
Abstract
Tissue plasminogen activator (tPA), a fibrin specific activator for the conversion of plasminogen to plasmin, stimulates thrombolysis and rescues ischemic brain by restoring blood flow. However, emerging data suggests that under some conditions, both tPA and plasmin, which are broad spectrum protease enzymes, are potentially neurotoxic if they reach the extracellular space. Animal models suggest that in severe ischemia with injury to the blood brain barrier (BBB) there is injury attributed to the protease effects of this exogenous tPA. Besides clot lysis per se, tPA may have pleiotropic actions in the brain, including direct vasoactivity, cleaveage of the N-methyl-D-aspartate (NMDA) NR1 subunit, amplification of intracellular Ca++ conductance, and activation of other extracellular proteases from the matrix metalloproteinase (MMP) family, e.g. MMP-9. These effects may increase excitotoxicity, further damage the BBB, and worsen edema and cerebral hemorrhage. If tPA is effective and reverses ischemia promptly, the BBB remains intact and exogenous tPA remains within the vascular space. If tPA is ineffective and ischemia is prolonged, there is the risk that exogenous tPA will injure both the neurovascular unit and the brain. Methods of neuroprotection, which prevent tPA toxicity or additional mechanical means to open cerebral vessels, are now needed.
Keywords
The use of tPA is now an established stroke treatment but only for those patients presenting within 3 hours of ischemic stroke onset. In large multicentered, randomized, placebo-controlled studies, tPA has been studied up to six hours from the time of onset. The strongest benefit is seen in those treated in the under 3-hour time window, and even this benefit is more pronounced for those patients treated within 90 minutes of onset (NINDS Study, 1995). While the main concern has always been the risk of hemorrhage, approximately 5 to 6%, the overwhelming problem is the 60% of patients who, despite being seen early and having signs of salvageable tissue (a good CT or a Perfusion Weighted Image (PWI)/Diffusion Weighted Image (DWI) mismatch), do not achieve a good response to tPA. Part of this is the relative inefficiency of IV tPA, which only achieves reperfusion in 25 to 30% of patients with middle cerebral artery occlusions. An IV/IA rescue strategy has been reported in the IMS study (The IMS Study Investigators, 2004; Hill et al., 2002), and increases the efficacy with which vessels are opened. In non-responders, where reperfusion is not achieved and a persistent ischemic state ensues, neurotoxic actions of tPA may further exacerbate ischemic damage. This is the subject of this review. In the future, mechanical methods of clot disruption may avoid some of these problems, but for now, it is important to fully dissect and understand the pleiotropic actions of tPA in brain so that combination therapies can be developed to promote thrombolytic safety and efficacy.
The 1996 adoption by the American Academy of Neurology and American Heart Association of the results of the NINDS Trial translated the efficacy results published in 1995 to the first effective therapy for acute ischemic stroke (Adams et al., 1996). The NINDS rtPA Stroke Study (1995) suggests that approximately 160 patients for every 1,000 treated will benefit from IV thrombolysis providing the criteria for the trial are adhered to and patients are treated within three hours. While the effect was striking with a 50% relative improvement in those making a full recovery (20 to 30%), the absolute provement in the avoidance of disability was from 26 to 42% or an absolute risk reduction of 16% resulting in a number needed to treat (NNT) of 6 (Wardlaw et al., 2000). One difference between the efficacy results of the NINDS study and what is happening in clinical practice, is that half of the patients treated in the trial were randomized within 90 minutes of the onset of ischemia, which does not happen in the real world. It will always be very difficult for patients in the community to be assessed, scanned and have treatment started within 90 minutes. While the overall results of the NINDS study can, and have been, translated to community practice, the magnitude of the benefit is attenuated since most patients are being treated towards the end of the 180-minute time frame; very few are treated within 90 or 120 minutes.
Experience in the real world has been published with the assessment of studies from Houston (Chiu et al., 1998), Calgary (Buchan et al., 2000) and Cologne (Grond et al., 1998). These studies were positive and most patients adhered to the protocol. The dangers of not adhering to the protocol were examined in the study from Calgary, which compared patients who adhered with those who did not and showed the excess risk when patients were treated inappropriately. This is underscored by the study from Cleveland (Katzan et al., 2000) that suggested that within the non-academic community many patients were being treated off label, i.e. patients who did not fit the NINDS protocol, and are therefore put at risk without likely benefit. The results of the STARS (Albers et al., 2000) study in the U.S. and CASES (Hill and Buchan, 2001)—a larger study from Canada of 1,135 consecutively treated patients, suggests that with adherence to protocol symptomatic hemorrhage rates can be maintained in the 4 to 6% range and outcomes can be achieved that match the NINDS study. Both the STARS (Albers et al., 2000) and the CASES (Hill and Buchan, 2001) series emphasize that strict adherence to protocol affords the best chance of a good outcome and minimizes the risk of hemorrhage.
The generalizability of IV-tPA remains limited. Ten years on, across the U.S., Canada, and Europe, only 1% of stroke patients receive IV-tPA. In centers that are highly organized and well coordinated, this number has now surpassed 10%. The goal is to treat 20% of all stroke patients. The availability of the therapy is dependent on concentrating resources, personnel and imaging equipment so that 24-hour/7-day availability with instant service can be provided (Hill et al., 2000).
Improvements in utilization will depend on organization of clinical service, enhanced brain and vascular imaging, and patient selection. Improved selection will require further therapeutic trials: those that may help extend the window by moving from a time window to a tissue window. It is hoped that a new generation of neuroprotectants such as those drugs that block excitotoxicity, inflammation or apoptosis may be used as adjuvant therapies in combination with tPA.
The efficacy studies, to date, have led to an analysis of results published in the Cochrane Meta Analysis of IV thrombolysis (Wardlaw et al., 2000). The NINDS study suggests that among patients treated within 3 hours (50% were treated within 90 minutes) there was an absolute risk reduction of 11% to reach an NIHSS of 0–1 or a RANKIN of 0–1 and a 16% benefit to achieve a 3-month RANKIN of 0-2. The ECASS II (Hacke et al., 1998) study showed a statistically significant 8% improvement (for RANKIN 0-2), suggesting that there is benefit beyond three hours, the subject of further trials including ATLANTIS (Clark et al., 1999), which failed to showed benefit in those treated between 3 to 5 hours. Currently, two further IV-tPA trials, ECASS III (3 to 4 hours) and IST 3 (0 to 6 hours), are being conducted. The results of the meta-analysis including both the benefits and the risks of hemorrhage across time have recently been published (Hacke et al., 2004) (Fig. 1). Further attempts to extend the time window using DWI/PWI mismatch are ongoing but the current recommendation is to treat as quickly as possible, and the closer to 90 minutes the better.
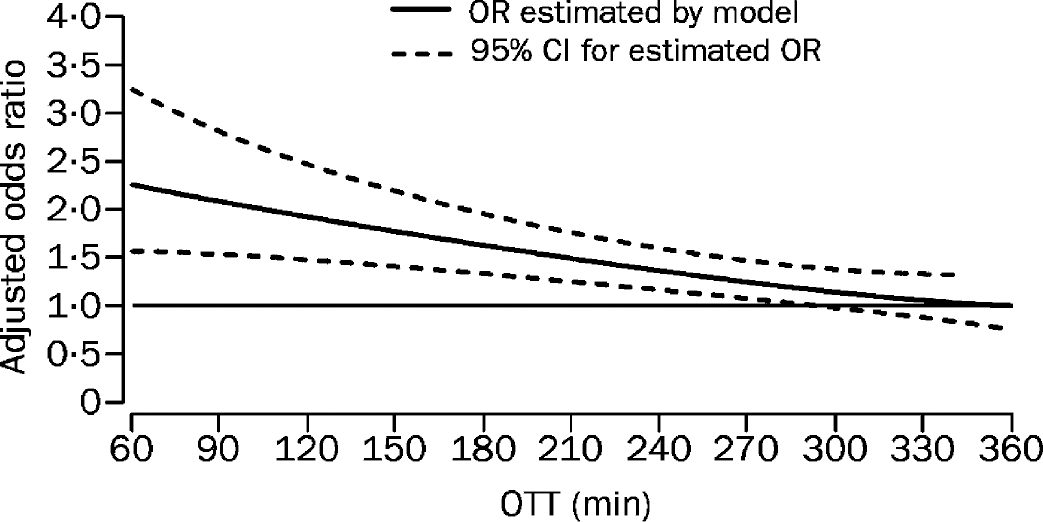
Estimating odds ration for favorable outcome. Model estimating odds ratio for favorable outcome at 3 months in rtPA-treated patients as compared with controls by OTT. Reprinted with permission from Elsevier (The Lancet, 2004, 363(9411): 768–774). OTT, Onset to treatment time.
RISK OF HEMORRHAGE
The major deterrent to using tPA in acute ischemic stroke is the fear of inducing intracerebral hemorrhage. The hemorrhage rate in the ECASS studies and the NINDS study suggested there would be a 5 to 6% increase in the rate of hemorrhage with careful control and adherence to protocol. The hemorrhage rates are maintained at 4% or less in large community-based studies (STARS, CASES). If protocol is violated and patients are treated with concurrent risks such as anticoagulation, low platelets and the like, the risk is higher. In the CASES study, treating patients slowly and treating those with high blood sugars posed incrementally greater risk. In the Lancet meta-analysis (Hacke et al., 2004), there was no clear-cut increase in the rates of intracerebral hemorrhage in those patients treated in the 3 to 6 hour window compared to those being treated under 3 hours. Although it was shown that patients with more severe strokes arrived more quickly (higher baseline NIHSS scores at earlier timepoints), the baseline NIHSS score was neither an effect modifier nor an important predictor of intracerebral hemorrhage. Only older age and tPA treatment predicted intracerebral hemorrhage.
While the risks of hemorrhage (surprisingly) do not appear to increase across time, the chances of making a reperfusion-induced recovery deteriorate rapidly (Fig. 1). Although the randomized NINDS trial indicates that people should be treated within 3 hours, the newly published meta-analysis proposes that the window be opened out to 4.5 hours (NINDS Group, 1995). This proposed increase in the time window may lead to more patients being treated with potentially deteriorating integrity of the blood-brain barrier, thus increasing the importance of the debate about the direct neurotoxic effects of exogenous tPA. Evidence comes not just from animal models but is also emerging from clinical experience.
NEUROTOXICITY IN ANIMAL MODELS
Whether tPA aggravates brain injury or not has been the subject of many studies and discussions (Treynalis and Lipton, 2001; del Zoppo, 1998). tPA neurotoxicity has been documented in cell culture experiments as well as animal models of focal cerebral ischemia although the overall data are not unequivocal.
Primary cortical neurons cultured from tPA knockout (KO) mice are resistant to oxygen-glucose deprivation (Nagai et al., 2001). Exogenously added tPA potently induced apoptosis in cultured neurons (Flavin et al., 2000) and amplified neurotoxicity after hemoglobin exposure (Wang et al., 1999). Perhaps the most compelling demonstration of the neurotoxic potential of tPA comes from the study by Wang and colleagues showing that tPA knockout mice were resistant to focal cerebral ischemia (Wang et al., 1998). After 3 hours transient focal cerebral ischemia followed by 21 hours of reperfusion, infarct volumes were smaller in tPA deficient mice compared with wildtype (WT) littermates. In WT mice, adminstration of 1 mg/kg of exogenous tPA administered intravenously significantly increased infarction after 2 hours of transient focal ischemia followed by 22 hours of reperfusion (Wang et al., 1998). However, contradictory data was collected in another study, where tPA deficient mice had larger infarcts after transient focal cerebral ischemia (Tabrizi et al., 1999). Although the exact reason for these variable results is not known, differences in genetic background of the mice used are likely to play a significant role (Ginsberg, 1999).
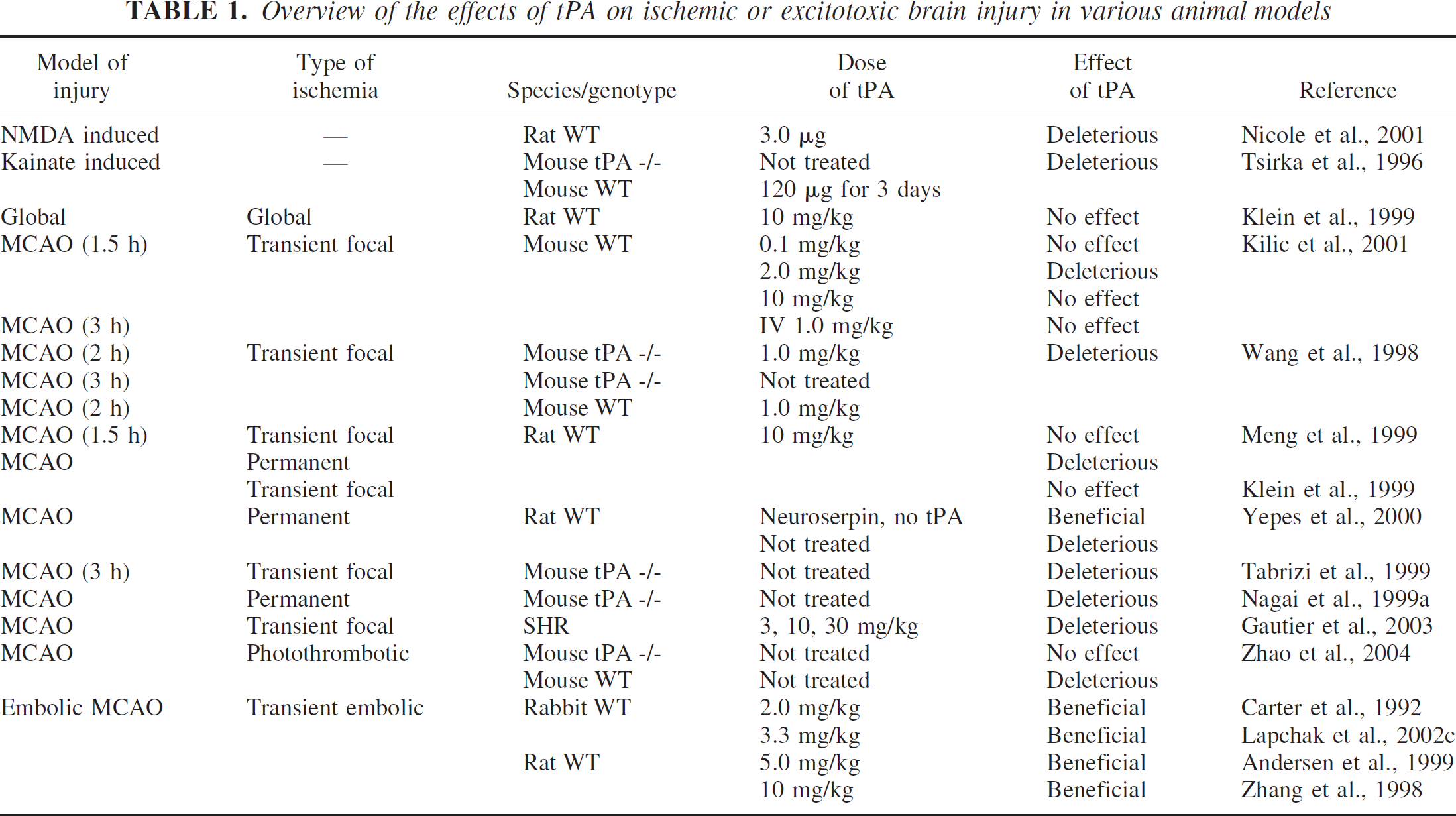
Equivocal data were also reported in stroke models where tPA was given immediately after global and mechanically induced transient focal ischemia. There was no increase in either CA1 selective neuronal injury (cytotoxicity) or infarct volume (Klein et al., 1999) even at doses 10x more than those used in humans and twice as much as the known effective thrombolytic dose for rodents. Kilic et al. (2001) observed a slight but nonsignificant reduction of the infarct volume in animals treated with recombinant tPA (rtPA) during ischemia, independent of the rtPA dose applied. Their data indicates that rtPA administration during ischemia reduces rather than increases the degree of injury, an effect most likely related to improving the microcirculation in this model. Furthermore, brain edema in their study was not affected in animals receiving rtPA during ischemia compared with saline-treated control animals. (Table 1)
Overview of the effects of tPA on ischemic or excitotoxic brain injury in various animal models
In a mouse model of transient focal cerebral ischemia, reduction of tPA activity (tPA gene inactivation) reduced infarct size, but its augmentation increased infarct size (Nagai et al., 1999a). In the same series of experiments, reduction of plasminogen activity increased infarct size, whereas its augmentation had the opposite effect. Thus tPA may exert its neurotoxic effects via its interaction with plasminogen (i.e. its ‘thrombolytic’ mode of action resulting in local tissue damage outside of the vascular compartment). These experiments lend support to the concept that the extravascular protease actions of tPA itself can damage the central nervous system. Thrombolytic agents other than tPA have also been shown to cause a dose related increase of focal infarction in a hamster model of stroke (Nagai et al., 1999b).
Another body of evidence comes from the interrelationship between tPA and seizures. Injection of the glutamate analog kainate into the amygdala resulted in upregulation of tPA in the amygdala and hippocampus along with seizures causing neuronal death in the hippocampus (Yepes et al., 2002). In tPA knockout mice, seizure propagation was attenuated. However in plasminogen knockout mice no such effect was observed. These results point towards a plasminogen independent effect of tPA on kainate induced seizures. It has been proposed that this effect could be a consequence of tPA mediated cleavage of the NR1 subunit of the NMDA receptor, which may lead to increased neuronal electrical activity, and seizures (Pawlak and Strickland, 2002).
Meng and colleagues compared the effects of tPA in mechanical and clot models of focal cerebral ischemia in rats (Meng et al., 1999). When tPA was infused after mechanical reperfusion, no increase in infarct size was observed. When tPA successfully dissolved the clots in the embolic models and reperfused ischemic brain, infarcts were reduced. Basically, tPA did not appear to have detectable neurotoxic effects in those models, where reperfusion was guaranteed. The apparent differences between these results and other published studies may be due to variations between species and/or experimental paradigms, as Meng and colleagues (1999) did not aim their study at the direct effects of tPA in permanent ischemia. Other studies using a mechanical MCA clip occlusion in a rat model of permanent focal ischemia (Buchan and Warren, 2002; Klein et. al., 2002) looked directly at whether tPA increased the infarct size or not. The detrimental effects of tPA in this model could also be due to the tPA carrier. This carrier contains large amounts of L-arginine, a substrate for all isoforms of Nitric Oxide Synthase (NOS). Nitric Oxide (NO), acting through the NMDA receptor, may be a link in the observed toxic effects of tPA. Mice deficient in neuronal NOS have smaller infarcts after middle cerebral artery (MCA) occlusion (Huang et al., 1994). This observation could be due to an effect of NO which, acting as a modulator of gene expression (Beck et al., 1999), may reduce tPA messenger RNA (mRNA) levels (Eberhardt et al., 2002).
tPA Knockouts
The neurotoxic potential of tPA in brain injury has also been supported using tPA knockout (tPA -/-) mice in various models. Mice lacking functional tPA (Carmeliet et al., 1994) are resistant to excitotoxic neuronal death induced by unilateral intrahippocampal injection of kainic acid (KA) (Tsirka et al., 1995; Tsirka et al., 1996), suggesting that tPA mediates neuronal destruction through plasmin proteolysis. Plasminogen deficient mice (Bugge et al., 1995) are also resistant to excitotoxic death (Tsirka et al., 1997). Microglia from tPA -/- mice show attenuated activation after KA injections. Infusions of either catalytically active or inactive tPA into these mice prior to KA injection restored microglial activation (Rogove et al., 1999). This microglial activation seems to be mediated by mechanisms other than the activation of plasminogen by tPA, achieved probably via its finger domain through binding to annexin II on the microglial cell surface (Siao and Tsirka, 2002).
tPA deficient mice were also protected against traumatic brain injury (TBI) and showed reduced cortical lesion and an overall amelioration in edema volumes compared to their wildtype littermates (Mori et al., 2001). MCA occlusion by intravascular filament produced a 50% smaller infarct in tPA deficient mice than in the wildtype and intravenous injections of tPA into WT and KO mice produced even larger infarcts, indicating that tPA can increase stroke related injury (Wang et al., 1998) (Fig. 2). In a mouse photothrombotic MCA occlusion model, delayed administration of heparin (3 hours after insult) failed to cause cerebral hemorrhage in tPA KO mice but significantly increased hemorrhage in WT (Zhao et al., 2004). Activity and mRNA levels of tPA were increased in microglial cells in the ischemic border of WT mice after heparin administration. Concomitant exacerbation in Matrix Metalloproteinase-9 (MMP-9) mRNA expression and its conversion to active form after heparin treatment were observed only in the WT and not tPA KO mice. MMP-9 expression was also localized in microglial and endothelial cells in WT. The authors suggest that endogenous tPA through enhancement of MMP-9 expression and proteolytic activation can play an essential role in the pathogenesis of cerebral hemorrhage produced by heparin. It has been suggested that absence of plasminogen activators in tPA deficient mice can retard the migration of neutrophils and macrophages, the predominant source of MMPs, to the site of injury and thus reduce the infarct or lesion volumes after ischemic insults (Siao and Tsirka, 2002).
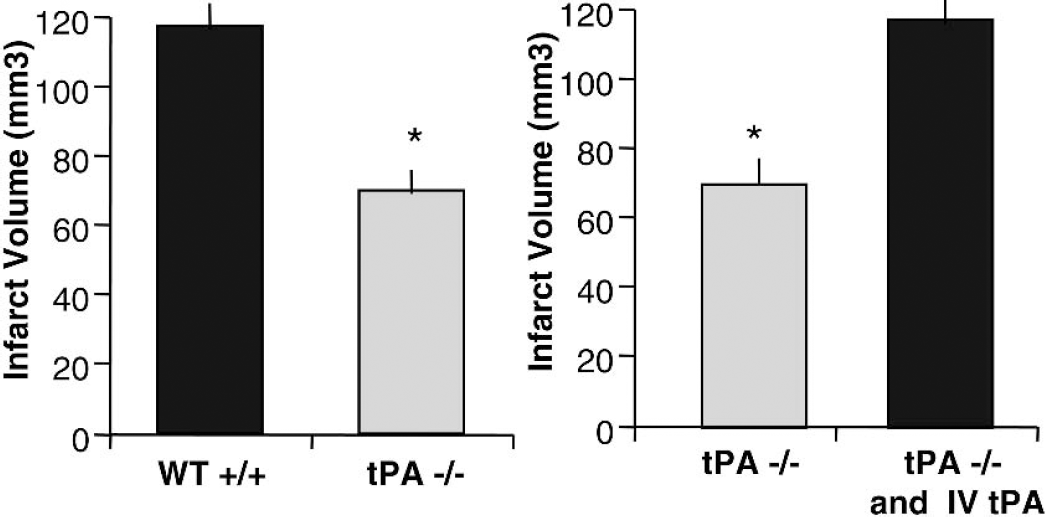
tPA knockout mice have reduced infarct volumes. (
Changing the duration of ischemia results in contrasting effects of tPA on the brain. Nagai et al. (2002) showed that after mild damage, injury was more extensive in tPA KO mice, but after severe damage the reverse was true. The authors postulated that endogenous tPA is protective after mild damage because of thrombolysis of local micro-thrombi. In large areas of damage the local toxic effects of tPA overwhelmed its benefits. In the focal clip model, permanent but not transient ischemia was associated with tPA induced increases in the volume of infarction (Buchan and Warren, 2002).
The beneficial effects of tPA are vascular; lysis should restore the blood flow to the parenchyma. Potentially deleterious side effects include amplification of neurotoxicity through interactions with NMDA receptors and MMP-9 dysregulation leading to a compromised BBB, neurovascular matrix, increased edema and the risk of hemorrhage (Fig. 3).
Tissue Plasminogen Activator in Stroke-Induced Injury: A Double-Edged Sword? Thrombolysis forms an important theory in acute stroke. (
Mechanisms of tPA Neurotoxicity
Various intracellular mechanisms such as excitotoxicity, free radical injury, and apoptosis (Bondy, 1995; Lipton, 1999; Chan, 2001; Graham and Chen, 2001; Lo et al., 2003) have been advanced in an effort to explain neuronal injury in ischemia.
During ischemia, levels of extracellular glutamate rise as high as 100μM. Glutamate receptors are activated, and even more calcium ions enter the cell (Benveniste et al., 1984). The entry of calcium ions into cells triggers cell death. Thus, any interaction between the NMDA receptor and exogenous serine protease tPA, whether direct or indirect, is of relevance to neurotoxicity when considering tPA as a potential threat, particularly in the face of injured blood-brain barrier integrity, when tPA, ordinarily acting in an intravascular fashion, will gain access to the extravascular neural parenchyma.
Studies suggest that extracellular pathophysiology, e.g. disturbances in the extracellular matrix (ECM) (Chen and Strickland, 1997), also play a major role in addition to the above outlined mechanisms. To this effect, the zinc-dependent MMPs, abundantly expressed in the ECM, are increasingly being studied in various animal models of stroke. Of the many MMPs, the gelatinase MMP-9 is specifically implicated in attacking important components of the basal lamina around the cerebral blood vessels and its activation during cerebral ischemia can cause microvascular damage leading to disruption of BBB, edema and hemorrhagic transformations (Rosenberg et al., 1998; Hamann et al., 1996; Hamann et al., 1995; Mun-Bryce and Rosenberg, 1998; Lo et al., 2002). The increased risk of hemorrhagic transformations seen in stroke patients following tPA therapy may therefore be due to tPA induced upregulation in MMP-9.
Plasmin is a potent protease that cleaves blood fibrin and is also implicated in microvascular and extracellular matrix degradation (Parry et al., 2000; Marder et al., 2001). It can bind to a variety of cells, including monocytes, and triggers aggregation of neutrophils and platelet degranulation. Plasmin is also known to activate MMP-9 and MMP-2, thereby forming another important mechanism of tPA induced hemorrhage (Murphy et al., 1992; Mun-Bryce and Rosenberg, 1998). tPA can produce abnormally high levels of plasmin and thrombolysis products (TLPs) that can trigger intracranial hemorrhage and can modulate the severity of hemorrhagic transformation (Gautier et al., 2003). Plasmin contained in TLPs can act on non-fibrinogenic substrates (Tsirka et al., 1997) and compromise cell viability. Plasmin is also known to potentiate NMDA receptor function (Junge et al., 2003). Plasminogen deficient (plg -/-) mice are shown to be resistant to excitotoxicity (Tsirka et al., 1997) and intrahippocampal infusion of plasminogen has been shown to restore kainate induced damage in plg -/- mice. However Nagai et al. (1999a) showed exacerbated injury following ischemia in a permanent MCA occlusion model using plg -/- mice. These results are indicative that the plasmin/plasminogen system can have opposing effects both in the CNS and vasculature (Gingrich and Traynelis, 2000).
PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL ROLES OF tPA
Endogenous tPA is synthesized and released by cells within the vascular system, and its major role is the conversion of plasminogen to plasmin, which breaks down intravascular blood thrombus and fibrin deposits (Liberatore et al., 2003) (Fig. 3). Both plasminogen and tPA are synthesized locally in the brain, and are expressed by glial and neuronal cells in the CNS (Krystosek and Seeds, 1984; Sappino et al., 1993; Chen et al., 1995; Nishibori et al., 1995; Backstrom et al., 1996; Geschwend et al., 1997; Davies et al., 1998; Scarisbrick et al., 2001; Siao et al., 2003). Activated glial cells can also release tPA (Rogove et al., 1999). Plasminogen and tPA have functions varying from synaptic transmission (Hoffman et al., 1998; Gingrich et al., 2000) to synaptic plasticity (Baranes et al., 1998; Neuhoff et al., 1999). tPA knockout mice show deficiencies in long term potentiation (Baranes et al., 1998) and also show distorted and punctated mossy fibers (Wu et al., 2000).
Beneficial effects of endogenous tPA activity in ischemia are confined to the presence of fibrin as tPA can restore blood flow in the brain by degrading fibrin and limiting damage (Strickland, 2001) (Figs. 4, 5). Deficiency of tPA augments harmful cerebrovascular fibrin deposition that correlates with enhanced brain injury, thus supporting the view that endogenous tPA protects the brain from an ischemic insult through its thrombolytic action (Tabrizi et al., 1999). Endogenous tPA activity was found to be significantly different between young and middle-aged mice brains, therefore it is possible that the absence of endogenous tPA production within the older brain may compromise brain fibrinolysis, predisposing it to larger and more disabling strokes (Ahn et al., 1999).
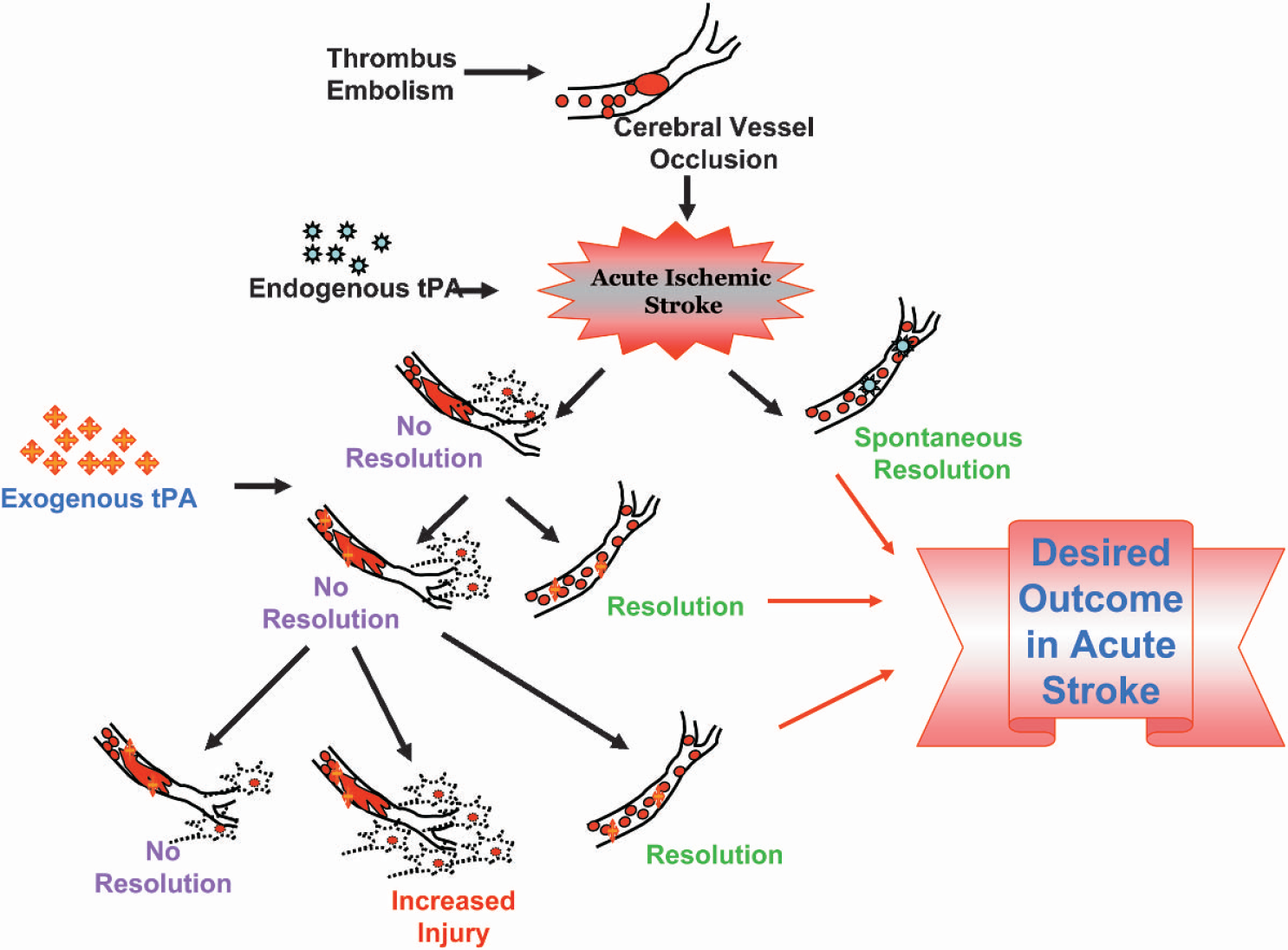
Following acute ischemic stroke, endogenous tPA reduces damage to the ischemic tissue through resolution by thrombolysis. However, in the absence of spontaneous resolution by endogenous tPA, exogenous tPA can either resolve the clot, or in the case of failed resolution, can enter the brain through compromised BBB, and thus can exacerbate ischemic injury. tPA, tissue plasminogen activator; BBB, blood brain barrier.
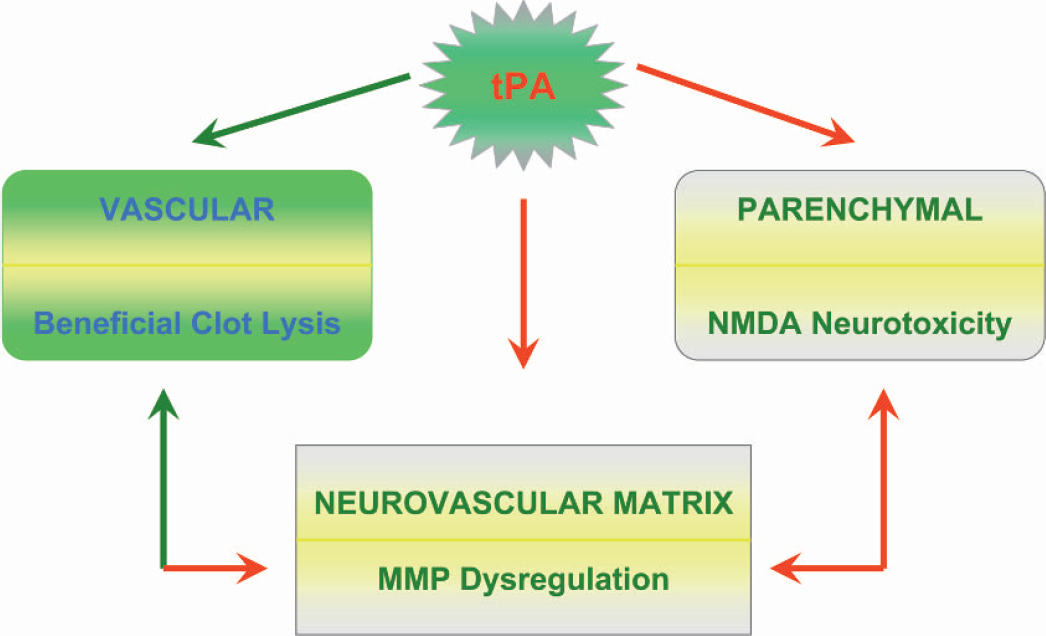
Pleiotropic effects of tPA. If tPA remains within the vasculature, its effects are beneficial, i.e., clot lysis and successful reperfusion. If tPA leaks into the parenchyma, it causes neurotoxicity through the NMDA receptors. It can also cause disruption of the neurovascular matrix through MMP dysregulation. tPA, tissue plasminogen activator; NMDA, N-methyl-D-aspartate; MMP, matrix-metalloproteinase.
Based on evidence in the literature, Salles and Strickland (2002) have outlined the sequence of events in tPA expression. tPA is stored in intracellular vesicles (Baranes et al., 1996; Gualandris et al., 1996; Parmer et al., 1997; Lochner et al., 1998) following its synthesis. Membrane depolarization would then induce a Ca++ dependent secretion of tPA (Gualandris et al., 1996; Parmer et al., 1997) and its release into the extracellular matrix. Release of tPA is also mediated by NMDA receptor signaling (Nicole et al., 2001; Fernandez-Monreal et al., 2004).
Normal physiological functions of tPA include long term potentiation (LTP) (Frey et al., 1996; Huang et al., 1996; Baranes et al., 1998; Madani et al., 1999), long term depression (LTD) (Calabresi et al., 2000), synaptic remodeling (Baranes et al., 1998; Neuhoff et al., 1999; Wu et al., 2000), neuronal regeneration (Salles et al., 1990), migration of cerebellar granule cells during development (Friedman and Seeds, 1995), and the induction of cerebellar motor learning (Seeds et al., 1997; Seeds et al., 2003). tPA also influences pathological situations like excitotoxic injury (Tsirka et al., 1995; Chen and Strickland, 1997; Wang et al., 1998), peripheral nerve injury (Akassoglou et al., 2000), and Wallerian degeneration (Bignami et al., 1982). Upregulation of tPA mRNA occurs during performance of complex motor tasks (Seeds et al., 1995). tPA also plays a role in functional recovery in models of sciatic nerve crush in mice, with tPA -/- mice displaying significant delays in hind limb response to sensory stimuli (Siconolfi and Seeds, 2001; Siconolfi and Seeds, 2003).
Salles and Strickland (2002) demonstrated concentration of the tPA protein and activity in the mossy fiber pathway within the mouse hippocampus under basal conditions. They observed robust tPA localization in vascular tissue, meninges and in the neuropil of the central nucleus and the hypothalamus. Localization of tPA mRNA has been observed in all neuronal layers of the hippocampus by other researchers (Qian et al., 1993; Sappino et al., 1993; Carroll et al., 1994; Tsirka et al., 1997). The authors (Salles and Strickland, 2002) could not find any significant tPA localization in the CA1 region under basal conditions. However, tPA antigen upregulation in CA1 neuronal cell bodies occurred after unilateral hippocampal kainic acid (KA) injection. Similar upregulation of tPA was observed with subcutaneous administration of KA (Salles and Strickland, 2002). tPA was also induced as an immediate-early gene after high frequency stimulation of the perforant pathway and induction of seizures (Qian et al., 1993).
The mossy fiber pathway within the hippocampus has large axon terminals that make contact with multiple synaptic spines. Furthermore, this pathway is unique, as it is characterized electrophysiologically by NMDA-independent LTP. Abundant tPA expression could be a prerequisite for structural reorganization necessary within these types of synaptic terminals and may be crucial in imparting specific signaling properties to these synapses. Presence of tPA induced proteolytic activity could underlie the NMDA-independent character of mossy fiber LTP (Salles and Strickland, 2002). Frey et al. (1996) and Huang et al. (1996) showed that in the hippocampus of tPA -/- mice, late-phase LTP was affected. Inhibitors of tPA were also able to inhibit late-phase LTP (Baranes et al., 1998). Application of tPA and overexpression of tPA increased and prolonged the late-phase LTP (Baranes et al., 1998; Madani et al., 1999).
The influence of tPA on LTP appears to be mediated by a variety of mechanisms. LTP induction (Baranes et al., 1998) and synaptic plasticity may require tPA to degrade the extracellular matrix and destroy cell adhesion molecules (Hoffman et al., 1998) in the formation and elongation of axons. Binding of tPA to low density lipoprotein receptor related protein (LRP) can activate protein kinase A (PKA), which is known to be a key factor in induction of late-phase LTP (Zhuo et al., 2000). Cleavage of the NR1 subunit of the NMDA receptor by tPA can enhance intracellular Ca++ influx thereby influencing LTP (Nicole et al., 2001; Fernandez-Monreal et al., 2004).
Salles and Strickland (2002) hypothesized that tPA mRNA may be under a translational control. Under basal conditions, tPA mRNA may be continually present but its translation into protein may occur after some significant injury. Various inhibitors of tPA such as type 1 plasminogen activator inhibitor (PAI-1) or neuroserpin can block the protease activity of tPA (Gautier et al., 2003). Recently, Fernandez-Monreal et al. (2004) demonstrated that astrocytes can scavenge extracellular tPA by a receptor dependent mechanism. Therefore, an understanding of the physiological regulation of tPA within the CNS will be crucial in developing newer strategies of stroke therapy.
tPA NEUROTOXICITY VIA NMDA RECEPTORS
Both neurons and microglial cells can release tPA (Siao et al., 2003). It has been proposed that tPA released from hyperdepolarized neurons can activate microglial cells. These activated microglia have a potential dual role. They clean up the dead and dying neurons, but also contribute to a neurotoxic cascade by releasing additional toxins, including more tPA.
The relationship between tPA and the NMDA receptor is of particular interest for understanding its role in neuroexcitotoxicity since tPA appears to increase neuronal cell death mediated by glutamatergic receptors. tPA cleaves a 15 to 20kDa fragment from the NR1 subunit of the NMDA receptor (Nicole et al., 2001) (Fig. 6). As a result, tPA treatment amplifies the NMDA induced increase in intracellular calcium concentration and potentially could provoke cell death. In the same series of experiments, injection of tPA into the striatum was not toxic, but co-injection of tPA with NMDA increased NMDA-induced lesion size by 50%, suggesting that tPA potentiates the consequences of NMDA activation. Cleavage of the NR1 sub-unit is responsible for this potentiation. This interaction of tPA with the NR1 subunit has been challenged, although the notion that tPA potentiates the NMDA receptor was not (Matys and Strickland, 2003). Post-ischemic treatment with an antagonist of the NR2 subunit has also been shown to be neuroprotective in a rat model of focal ischemia (Yang et al., 2003). Combination of NR2 antagonist and tPA had a worse outcome than the use of NR2 antagonism alone. However, this combination therapy still had a better outcome than the control group. A logical explanation for these results is that tPA can act in more than one way to cause damage. An interaction with the NR2 subunit could be one such mechanism of neuronal death (Fig. 2).
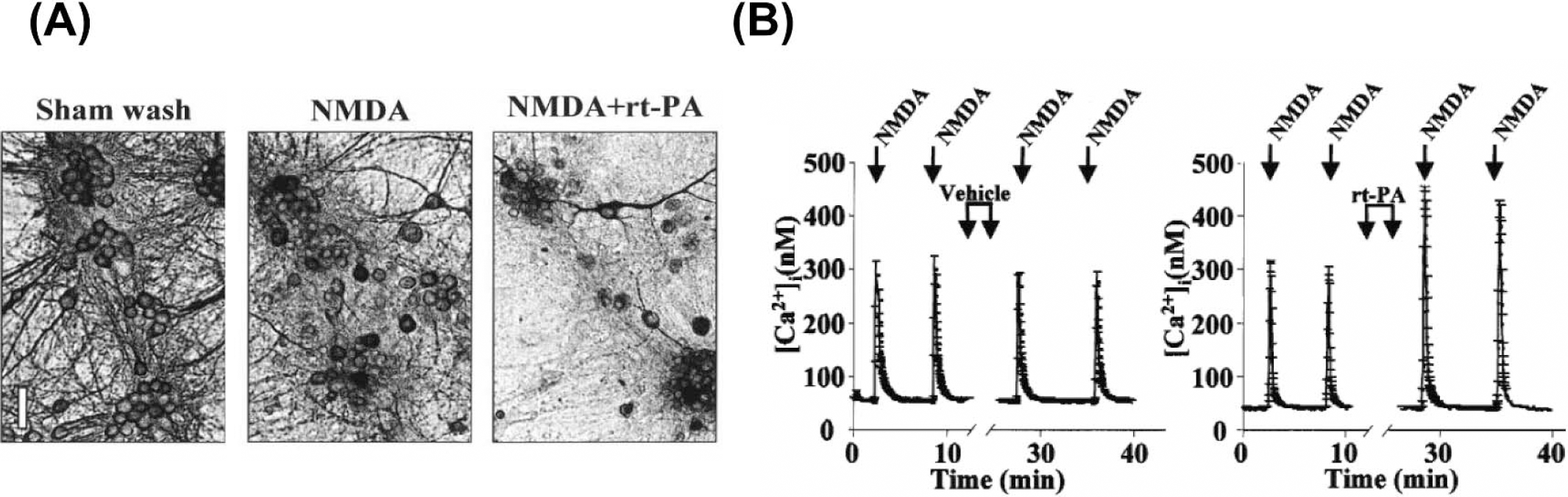
Deleterious effects of tPA. (A) Exacerbation of NMDA-induced neuronal death occurred when tPA was added to mixed cortical cultures; and (B) enhanced NMDA-evoked Ca++ increase in cortical neurons. tPA, tissue plasminogen activator; NMDA, N-methyl-D-aspartate Adapted (permission from author), and reprinted with permission from Nature Med (Nature Med, 2001, 7:59–64).
MK-801, a non-competitive antagonist of the NMDA receptor, blocks the neurotoxic effects of tPA (Buchan and Warren, 2002; Kilic et al., 2001; Warren et al., 2003). In both transient and permanent focal ischemia, MK-801 reduced infarct size. In these experiments, thrombolysis was not employed. This effect was believed to be secondary, at least in part, to increased cerebral blood flow (CBF), an effect that was shown in both models. In different models, CBF was increased in the penumbra or the ischemic core. This would imply that the beneficial effect of MK-801 is independent of its known action of ion channel antagonism, which reduces calcium entry into ischemic neurons. MK-801 blocks the neurotoxic effects of tPA by blocking the receptor, thereby inhibiting calcium overload. Recently, it has been shown that tPA can have complex vasoactive roles in brain; at low 1 nM concentrations tPA inhibits phenylephrine-induced vasoconstriction, whereas at high 20 nM concentrations tPA augmented phenylephrine's effects on vessel tone (Nasser et al., 2004).
Finally, tPA may also promote excitotoxic cell death through a receptor mediated apoptotic pathway. This evidence comes from a study of apoptosis in the Lurcher mutant mouse model (Lu and Tsirka, 2002). Lurcher is a spontaneous mouse mutant in which premature aberrant apoptosis occurs in the cerebellum. This is known to be secondary to a mutation in a glutamate receptor subunit gene that results in a large inward calcium current. Elimination of tPA delays the apoptotic death of Purkinje and granule neurons.
tPA AND INJURY TO NEUROVASCULAR UNIT
Unless exogenously administered tPA can cross the BBB and enter the brain, its relevance as a neurotoxic agent is debatable. It has been shown in animal models that the BBB opens in a biphasic manner after ischemia, immediately after reperfusion and again after a latency (Kuroiwa et al., 1985; Huang et al., 1999). The initial opening of the BBB is related to hemodynamic factors. Vasodilatation caused by acidosis and loss of autoregulation results in luxury perfusion. Raised pressure in the cerebral vessels changes transport across endothelial cells and opens interendothelial tight junctions. The second phase of BBB opening is the result of injury to the neurovascular unit and vasogenic cerebral edema (Huang et al., 1999).
It is the first phase of BBB opening that raises concern in the context of tPA neurotoxicity. Exogenously applied tPA has the potential to enter the brain. The second phase also raises clinical concerns: cerebral edema is a major and potentially fatal complication of acute ischemic stroke (AIS). This may be exacerbated by reperfusion so the combination of reperfusion with tPA in an already injured brain may make it more prone to edema. Analysis of clinical data shows that there is no evidence for an increased risk of herniation and brain edema among patients treated with tPA. This observation was in contrast to the placebo group who showed an increase in cerebral edema.
An emerging concept in cerebrovascular biology ties BBB function to the integrity of the associated neurovascular matrix of the basal lamina. MMPs comprise a large family of endopeptidases associated with basal lamina and extracellular matrix remodeling and are involved in both physiological and pathological CNS processes (Pagenstecher et al., 1998; Yong et al., 1998). They are secreted in an inactive form and require activation via autocatalytic mechanisms or through mediation by other proteases (Nagase, 1997; Nagase and Woessner, 1999). These metalloproteinases not only cleave extracellular matrix proteins (Matrisian, 1990) but membrane bound receptors and various cytokines as well (Nagase and Woessner. 1999). MMPs can degrade most constituents of the neurovascular matrix: collagen, elastin, fibronectin, vitronectin, and gelatin (Yong et al., 2001). The gelatinases MMP-2 and MMP-9 specifically attack the important components of the basal lamina around the cerebral blood vessels that precede microvascular damage in cerebral ischemia. This leads to disruption of the blood brain barrier, edema, and hemorrhagic transformation in animal models of ischemia (Rosenberg et al., 1998; Hamann et al., 1996; Hamann et al., 1995; Mun-Bryce and Rosenberg, 1998; Lo et al., 2002). Plasmin, activated by tPA or urokinase type plasminogen activator (uPA), activates MMP-9 and MMP-2 (Murphy et al., 1992), suggesting molecular mechanisms underlying the phenomenon of tPA induced hemorrhage.
Significant increases in MMPs have been reported after ischemic onset in mouse, rat and non-human primate models (Heo et al., 1999; Rivera et al., 2002; Zalewska et al., 2003; Chan, 1996; Lee et al., 2004) as well as in human stroke (Clark et al., 1997). Most recently, Montaner and colleagues (2003) have shown a correlation between the pretreatment levels of MMP-9 and intracranial hemorrhagic complications in stroke patients after tPA therapy. This implicates MMP-9 as a negative prognostic factor in human stroke. These authors suggested the eventual possibility of developing a screening test prior to the use of tPA to identify patients at high risk of hemorrhage. In rodent models of embolic focal ischemia, delayed treatment with tPA has been observed to result in hemorrhagic transformations (Brinker et al., 1999; Chopp et al., 1999; Kano et al., 2000). Various reports on the activation of MMP-9 and degradation of critical protein components of cerebral vasculature have been correlated with the development of edema and hemorrhage (Sumii and Lo, 2002; Asahi et al., 2001; Hamann et al., 1996; Heo et al., 1999).
Lo and colleagues (2002) consider MMP-9 to be a putative molecular trigger in the context of hemorrhagic transformations since it targets the neurovascular matrix and weakens the vessel integrity. Interactions between the plasminogen activator system (PAS) and MMP-9 may underlie the phenomenon of tPA induced hemorrhages. In a rabbit multi-embolic stroke model, combination of the broad-spectrum metalloproteinase inhibitor, Batimastat (BB-94), and tPA significantly reduced the incidence of hemorrhage as compared to tPA alone (Lapchak et al, 2000). In a model of focal embolic stroke in spontaneously hypertensive rats, BB-94 combined with tPA reduced the volume of tPA-induced hemorrhagic conversion (Sumii and Lo, 2002). Aoki et al. (2002) compared MMP-9 profiles in rat models of focal cerebral ischemia achieved either with mechanical occlusion and reperfusion or embolic clot occlusion and tPA thrombolysis. Although levels of ischemia and infarct volumes were similar in both models, there were significant differences in MMP-9 responses. Both central and penumbral zones showed elevated cellular and extracellular MMP-9 levels in the embolic model where reperfusion was achieved with tPA. Sumii and Lo (2002) showed that pro-MMP-9 and cleaved MMP-9 were upregulated after embolic focal ischemia in rats and delayed treatment with tPA significantly enhanced this increase. This implicates the involvement of MMP-9 in the process of tPA-associated hemorrhage. Dysregulation of MMP-9 may also be mediated by upregulation of mitogen activated protein (MAP) kinase, especially extracellular signal regulated kinase (ERK) (Wang et al., 2002). Thus tPA may contribute to neurovascular injury by activating MMPs, and the interaction between the two proteases is important in understanding the full implications of all the mechanisms of tPA neurotoxicity.
MMP Knockouts
Compared with wildtype mice, MMP-9 knockout mice are protected against brain trauma (Wang et al., 2000), focal cerebral ischemia (Asahi et al., 2000a; Asahi et al., 2001) and transient global cerebral ischemia (Lee et al., 2004). MMP-9 was upregulated in cortex and striatum of wildtype mice following an ischemic insult. Its expression was predominantly observed in endothelial cells of cerebral blood vessels and to some extent in the parenchymal cells. The protein ZO-1 associated with the expression of tight junctions in BBB was also significantly degraded following ischemia in wildtype mice, but its degradation was significantly reduced in the MMP-9 knockouts (Asahi et al., 2001) (Fig. 7). This effect may underlie the protection against cerebral ischemia observed in MMP-9 deficient mice. In addition to BBB matrix substrates, MMP-9 is also known to degrade myelin basic protein (MBP). Ischemic degradation of myelin basic protein isoforms was significantly ameliorated in MMP-9 knockouts (Asahi et al., 2001). In addition to the attenuated proteolysis of critical BBB and white matter components, Asahi and colleagues also reported significantly reduced infarct volumes after transient and permanent focal ischemia in MMP-9 knockouts (Asahi et al, 2000a; Asahi et al, 2001).
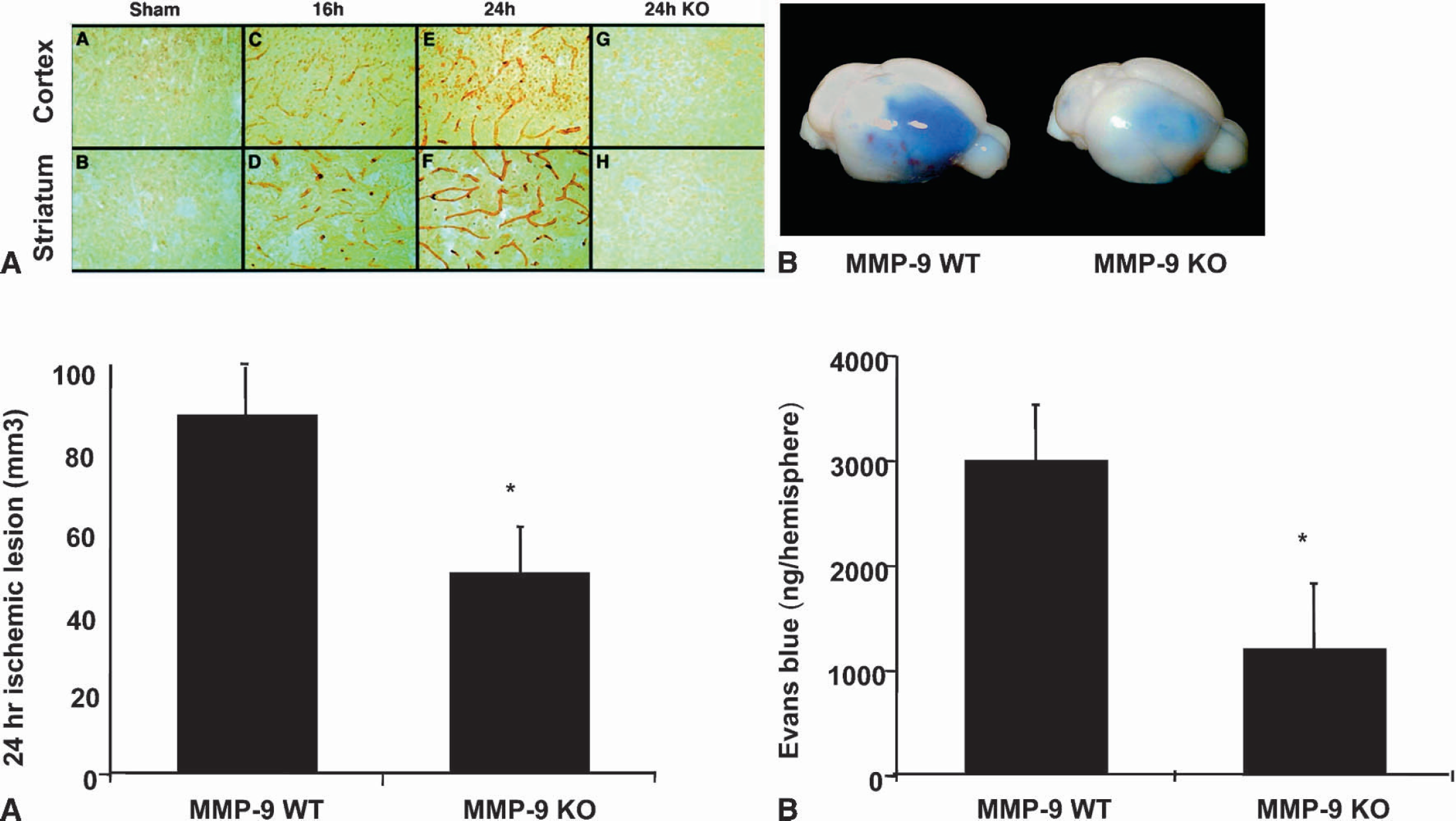
MMP-9 may underlie tPA-induced disruption of BBB: (A) (top) Increased endothelial and parenchymal MMP-9 immunoreactivity was observed in ischemic cortex and striatum in WT mice, whereas no such immunoreactivity was seen in sham-operated and MMP-9 KO littermates; (A) (bottom) smaller ischemic lesion in MMP-9 KO than in WT; (B) (top and bottom) MMP-9 KO mice had attenuated BBB disruption as seen in leakage of Evans blue. tPA, tissue plasminogen activator; KO, knockout; WT, wildtype; MMP, matrix-metalloproteinase. Reprinted with permission, from Society for Neuroscience (J Neurosci, 2001, 21:7724—7732)
LOW DENSITY LIPOPROTEIN RECEPTOR RELATED PROTEIN (LRP) SIGNALING
Because tPA may upregulate MMP-9 in cerebral ischemia, thus accounting for some of the neurovascular complications associated with thrombolytic therapy, it is important to dissect the signaling pathways involved. The low-density lipoprotein (LDL) receptor family comprises a group of cell surface receptors that transport a number of macromolecules into cells by receptor-mediated endocytosis. In particular, LRP is a multifunctional receptor expressed abundantly in astrocytes and brain endothelial cells. LRP has been implicated in various neuronal functions as well as in the pathogenesis of various neurodegenerative diseases (Kang et al., 1997; Lambert et al., 1998). Because LRP is abundantly expressed in brain, possesses cell-signaling properties, and avidly binds tPA, it is a reasonable candidate as a receptor mechanism to explain the phenomenon of tPA-induced MMP-9 and BBB perturbations (Herz, 2003).
Bacskai et al. (2000) demonstrated that activated a2-macroglobulin (α2M*), an LRP ligand, induces NMDA receptor mediated calcium influx in cultured primary neurons. They also postulate ligand-induced LRP stimulations serve as a neuronal sensor for proteolytic activity. This influx of calcium can also influence a wide variety of downstream signaling cascades that may include inositol triphosphate (IP3), protein kinase C (PKC), etc. (Alkon et al., 1998), which may affect dendritic excitability and synaptic efficacy. The association of tPA and LRP seems to be important in generating signaling response in neurons. Hippocampal neurons abundantly express LRP and its interaction with tPA plays an important role in induction of late phase LTP (Zhuo et al., 2000), probably because their interaction increases protein kinase A.
LRP serves as a crucial regulator of extracellular proteolytic activity through its effects on serine proteases and MMPs (Herz and Strickland, 2001). It is involved in the catabolism of MMP-2 (Yang et al., 2000), MMP-13 (Barmina et al., 1999) and MMP-9 (Hahn-Dantona et al., 2001). The ability of this receptor protein to modulate the activities of these MMPs is indicative of its role in removal of deleterious proteolytic activity. Kancha et al. (1994) have reported decreased LRP levels in tumors. This would mean a decreased catabolism and increased levels of MMP-9 at the tumor sites, leading to an enhanced proteolytic activity.
Wang et al. (2003) recently demonstrated a direct link between tPA, LRP, and MMP-9. After transient focal cerebral ischemia, brain MMP-9 levels were significantly lower in tPA knockout mice compared to wildtype mice. In cultured human brain endothelial cells, addition of exogenous tPA increased MMP-9 mRNA and protein. When LRP receptor levels were decreased by RNA interference or eliminated by gene knockout, the tPA-induced MMP-9 response was significantly reduced. Yepes and colleagues (2003) further showed that tPA infused intraventricularly into mouse brain opened the BBB, and this opening was ameliorated by the LRP antagonist RAP, but not the NMDA antagonist MK-801. Interestingly, they also showed that BBB leakage after focal cerebral ischemia was also dramatically reduced with the tPA inhibitor neuroserpin, both in WT and tPA -/- mice, but not in MMP-9 knockout mice, suggesting that in addition to MMP protease effects, tPA may also have direct actions at the BBB per se. Taken together, these data all point to new roles for tPA within the neurovascular unit beyond its intended clot lysis effects within the blood vessel in stroke (Lo et al., 2004).
COMBINATION WITH NEUROPROTECTION
The rationale for combination therapy with tPA thrombolysis is based on the increasing knowledge of its physiological and pathophysiological effects. The possible mechanisms of combination therapies in acute ischemic stroke should include the following: reduced tPA neurotoxicity, reduced risk of hemorrhage, decreased reperfusion injury, amplified neuroprotective effect and increased therapeutic time window. An effective postischemic neuroprotective agent would permit the benefits of tPA and block all the negative effects.
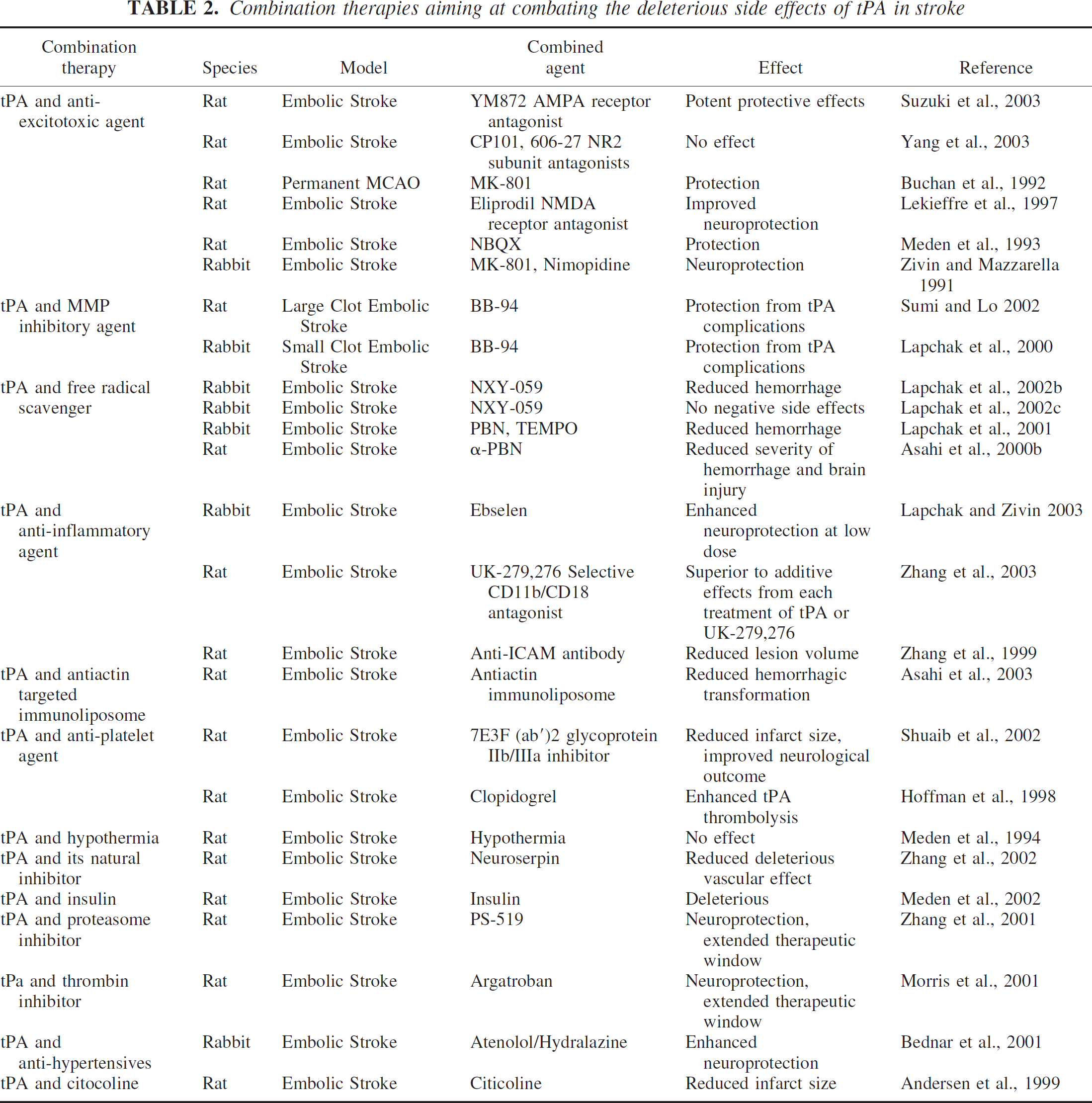
To this end, a series of studies have appeared in the literature in an attempt to come up with an optimal combination treatment to accompany tPA therapy (Table 2). Lo and colleagues (Asahi et al., 2000b) illustrated reduced injury using α-PBN, a free radical spin trapping agent, in combination with tPA, in a rat thromboembolic model of stroke. Another biotechnical approach utilized targeted immunoliposomes to reseal vascular disruptions thus ameliorating tPA-induced hemorrhagic conversions in a rat model of embolic stroke (Asahi et al., 2003). Use of MK-801 in a rat permanent MCA occlusion model caused a decrease in tPA induced injury (Warren et al., 2003). Reduction in infarct volume was also achieved after combining tPA and proteasome inhibitor PS-519 (Zhang et al., 2001). Anti-hypertensives, such as Atenolol and Hydralazine, were also protective in an embolic MCA occlusion model (Bednar et al., 2001). Sumi and Lo (2002) using BB-94, a broad spectrum MMP inhibitor, were able to achieve a decreased hemorrhage rate in a rat embolic stroke model. However, their study could not demonstrate any reduction in infarct volumes. Shuaib et al. (2002) reported reduced infarct volumes but an increased hemorrhage incidence when they combined 7E3-F(ab)2, a glycoprotein receptor antagonist, with tPA. Their study illustrated the protective effect of this antagonist against tPA induced complications, but only at a low dose.
Combination therapies aiming at combating the deleterious side effects of tPA in stroke
A combination of ethanol plus caffeine (Caffeinol) has been shown to be safe in humans, is effective in a rat model of focal stroke (Piriyawat et al., 2003, Aronowski et al., 2003), and is the focus of a current pilot clinical trial. However, in a rabbit multiple emboli model, the combination of caffeinol and tPA showed no benefit (Lapchak et al., 2004a), and might possibly increase the risk of hemorrhagic conversion.
The mechanism of action of caffeinol is still unknown. However, a number of mechanistic studies have looked at the mode of action of these agents individually. Caffeine, a competitive adenosine receptor antagonist, augments the brain adenosine receptor numbers when administered chronically (Rudolphi et al., 1989). This upregulation of adenosine receptors can lead to increased stimulation of inhibitory adenosine transduction pathways and hence can be neuroprotective (Miller and Hsu, 1992). Mechanistic studies on ethanol point towards adverse effects on ischemic outcome and do not provide any insight into the mechanism of caffeinol-induced neuroprotection. As with all combination therapies, responses in different stroke subtypes should be carefully assessed.
THE ROLE OF NEW THROMBOLYTIC AGENTS
An alternative approach is to use other modalities for thrombolysis. Initial attempts to use alternative thrombolytics in clinical trials, for instance streptokinase, resulted in increased mortality and all the trials ended prematurely (MAST-1 Group, 1995; MAST Group, 1996; Donnan et al., 1995). Despite this, there has been ongoing interest in the use of alternate thrombolytic agents.
In a direct comparison of tPA and staphylokinase (rSak) in a rabbit model of embolic stroke, rSak was significantly more fibrin-specific than tPA and at least as effective in lysing arterial emboli and limiting ischemia and neurological impairment (Vanderschueren et al., 1997). Another study directly compared tPA, streptokinase and rSak in a hamster model (Nagai et al., 1999b). Both focal cerebral ischemia and pulmonary thrombolysis were studied, and it was concluded that all three agents caused similar dose related extension of focal infarction.
Novokhatny and colleagues (2004) have recently discussed the potential of using plasmin as a new generation thrombolytic. Plasmin has direct fibrinolytic activity and can efficiently dissolve long, retracted thrombi deficient in plasminogen, thus omitting the need to use either plasminogen or its activators. Preclinical studies, in a rabbit model of abdominal aorta thrombosis (Marder et al., 2001), have been successful in demonstrating that plasmin could be a better and safer intervention than tPA in acute stroke. Nagai et al. (2003) have outlined the potential therapeutic properties of microplasmin, a truncated form of plasmin. Improved behavioral scores and decreased incidence of hemorrhage were observed following microplasmin administration in a rat embolism model (Lapchak et al., 2002a). Given its thrombolytic and neuroprotective properties, microplasmin is an excellent new candidate for developing treatments in stroke.
The increased incidence of hemorrhagic transformation after tPA therapy in acute stroke has led to the development of so-called third generation thrombolytics (Qureshi et al., 2002). By virtue of genetic and structural manipulations, these thrombolytics have longer half-lives and have a greater penetration into the thrombus matrix. TNK-tPA (tenecteplase) is one such third generation thrombolytic that has been genetically modified to not only prolong its half-life, but also exhibit fibrin specificity. It also has improved resistance to plasminogen activator inhibitor (Werner, 2001). Longer half-life and reduced systemic clearance rate allows TNK-tPA to be given by rapid bolus injection. tPA currently in use in stroke therapy has a half-life of 6 to 8 minutes from plasma to liver in humans. Evaluation of TNK-tPA in experimental animal models (Thomas et al., 1994; Collen et al., 1994) has resulted in faster and complete recanalization of occluded vessels. Postischemic intracarotid TNK-tPA treatment of rats with embolic stroke was shown to be effective in reducing neurological deficits and lesion volumes without causing any increase in hemorrhagic transformation, even when administered 4 hours after onset of ischemia (Zhang et al., 2000). However, Chapman et al. (2001) did not find any statistically significant differences in thrombolytic efficacy or hemorrhage rates between TNK-tPA and tPA interventions in a similar model of stroke. Lapchak et al. (2004b) compared TNK-tPA and tPA in rabbit embolic stroke model and have postulated TNK-tPA to be an effective therapeutic since it significantly improved behavioral scores over a wide dose range and with a long (3 hour) therapeutic window. The authors further hypothesize that since tenecteplase is effective up to 3 hours following embolization in rabbits, the time window in humans may be much longer than three hours. This underscores the importance of delivery of this compound in scenarios where stroke patients are unable to receive treatment under the current stipulated 3-hour time limit. Efficacy of TNK-tPA has also been demonstrated in various clinical trials and has shown reduced mortality among patients receiving late treatment and decreased incidence of non-cerebral hemorrhages (Armstrong et al., 2003; Van de Werf et al., 2001; Serebruany et al., 2003).
Vampire bat salivary plasminogen activator (desmoteplase or DSPA) has been studied in mouse models of ischemia (Liberatore et al., 2003). DSPA was compared to tPA in two different animal models of neurodegeneration. In both models, DSPA did not cause additional neurotoxicity. The authors suggested that the observed differences were because of different biochemical properties of the two proteases. The catalytic activity of DSPA is dependent on fibrin and independent of fibrinogen as a cofactor. DSPA is not normally expressed in the mammalian brain and therefore may not have the same properties as tPA in terms of the other physiologic actions discussed above. It would then potentially lack the ability to interact with neurons in the same way. Increasing attention is turning to early intervention because of the concerns of using these proteolytic agents that have protease activity. New approaches are being considered to open up the occluded vessels and minimize injury to the BBB. These also include use of non-thrombolytic strategies, for instance mechanical obliteration of the thrombi. The IMS II study (The IMS Study Investigators, 2004) involves using an ultrasonic vibrating device at the end of a catheter. Thrombus retrieval devices such as the Concentric device are also being employed to remove or extract obstructing thrombi from vessels without having to resort to the use of thrombolytics.
Reductions in brain temperature may be useful in reducing injury but may increase the risk of re-thrombosis, and dropping temperature will inactivate tPA whose activity is exquisitely temperature sensitive (Yenari et al., 1995). A recent approach is the use of continuous transcranial doppler (TCD) as a way of improving tPA's ability to induce reperfusion. TCD improves tPA's ability to reperfuse by: 1) increasing the surface area of the clot and thereby allowing more exposure to fibrin, 2) increasing temperature, and 3) increasing vasodilation. A positive trial, positive in terms of the proportion of patients with reperfusion and good clinical outcomes, has been reported. tPA plus continuous TCD is an improvement on tPA alone among patients treated under 3 hours (Alexandrov et al., 2004) not only in terms of the proportion of patients with reperfusion but also the clinical outcomes.
FUTURE: IMAGING OF BRAIN, BRAIN ISCHEMIA, AND INTRACRANIAL VESSELS
The ECASS study suggests that if more than 1/3 of the MCA territory of the brain was injured, hemorrhage was more likely. This 1/3 rule was hampered by the difficulty in the reliability of judging the 1/3 rule in terms of intra-and inter-observer reliability. A quantitative score, ASPECTS, has been able to select patients likely to benefit and to minimize risk (Barber et al., 2000). In the PROACT study (Hill et al., 2003), for patients with a MCA occlusion and a good scan (ASPECTS >7), there was a 50% absolute risk reduction compared to those assigned to placebo. This proves the point that if the scan shows the integrity of the brain is maintained, i.e. the integrity of the brain is preserved, but the MCA is reopened, then reperfusion will be successful. The corollary is that if the MCA remains occluded, whatever the state of the scan, the result will be uniformly bad, i.e. persistent MCA occlusion goes on to recruit extensive injury, which it did in all those assigned to placebo treatment.
Newer imaging modalities including perfusion and diffusion weighted imaging advance a perfusion/diffusion mismatch concept, leading to a putative tissue window. Newer trials that go beyond three hours are using mismatch to determine who might benefit if there is a large perfusion deficit, in the absence of much in the way of diffusion injury, when patients are being randomized to tPA (Diffuse, Epithet) or Desmoteplase (Butcher et al., 2003).
The initial study of IV/tPA using duteplase (Burroughs Wellcome Co., Research Triangle Park, NC, U.S.A.) was angiographically controlled; an angiogram was performed before and after treatment to determine the effect of treatment (del Zoppo, 1998). By imaging arteries we also get a sense of just how effective intravenous thrombolysis is at opening up vessels and IV/tPA seems to have a 25 to 40% rate of inducing recanalization compared to the 70 to 80% efficacy seen with intra-arterial therapy such as in the PROACT study. Vascular imaging is becoming more readily available with the increasing availability of non-invasive MRA or CTA imaging. Transcranial Doppler is also helpful if bone windows are adequate and good sonographic signal can be obtained. The ability to image vessels allows one to see whether or not treatment should be continued (or perhaps started in the first place), whether IV therapy is successful, or whether there should be some rescue therapy with IA intervention following the IV protocol. Trials such as IMS 1 have tested IV followed by IA-tPA. IMS 2 tests IV-tPA followed by intraluminal intervention using the EKOS catheter. Other trials are been developed, which will use methods that avoid thrombolysis (and its potential neurotoxic consequences) using obliteration or retrieval such as with the MERCI concentric device.
CONCLUSIONS
Clinically, tPA is the only definitive therapy for acute ischemic stroke. It must be given within hours, preferably 90 minutes. Although hemorrhage is the main concern, other concerns include its poor ability to open up vessels, (less than 25% efficient) and its potential neurotoxicity, particularly in the face of continuing obstruction to the vessel or failed thrombolysis. While it is a protease and works as a thrombolytic, it is also directly neurotoxic to the neuronal parenchyma when it crosses the blood brain barrier and, to make matters worse, its amplification of calcium currents through the NMDA receptor reflects properties that are the opposite of what we desire in a neuroprotectant. Toxicity is further compounded by tPA directly activating MMPs, perhaps through an LRP signaling mechanism, which disrupts the neurovascular matrix, and leads to breakdown of the blood brain barrier with increased risk of cerebral hemorrhage and edema.
The NINDS study was the first step in establishing acute stroke care in addition to the use of stroke units. Trials using imaging of both brain and vascular lesions will allow us to put tissue windows into place with effective new agents and new intraluminal techniques. A key breakthrough will be the addition of neuroprotective agents, which might come about through the understanding of the toxicity of tPA and the invention of agents that will block the toxicity without affecting its ability to thrombolyse.