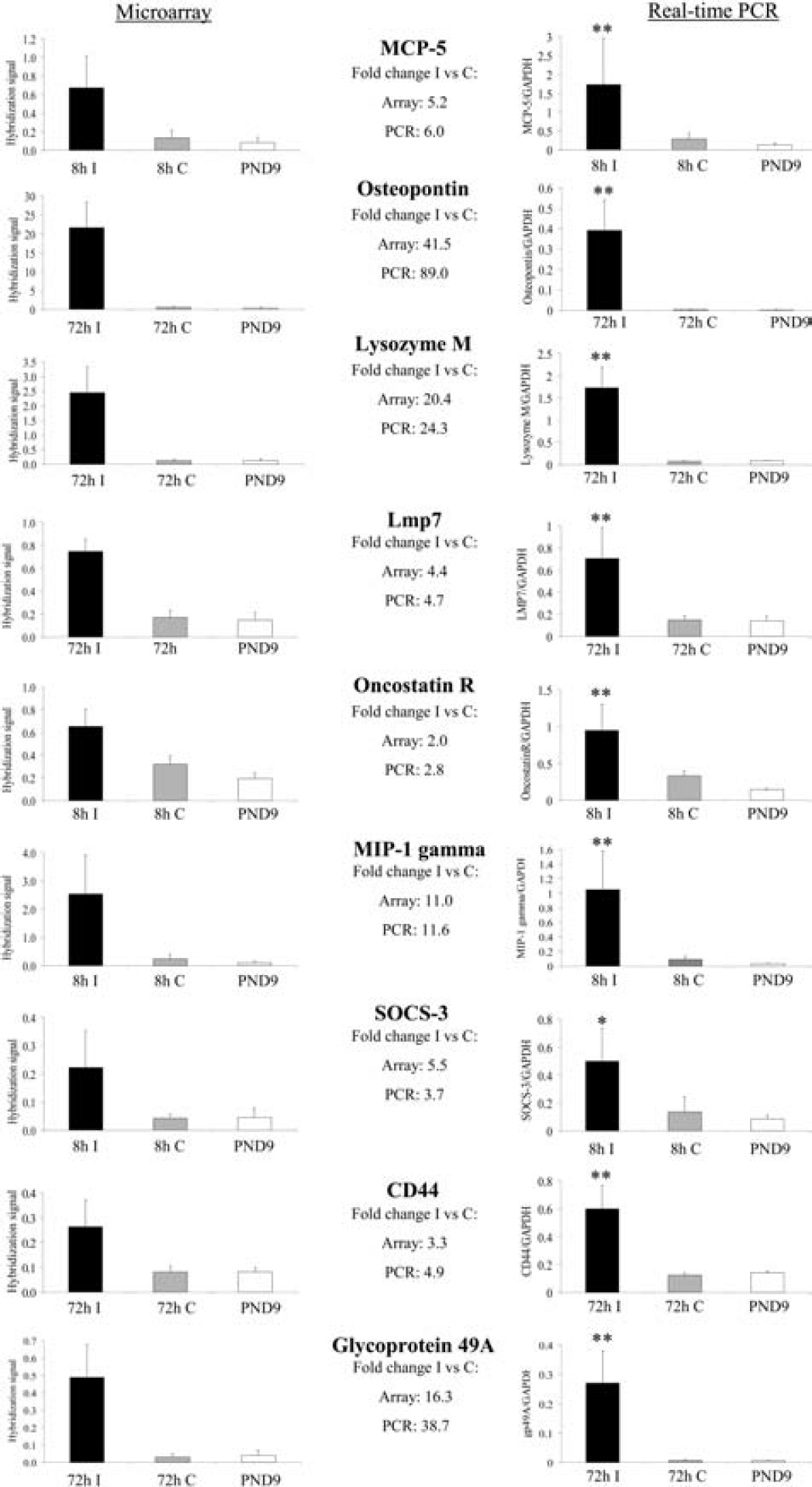
Confirmation of upregulated genes by real-time PCR. In the left column the results from the microarray analysis are shown, and the right column displays the PCR results for the same genes. Error bars indicate ± SD; **P < 0.01 and *P< 0.05 versus contralateral hemisphere using (Mann-Whitney U-test). I, ipsilateral hemisphere; C, contralateral hemisphere; PND9, control at postnatal day 9; n = 5 in each group.
Immunohistochemistry
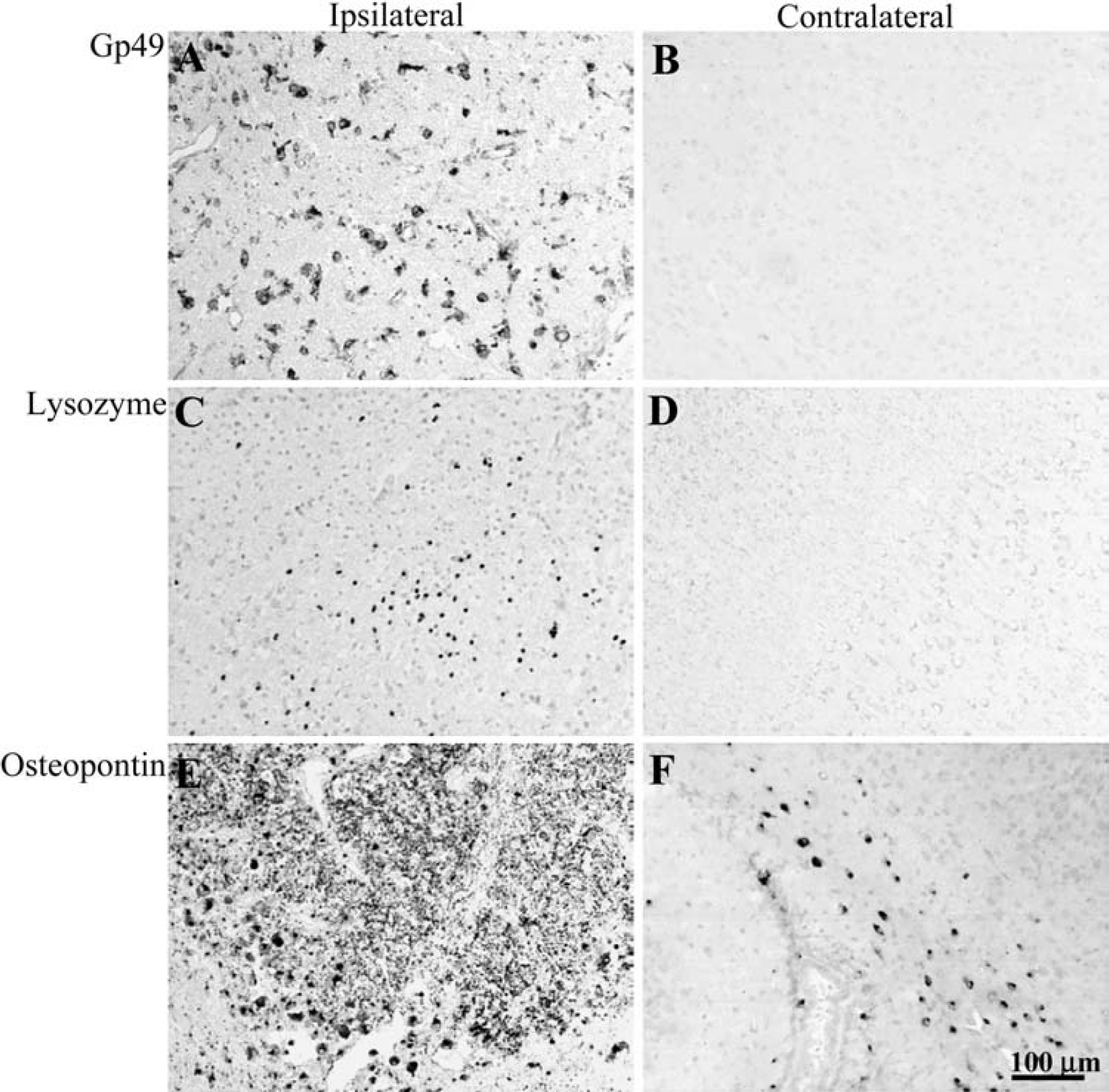
Immunoreactivity of glycoprotein 49 (gp49), lysozyme, and osteopontin after HI. (
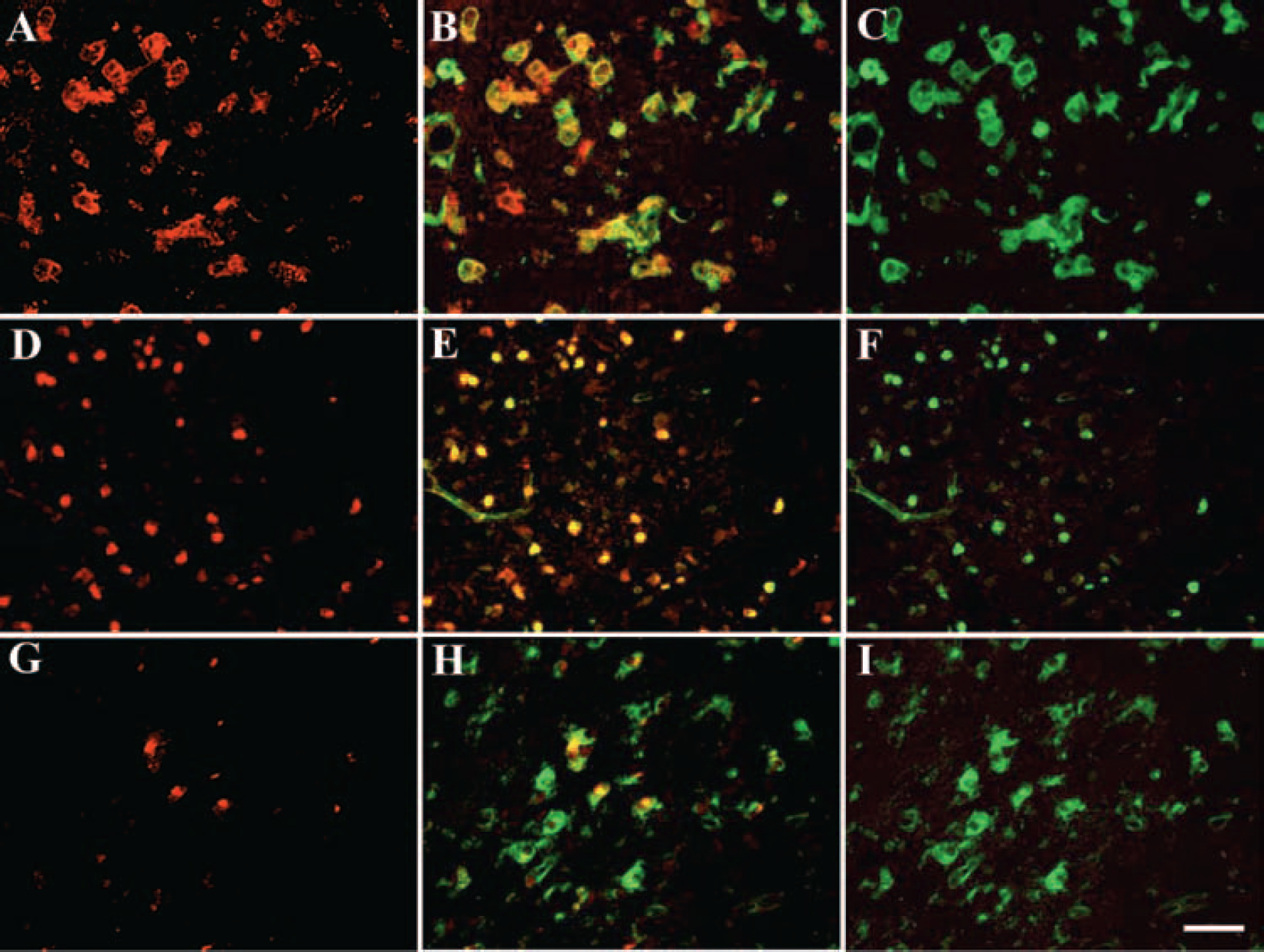
Double-labeling immunofluorescence of gp49 (
Functional categories of differently expressed genes
The differentially expressed genes were broadly divided into nine different functional categories (Tables 1–9) based on their function and cellular expression. The genes that were already induced at 2 hours after HI belonged to the chemokine family. Otherwise, the upregulation of genes in all other functional groups started at 8 hours after HI, and the amount of upregulated genes in each category continued to increase further at 24 and 72 hours after HI in a quite similar pattern, with two exceptions. The interferon-regulated genes showed an elevated and quite stable expression from 8 to 72 hours after HI, and in the group of MHC class I– and II–related genes, only one gene was induced at 8 hours, whereas a quite extensive increase was detected at 24 and 72 hours after the injury (Fig. 1). The downregulated genes were found in the categories chemokines, “others,” and adhesion-related genes.
Chemokines
Several chemokines that are known to be induced by ischemia in both adult and immature brain, such as MIP-1 alpha, MIP-1 beta, MCP-1, and GRO, as well as some of their receptors (e.g., CCR2 and CCR5), were upregulated on the array. In addition, MCP-3, IP-10, and the chemokine receptor CX3CR1, all of which have previously been reported after ischemia in the adult but not the immature brain, were upregulated. MIP-1 gamma, MCP-5, eotaxin, and chemokinelike factor superfamily 3 were induced by HI, and have not previously been reported after ischemia in the brain (Table 1).
Complement-related genes
A number of genes involved in the complement system were induced by HI in the immature brain, none of which has previously been reported after neonatal HI. Complement component 3a receptor 1, complement component 5 receptor 1, complement component 1, q subcomponent, beta polypeptide, and complement C4 precursor have been reported after adult ischemia, but complement component 1, q subcomponent, c polypeptide, complement component 1, q subcomponent, receptor 1, and complement factor H-related protein are newly reported genes (Table 2).
Genes expressed by leukocytes
Twenty-one different genes and ESTs known to be expressed by leukocytes were induced by HI in the immature brain, and only two of them, lysozyme M and Fc gamma RIIB, have been reported after ischemia. At 8 hours after HI, an upregulation of cysteine-rich intestinal protein, glycoprotein 49A, L-plastin, cell surface glycoprotein 53, and retinoic acid–inducible E3 protein was detected, and these genes stayed upregulated until 72 hours after the injury. Ly-6A.2 and paired-Ig-like receptor A3 were transiently induced at 24 hours after HI, whereas glycoprotein 49B, lymphocyte cytosolic protein 2, TYRO protein tyrosine kinase binding protein, lysozyme P, CD52 antigen, and Fc gamma RI were induced at 24 hours and further upregulated at 72 hours after HI. At 72 hours after HI an upregulation of hemopoietic cell kinase, lymphocyte-specific protein 1, CSF-3 receptor, FcR-gamma, dedicator of cytokinesis 2, and immunoglobulin superfamily, member 7 was detected (Table 3).
Macrophage-related genes
Five macrophage-related genes were identified after HI that had not previously been reported after ischemia: capping protein gelsolin-like, macrophage metalloelastase, and natural resistance–associated macrophage protein 1 were induced 24 hours after HI and further upregulated at 72 hours, whereas macrophage scavenger receptor 1 and ADAM-8 were upregulated 72 hours after the injury. CD68 antigen, mac-2 antigen, M-CSF, and osteopontin, which previously have been reported after ischemia in the adult but not immature brain, were induced at 8 hours and stayed upregulated until 72 hours after HI; CD63 antigen was induced at 24 hours and lymphocyte antigen 71 and colony stimulating factor 1 receptor were upregulated at 72 hours. CD14 was the only gene that had previously been shown to be induced after ischemia in both adult and immature brain; its expression was upregulated at 8 hours after HI, and the levels stayed increased until 72 hours after HI (Table 4).
Genes related to B- and T-lymphocytes
Thirteen genes related to B- and T-lymphocytes were induced after HI, none of which had previously been reported after cerebral ischemia. CD72 antigen, which is important in B-cell proliferation and differentiation, was induced at 8 hours with a subsequent increase in expression at 24 and 72 hours after HI, whereas other B-cell–related genes were induced at 24 hours (e.g., Bruton agammaglobulinemia tyrosine kinase and lyn) and at 72 hours (CD22) after HI. CD45R, which is an important regulator of T- and B-cell antigen receptor signaling, and NF-ATc, a transcription factor involved in B- and T-cell activation, were induced at 8 hours after HI; vav-1 oncogene, which is involved in B- and T-cell development and activation, was induced at 24 hours after HI. Lymphocyte antigen 9, cytotoxic T-lymphocyte–associated protein 2 alpha and beta, cytoplasmic tyrosine kinase, CD48 antigen, and lymphocyte antigen 57 are other T-cell–related genes induced by neonatal HI (Table 5).
Chemokines
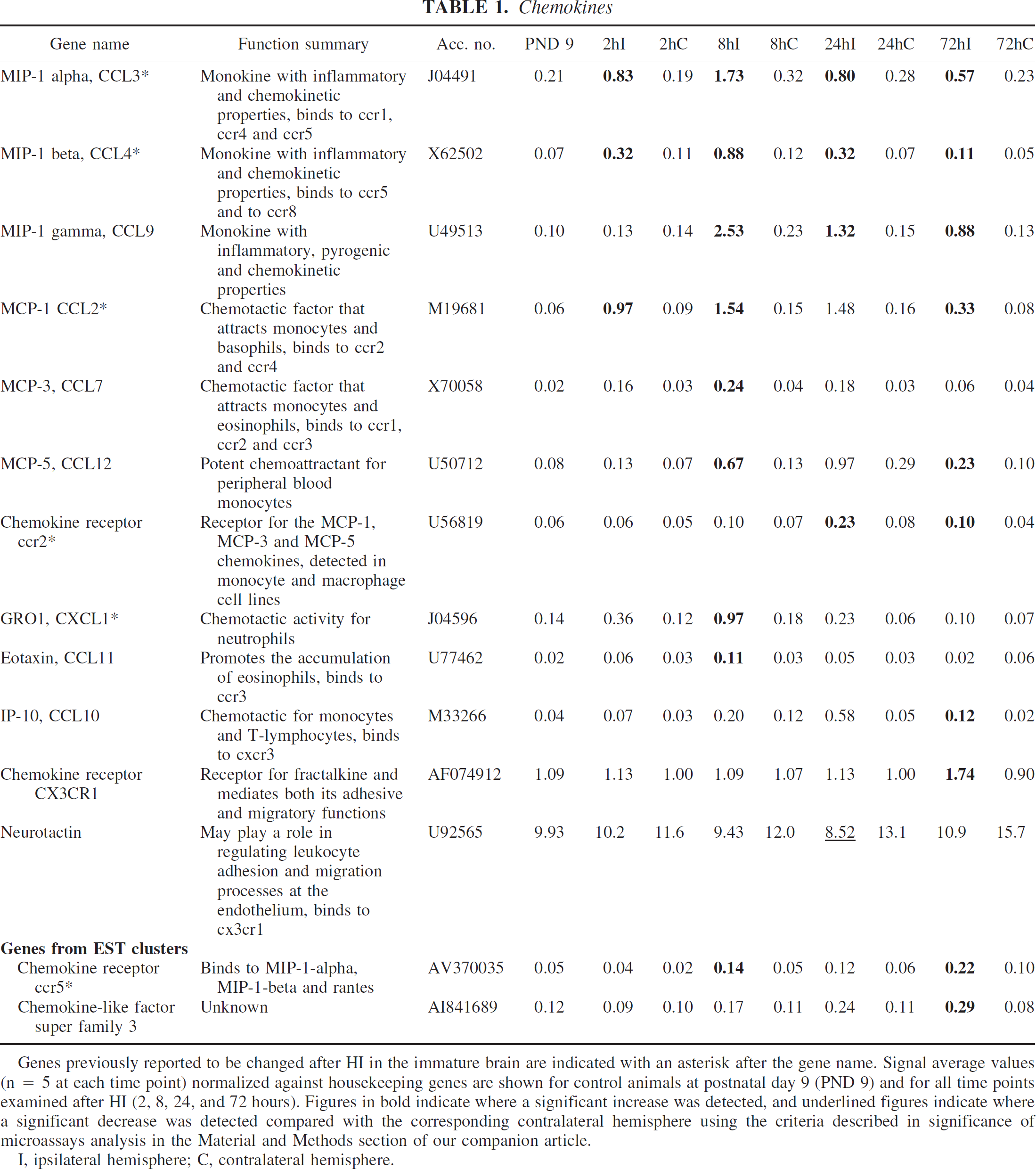
Genes previously reported to be changed after HI in the immature brain are indicated with an asterisk after the gene name. Signal average values (n = 5 at each time point) normalized against housekeeping genes are shown for control animals at postnatal day 9 (PND 9) and for all time points examined after HI (2, 8, 24, and 72 hours). Figures in bold indicate where a significant increase was detected, and underlined figures indicate where a significant decrease was detected compared with the corresponding contralateral hemisphere using the criteria described in significance of microassays analysis in the Material and Methods section of our companion article.
I, ipsilateral hemisphere; C, contralateral hemisphere.
Complement
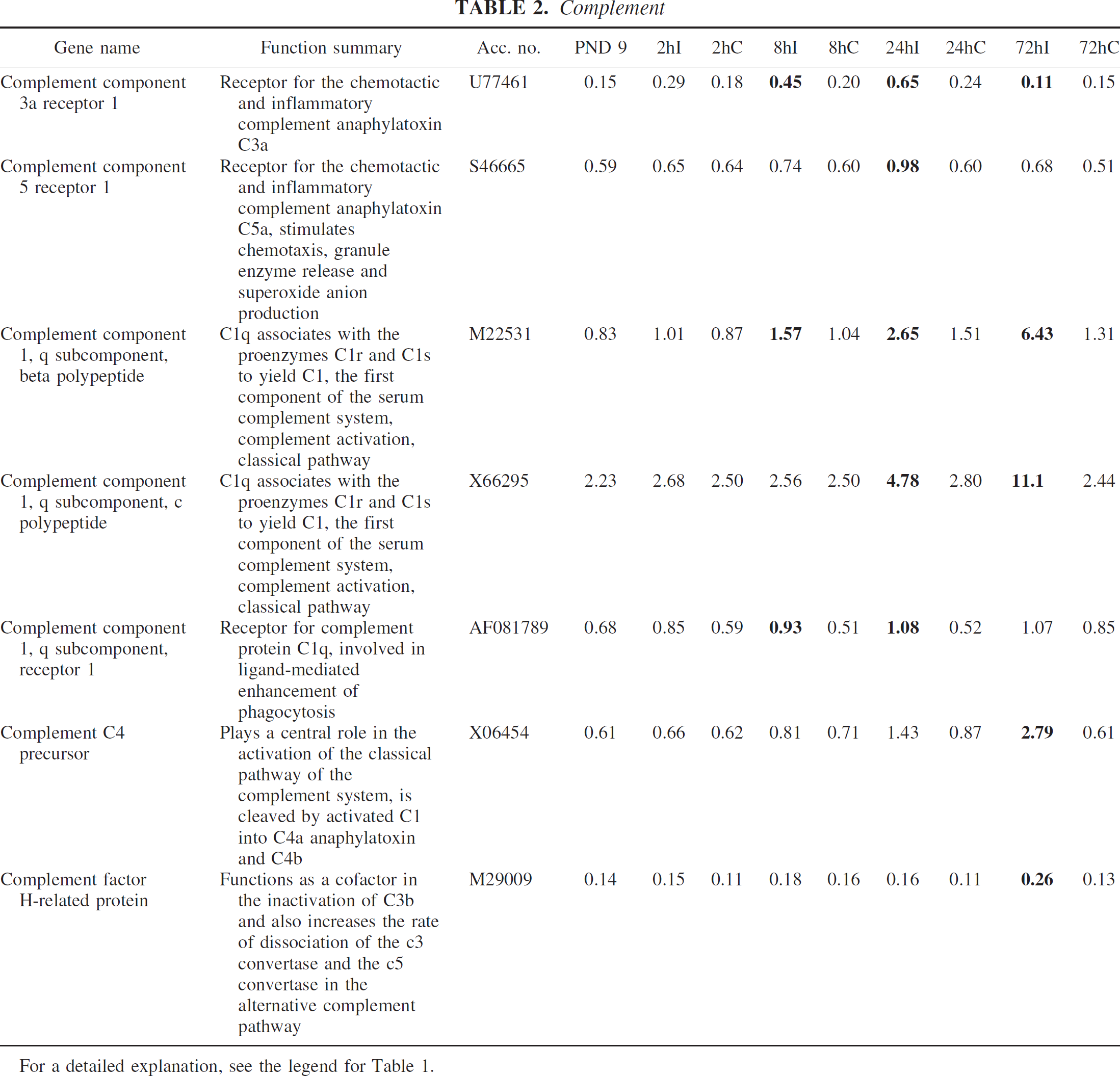
For a detailed explanation, see the legend for Table 1.
Genes induced or regulated by interferons
Of the 12 genes in this category, only one, interferon-related developmental regulator 1, had previously been described after ischemia in the immature brain; the others were previously unreported. Several genes, including interferon-γ-induced GTPase, interferon-induced protein with tetratricopeptide repeats (Ifit) 1, guanylate nucleotide binding protein (GBP) 2 and 3, and interferon-γ-inducible protein 30, were induced at 8 hours, and all except for interferon-γ-induced GTPase remained upregulated at 72 hours after HI. Interferon-activated gene 205, Ifit-2, fragilis, and interferon-inducible protein were transiently upregulated at 24 hours, whereas interferon-inducible protein 1, Ifit-3, and interferon-induced 35-kd protein homolog were induced 72 hours after the insult (Table 6).
Major histocompatibility complex class I– and II–related genes
Several genes related to major histocompatibility complex (MHC) class I receptors, such as MHC (A.CA/J (H-2K-f) class I antigen, histocompatibility 2, K region, T region locus 17, D region locus 1, Q region locus 2, beta-2 microglobulin, Q8/9d gene, MHC class I related, and hemochromatosis were upregulated after HI. In addition, lmp-2 and lmp-7, MDR/TAP, tapasin, and tap-binding protein, which are involved in the processing and transportation of antigens for presentation in these receptors, were also induced. Ia-associated invariant chain, histocompatibility 2, class II antigen A alpha and beta1 and histocompatibility 2, class II antigen E beta, which are involved in presentation of antigens in MHC class II molecules, were also induced. All genes except for Ia-associated invariant chain were previously unreported (Table 7).
Genes expressed by leukocytes
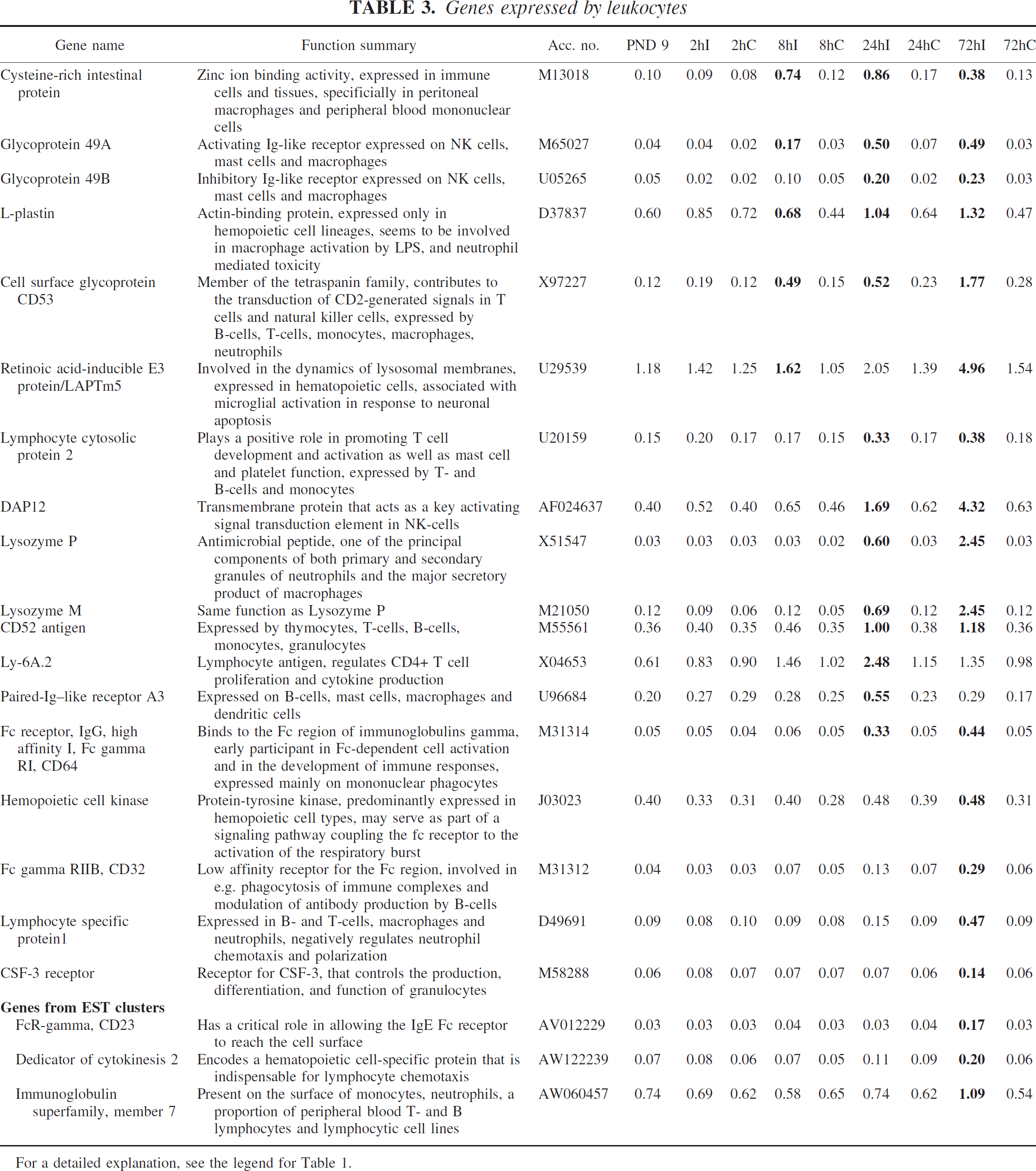
For a detailed explanation, see the legend for Table 1.
Other genes involved in immune and inflammatory responses
Several previously unreported genes were induced in this category, such as MD-1, RP105, Myd88, cyclophilin C–associated protein, lipopolysaccharide-induced TNF (involved in toll-like receptor signaling and/or lipopolysaccharide responses), oncostatin M receptor (interleukin-6–signaling pathway), p22 phox, gp91 phox and p67 phox (subunits of the NADPH oxidase of phagocytes), proteoglycan secretory granule, galectin-9, interleukin-4 receptor alpha, granulin, TNF intracellular domain–interacting protein, caveolin-1, GAPR-1, and VHSV-induced gene 1. Some genes previously reported after ischemia in the adult brain were TNF receptor superfamily member 1a, pentaxin-related gene, SOCS-3, lipocortin-1, tissue inhibitor of metalloproteinase 1, iba1, cox-1, and gp130 (Table 8).
Macrophage-related genes
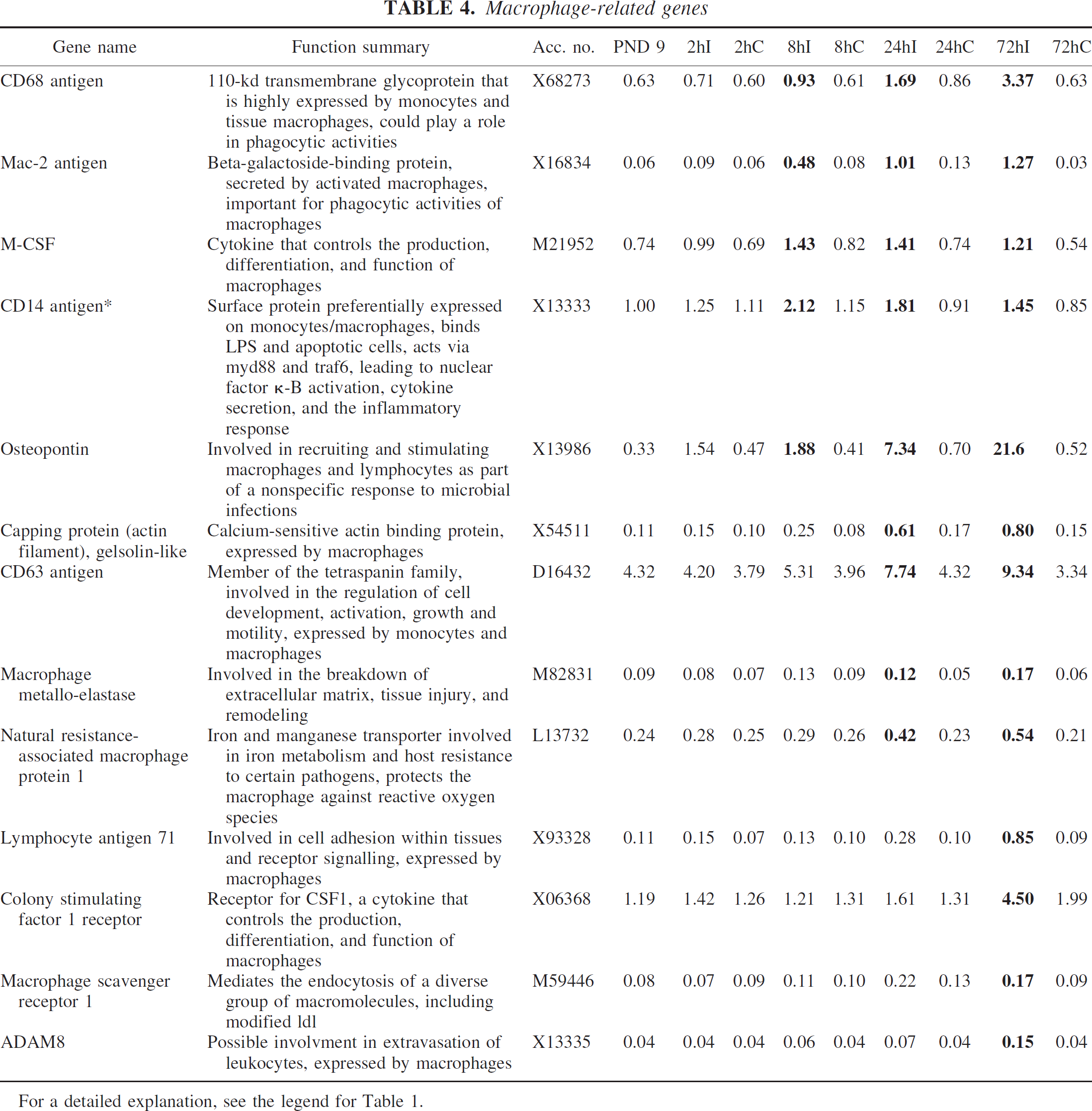
For a detailed explanation, see the legend for Table 1.
Genes related to adhesion
Hypoxia-ischemia in the immature brain induced several genes related to adhesion that previously had been reported after adult ischemia, such as CD44 antigen, ICAM-1, decorin, thrombospondin 1, integrin beta 2, and CD9 antigen. In addition, a number of new genes were also identified, such as VCAM-1, catenin alpha 1, cell adhesion kinase, c-Ccl–associated protein CAP, integrin beta 5, glycam-1, carboxypeptidase X1, and several different members of the procollagen family (Table 9).
DISCUSSION
In this study, 148 inflammatory genes were differentially expressed from 2 to 72 hours after neonatal HI. Of these genes, 30% had previously been shown to be induced by ischemia in the adult brain, whereas only 7% had previously been reported after neonatal HI.
Genes related to B- and T-lymphocytes
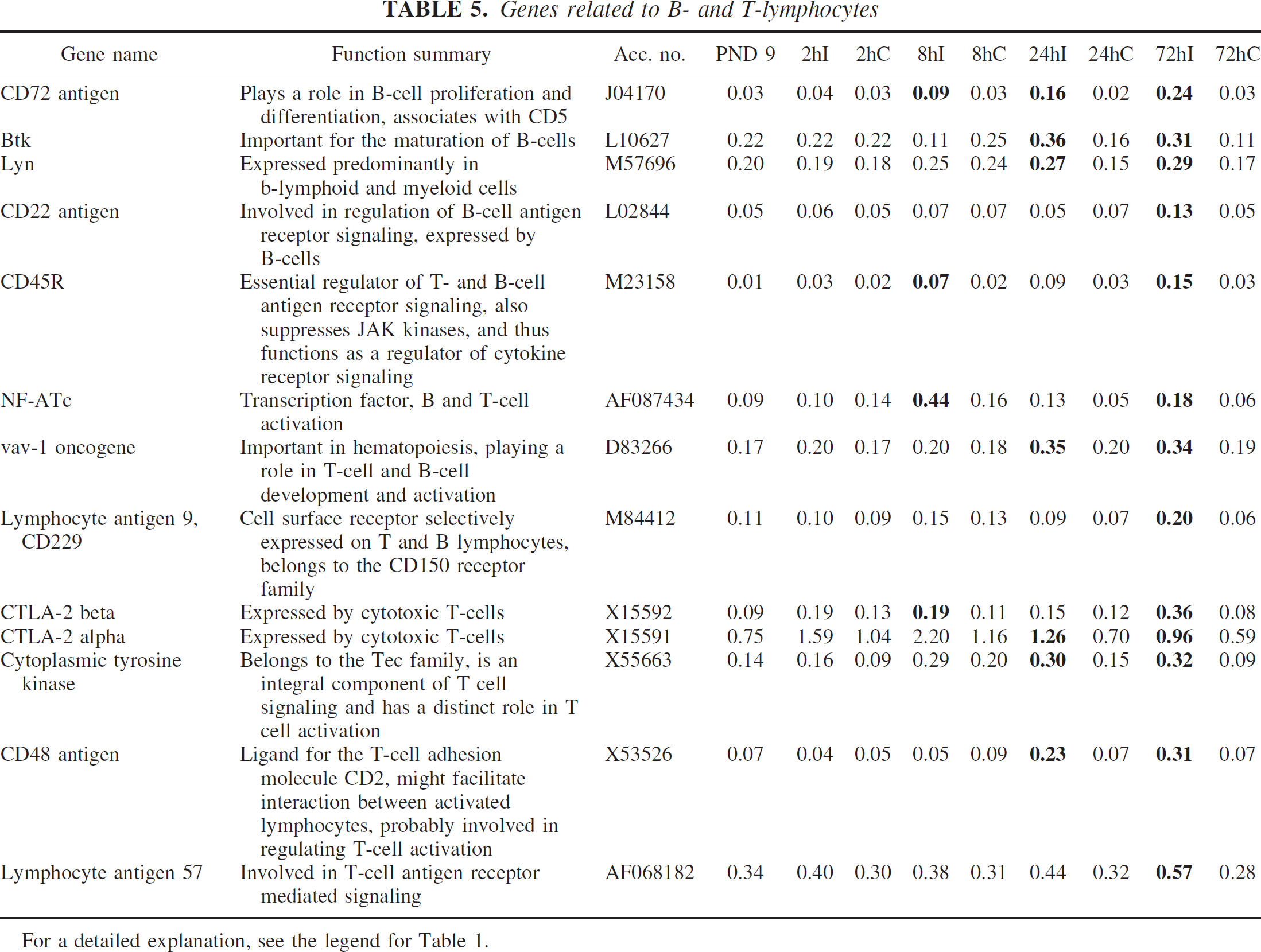
For a detailed explanation, see the legend for Table 1.
Because the injury after HI develops over 1 or 2 days (as discussed in our companion article), the changes in gene expression correspond to developing brain damage (including all cellular elements) in brain tissue sampled at 2 hours, 8 hours, and to some extent 24 hours, whereas tissue at 72 hours (and partly at 24 hours) corresponds to partly damaged tissue with fewer neurons and a higher contribution from glia in particular but also from other inflammatory cells. These cells are known to secrete a diverse subset of pro- and/or anti-inflammatory mediators, which are important in regulating the inflammatory response after ischemia. In addition to their important function in directing the inflammatory response either to sustained inflammation or to tissue repair, they may also influence neurons and glia in the nearby periinfarction area. Only investigating genes in the possible periinfarction area would leave out a lot of important information about changes in gene expression that may be involved in the inflammatory process after neonatal HI. The inclusion of injured (and partly injured) tissue in the analysis may be one further explanation for the large number of newly identified inflammatory genes in this study, in addition to the factors discussed in our companion article.
The results from this microarray analysis were supported by the fact that the induction of nine genes on the array was confirmed with real-time PCR; the protein expression for gp49, osteopontin, and lysozyme was confirmed by immunohistochemistry; and that some of the genes had previously been reported after neonatal HI. More than 97% of the genes were upregulated, showing that an extensive induction of genes belonging to the immune-inflammatory system occurs after HI. The induced genes belong to diverse categories, indicating that the inflammatory response after HI involves the activation of many different inflammatory mediators, which may be of importance in the injurious or repair process after HI. The functions and potential roles for some of the genes induced after neonatal HI will be discussed further.
Genes induced or regulated by interferons
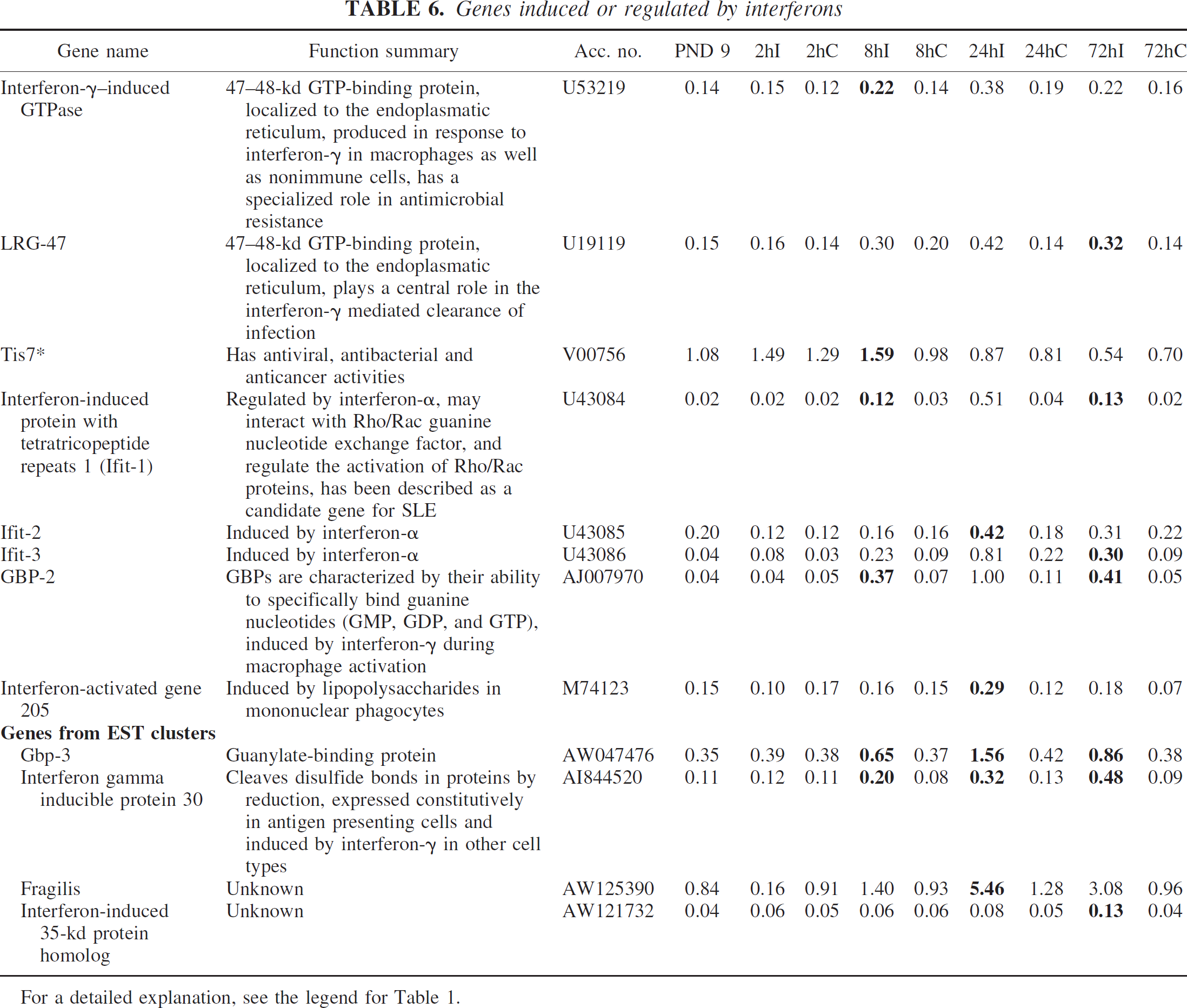
For a detailed explanation, see the legend for Table 1.
Macrophages/microglia
Retinoic acid–inducible protein E3/LAPTm5 was induced 8 hours after HI and increased even further at 24 and 72 hours after HI. LAPTm5 is a lysosomal protein expressed in microglia that is upregulated in response to neuronal apoptosis in vitro (Origasa et al., 2001). Interestingly, the increase in LAPTm5 seems to occur before the cells die, suggesting that it may be a prerequisite for phagocytosis of dead neurons. Speculatively, it could be involved in the process of converting resting microglia into amoeboid-activated microglia with phagocytic activity. Glycoprotein 49A and 49B are two closely related immunoglobulin-like receptors expressed on the cell surface of mast cells, NK cells, and macrophages. Glycoprotein 49B has immunoreceptor tyrosine-based inhibitory motifs and can inhibit mast cell and macrophage function (Katz, 2002; Matsumoto et al., 2001), whereas gp49A lacks these motifs and has stimulatory functions on mast cells (Lee et al., 2000). The effect of gp49A on macrophages is not known. We found expression of gp49 (A + B) in microglia at 24 and 72 hours after HI in the injured part of the brain. The increase in gene expression of gp49A preceded that of gp49B, suggesting that gp49A may have a stimulatory role on microglia, which subsequently is counteracted by gp49B. Capping protein, gelsolin-like (CapG), a protein involved in actin-based motility in nonmuscle cells, was induced after HI. CapG is required for receptor-mediated ruffling, facilitates phagocytosis, and accelerates the motility of vesicle rockets in macrophages (Witke et al., 2001), suggesting that it may be important in microglial function and activation after HI. Macrophage metalloelastase/matrix metalloproteinase 12 (MMP-12) was induced 24 hours after HI. MMPs are proteases involved in the remodeling of the extracellular matrix. MMP-12 upregulation after spinal cord trauma contributes to the development of secondary injury (Wells et al., 2003), indicating that its expression after HI may increase injury. Lysozyme, one of the principal proteins of phagocytes, is expressed in neutrophils (Cramer and Breton-Gorius, 1987) and macrophages (Gordon et al., 1974). Mice have two genes for lysozyme, lysozymes M and P, both of which were upregulated at 24 and 72 hours after HI. Lysozyme was expressed in a subpopulation of microglia with amoeboid and activated morphology at 24 and 72 hours after HI, implying that they were phagocytotic. Lysozyme is an enzyme with peptidoglycan as a common substrate (Ganz et al., 2003), but it also has bacteriocidal activity independent of its enzymatic activity (Ibrahim et al., 2001). The increased expression of lysozyme in the absence of bacteria suggests that under some circumstances it may also have tissue-damaging effects. Osteopontin is an adhesive glycoprotein that interacts with αvβ3 integrins (Liaw et al., 1995) and is upregulated in microglia after ischemia in the adult brain (Ellison et al., 1998; Lee et al., 1999; Wang et al., 1998). The function of osteopontin after stroke is not known, but it may be involved in the repair process (Ellison et al., 1998). A recent publication reports that osteopontin is transiently expressed in activated phagocytic microglia in the embryonic and early postnatal brain (Choi et al., 2004), suggesting that osteopontin is important for microglial migration, activation, and phagocytosis in the immature brain after HI.
MHC class I– and II–related genes
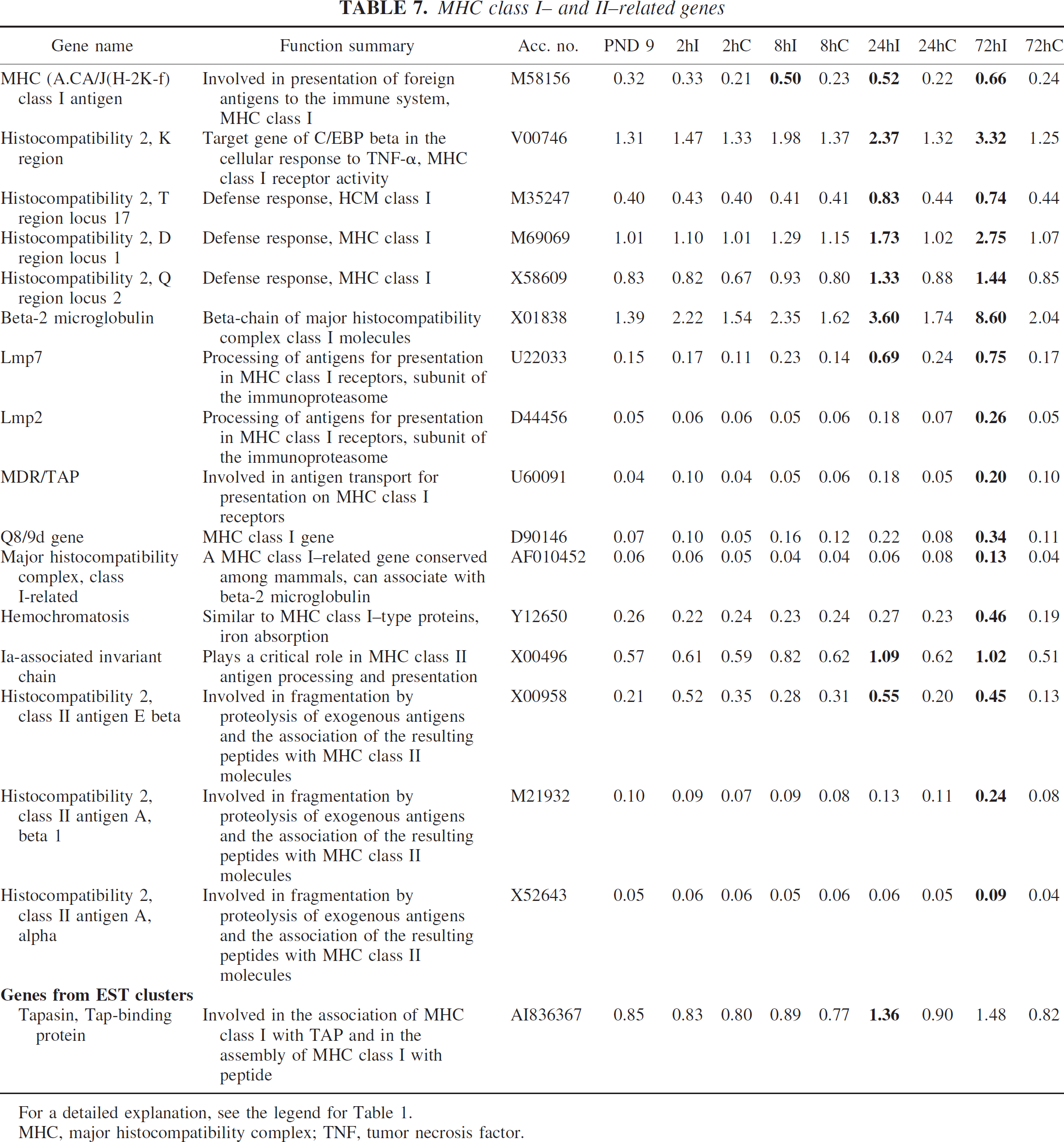
For a detailed explanation, see the legend for Table 1.
MHC, major histocompatibility complex; TNF, tumor necrosis factor.
Other genes involved in immune and inflammatory responses
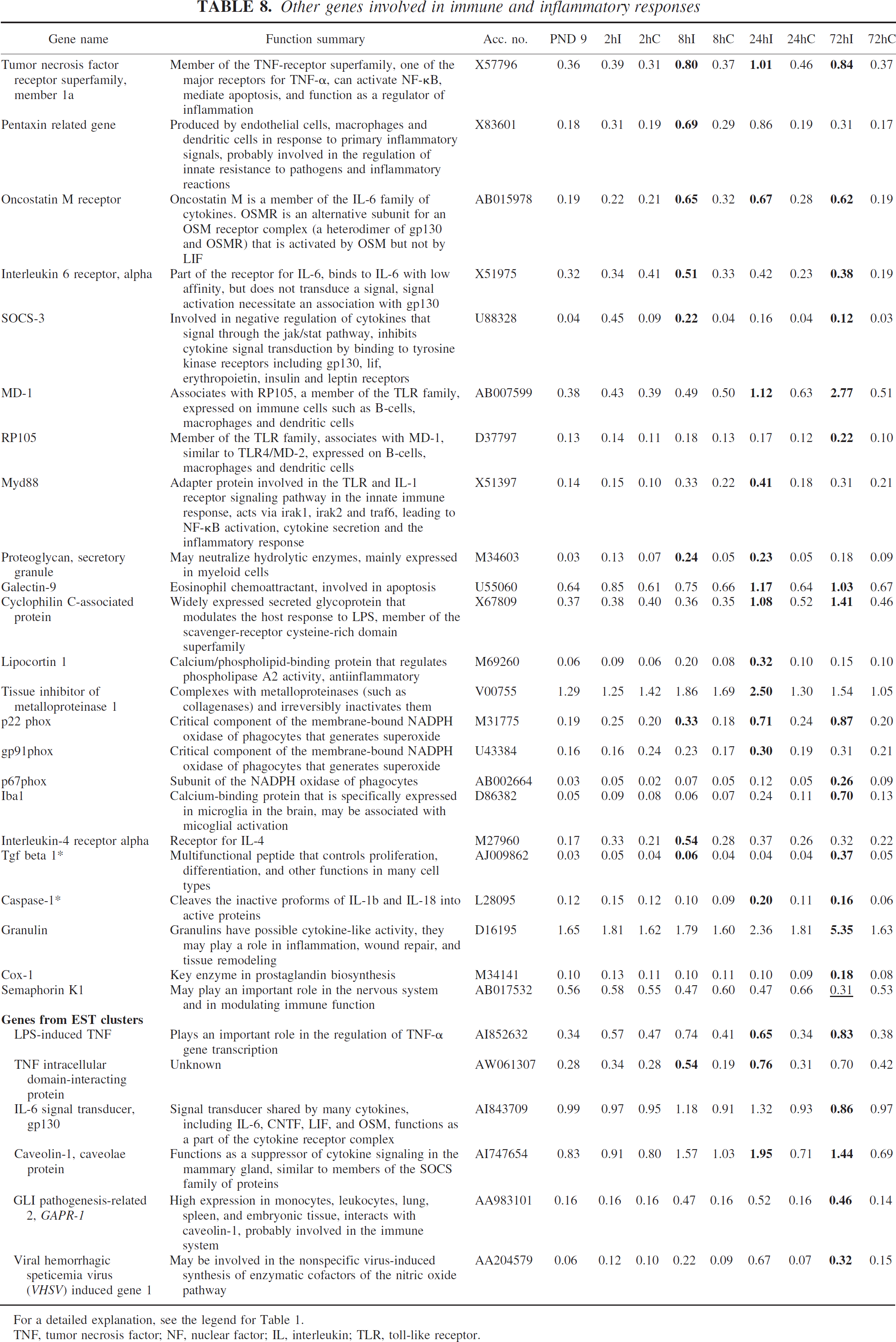
For a detailed explanation, see the legend for Table 1.
TNF, tumor necrosis factor; NF, nuclear factor; IL, interleukin; TLR, toll-like receptor.
Interferon-regulated genes
The response of cells to interferon-γ is dominated by the induction of two families of GTPases, the 47-kd family and the guanylate-binding proteins (GBPs) (Boehm et al., 1998). Members of both of these families were found to be upregulated after HI. Interferon-γ-induced GTPase, which seems to have a role in cell survival (Zhang et al., 2003), was induced 8 hours after HI, and LRG-47, which is involved in the defense against protozoan and bacterial infections (Collazo et al., 2001; MacMicking et al., 2003), was also induced. GBP 2 and 3 were upregulated 8 hours after HI, and their expression levels remained increased until 72 hours after the insult. GBPs are induced by lipopolysaccharide or interferon-γ in macrophages and microglia (Han, 1999; Vestal et al., 1996). The function of GBPs in microglia and macrophages remains obscure, but GBP-2 promotes growth in fibroblasts (Gorbacheva et al., 2002). In addition, the induction of GBP-3 by interferon-γ and lipopolysaccharide in macrophages seems to be mediated through different signal-transduction pathways (Han, 1999), suggesting a role for GBP 2 and 3 in microglial activation and proliferation after HI. Several interferon-α-inducible genes, such as interferon-induced protein with tetratricopeptide repeats (Ifit) 1, 2, and 3, were also induced by HI, but their function remains to be elucidated.
T- and B-cells
CD72, a transmembrane glycoprotein expressed by B-cells and involved in B-cell proliferation (Subbarao and Mosier, 1983), was induced 8 hours after HI and further upregulated at 24 and 72 hours. Bruton agammaglobulinemia tyrosine kinase (btk), which is important for normal B-cell development (Desiderio, 1997) and the genes lyn and CD22, which have been implicated in both positive and negative signaling pathways in B-lymphocytes, were also induced (Tsubata, 1999). CD48, a member of the immunoglobulin family that is expressed in almost all B- and T-cells and participates in T-cell activation (Blazar et al., 1998; Kato et al., 1992), vav-1 and NF-AT, factors involved in B- and T-cell activation/signaling (Deckert et al., 1996; Katzav, 2004), Ly-9, a T- and B-lymphocyte–specific cell surface receptor (de la Fuente et al., 2001), and cytotoxic T-lymphocyte–associated protein 2 alpha and beta were all induced, indicating that B-and T-cells are involved in the inflammatory process after HI.
Chemokines
In this study, we found that a number of chemokines (MCP-1, MIP-1 alpha, MIP-1 beta, GRO) and their receptors (CCR2, CCR5) are upregulated after HI, which confirms previous findings (Bona et al., 1999; Cowell et al., 2002; Ivacko et al., 1997). In addition, multiple beta-chemokines (MCP-3, MCP-5, MIP-1 gamma, IP-10, eotaxin) and the fractalkine receptor (CX3CR1) were induced in the immature brain, which has not previously been demonstrated. Chemokines seem to play a critical role in ischemic brain injury for example a broad-spectrum antagonist of chemokine receptors attenuates brain injury (Minami and Satoh, 2003). The mRNA for MCP-3 (CCL7) increased 12 times at 8 hours and MCP-5 (CCL12) increased eight times at 8 to 72 hours of recovery. MCP-5 binds preferentially to CCR2 (which was also upregulated) and is considered homologous to human MCP-1 (Sarafi et al., 1997), whereas MCP-3 binds to CCR1, CCR2, and CCR3 receptors (Tran and Miller, 2003). The mRNA expression of MIP-1 gamma (CCL9) increased 25-fold at 8 hours of recovery and remained elevated for at least 3 days after HI. Its significance in the immature brain remains uncertain, but it activates CCR1 just as MIP-1 alpha, and the latter contributes to ischemic injury in the adult brain (Takami et al., 2001). In summary, these beta-chemokines are emerging as key players in attracting and activating microglia/macrophages in the CNS, and the discovery that new members are induced may be important.
Complement
Transcripts of several previously undetected components of the classical and alternative pathways were induced after HI. This may indicate that the complement system is involved in HI; recently, Cowell et al. (2003) indeed found signs of complement activation after HI, and reported that a complement-depleting agent reduced the lesion size.
Genes related to adhesion
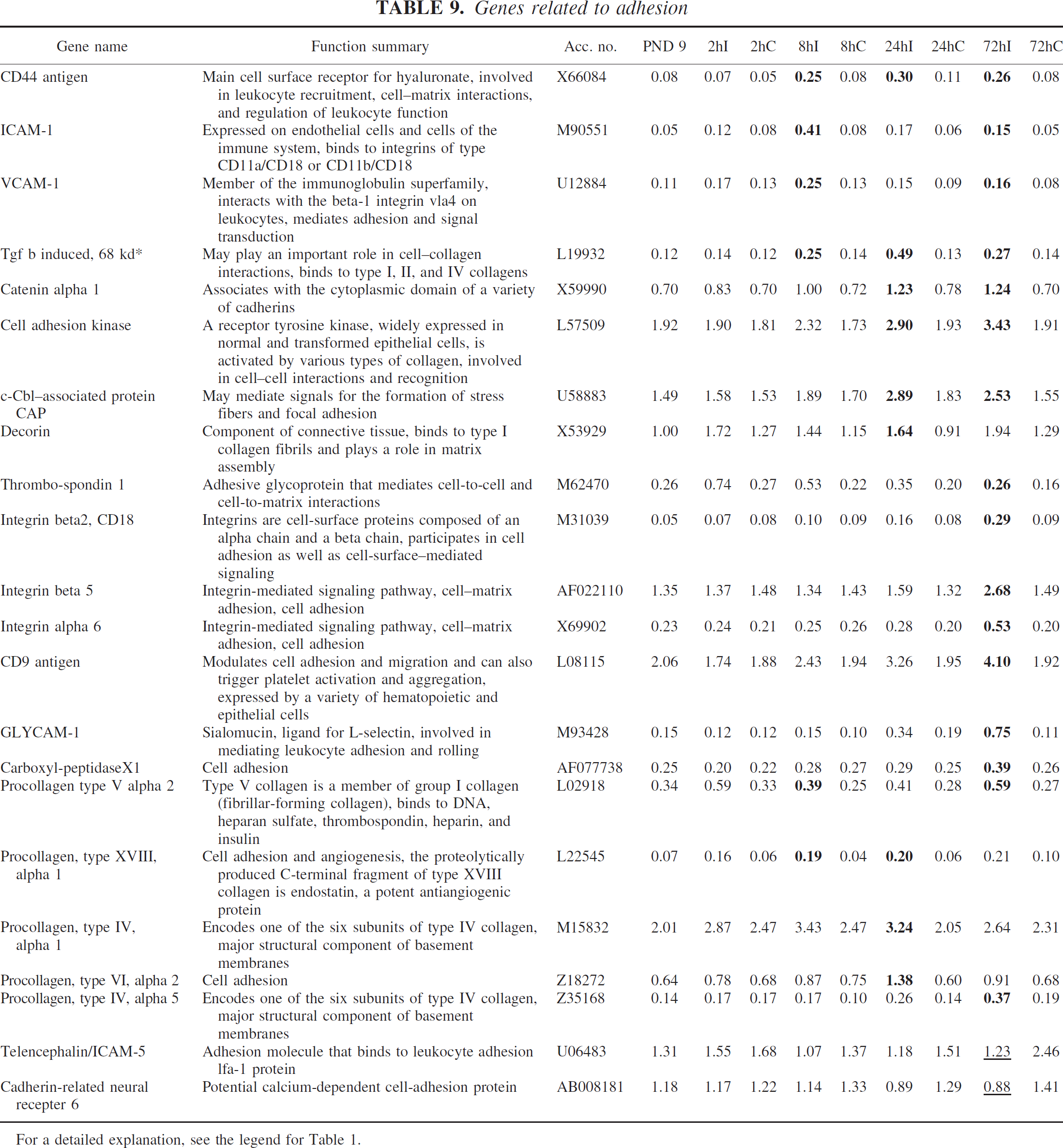
For a detailed explanation, see the legend for Table 1.
Activation of the TIR/nuclear factor-κB pathway
Interleukins 1 and 18 were produced (Hagberg et al., 1996; Hedtjarn et al., 2002), IkB (inhibitor of nuclear factor-κB) was induced (data not shown), and the expression of several of the molecular components of the TOLL-like receptor (TLR) complex (MD-1, RP105, MYD88) was increased after HI. These data suggest that the common cytoplasmic portion of TOLL-like and interleukin-1 receptors (called the TIR domain) is activated after HI, leading to the induction of nuclear factor-κB (Nguyen et al., 2002). Interleukins 1 and 18 seem to play important roles in HI in that genetic deficiency of caspase-1 or interleukin-18 and the administration of interleukin-1 R antagonist confer resistance in the immature brain (Hagberg et al., 1996; Hedtjarn et al., 2002; Liu et al., 1999). In addition, activation of TLRs (with lipopolysaccharide) before HI seems to enhance CNS vulnerability (Eklind et al., 2001) and it has been suggested that TLRs on microglia are responsible for this effect (Lehnardt et al., 2003). Besides bacterial products, TLRs can be activated by molecules released from injured cells (e.g., CG dinucleotide motif–rich DNA can induce neuronal injury through activation of TLRs on microglia; Iliev et al., 2003). DAP12, a tyrosine protein kinase binding protein originally described in NK cells (Lanier et al., 1998) but later also found in neutrophils, macrophages, and microglia (Bakker et al., 2000; Lucas et al., 2002), was upregulated at 24 hours (threefold) and 72 hours (sevenfold) after HI. Overexpression of DAP12 seems to initiate inflammatory reactions in the absence of microbial challenge (Lucas et al., 2002), and DAP12 signaling may augment the lipopolysaccharide response through the TLR4/Myd88 pathway (Nathan and Ding, 2001), suggesting that the increase of DAP12 after HI leads to enhanced inflammation and injury. Another example of activation of the TIR domain/nuclear factor-κB is the induction of pentaxin related gene (PTX3), a soluble pattern recognition receptor belonging to the lectin family (Kilpatrick, 2002). PTX3 is induced by nuclear factor-κB, preferentially in dendritic cells, during the early phase of inflammation by interleukin-1β, TNF-α, and by TLR activation (Doni et al., 2003). Besides amplification of innate immunity in the recognition of microbes, PTX3 has other functions, such as clearing self-components (prevention of autoimmunity) and decreasing dendritic cell recognition of apoptotic cells (Mantovani et al., 2003). Cysteine-rich intestinal protein (CRIP) is a double-zinc finger LIM protein, expressed among others in peritoneal macrophages and peripheral blood mononuclear cells (Hallquist et al., 1996), that was increased sixfold at 8 hours after HI. The expression of CRIP can be induced by lipopolysaccharide and interferon-γ, but lipopolysaccharide-stimulated induction of CRIP is eliminated in metallothionein-deficient mice (Cousins and Lanningham-Foster, 2000; Hallquist et al., 1996). Mice overexpressing CRIP show increased susceptibility to lipopolysaccharide and exhibit an altered cytokine production pattern in response to lipopolysaccharide, with a shift to an increase in interleukins 6 and 10 and a decrease in interferon-γ and interleukin-2 (Lanningham-Foster et al., 2002), suggesting an anti-inflammatory role for CRIP after HI.
The expression of CD53, which is a member of the tetraspanin membrane protein family, was also induced at 8 hours after HI and a further increase was detected at 72 hours. CD53 is mainly expressed by cells of the lymphoid-myeloid lineage, and ligation of CD53 can induce homotypic adhesion (Lagaudriere-Gesbert et al., 1997), the activation of N-terminal Jun kinase (JNK) (Yunta et al., 2002), and protection against apoptosis (Yunta and Lazo, 2003). This suggests that CD53 inhibits apoptosis in immune cells after HI, leading to sustained inflammation.
NADPH oxidase
NADPH-oxidase is composed of cytosolic (p40phox, p47phox, p67phox, and Rac2) and membrane-bound (gp91phox and p22phox) components. Upon activation, the cytosolic proteins are translocated to the membrane and fuse with the membrane bound proteins, leading to the formation of superoxide (Babior, 1999). Several substances induce activation of NADPH-oxidase, including galactins, chemokines, and proinflammatory cytokines (Karlsson and Dahlgren, 2002). We presently found induction of genes for both the membrane and cytosolic components of NADPH-oxidase, as well as an increase of chemokines (above) and a marked (20-fold) increase of galectin-3 (Mac-2 antigen), suggesting that NADPH-oxidase in phagocytes (polymorphonuclear leukocytes and/or microglia/macrophages) is activated after HI and that its importance in the immature brain injury has to be further explored.
Anti-inflammatory cytokines or receptors
Anti-inflammatory molecules (lipocortin-1, SOCS-3, transforming growth factor-β1, tissue inhibitor of metalloproteinase 1) and receptors (interleukin-4 receptor) were induced. Lipocortin-1 possesses anti-inflammatory effects probably through inhibition of phospholipase A2, and is proposed to be an endogenous neuroprotective molecule in a number of tissues including the brain (Rothwell and Relton, 1993). An early 10-fold increase of SOCS-3 (but not SOCS-5) was found in the immature brain, which basically agrees with the response to ischemia in the adult brain (Bates et al., 2001). This suppressor of cytokine signaling is induced by interleukin-6, interleukin-10, and interferon-γ, and may exert its anti-inflammatory effect through inhibition of JAK2 kinase (Larsen and Ropke, 2002). The induction of interleukin-4 receptor is interesting in that this cytokine has been shown to kill microglial cells and may thereby contribute to the termination of the powerful microglial response and prevention of chronic inflammation (Yang et al., 2002).
In summary, this study on the global pattern of gene expression after HI emphasizes the complexity and multifaceted nature of the immunoinflammatory response. Besides the existence of various subpopulations of microglia/macrophages, our data indicate that T-cells, NK-cells, mast cells, B-cells, dendritic cells, and polymorphonuclear leukocytes may participate in the response to HI. Furthermore, novel cytokines/chemokines, complement-related genes, interferon-regulated genes, components of the TIR/nuclear factor-κB pathway, and a number of anti-inflammatory or immunomodulatory genes are likely to be involved. Several of these genes may be of pathophysiologic significance, and their roles in immature brain injury need to be revealed in future studies.
Footnotes
Acknowledgements
The authors thank Eva Cambert for technical assistance.