
Abstract
Fibroblast growth factor (FGF)-2 is a potent neurotrophic and angiogenic peptide. To examine possible protective effects of FGF-2 gene expression against transient focal cerebral ischemia in rats, a replication defective, recombinant adenovirus vector expressing FGF-2, was injected intraventricularly 2 hours after middle cerebral artery occlusion (MCAO). The treatment group showed significant recovery compared with the vehicle-treated groups in terms of serial neurologic severity scores over the 35 days after MCAO. Further, 2,3,5-triphenyltetrazolium chloride staining showed that FGF-2 gene transfer decreased infarct volume by 44% as compared with that in the vehicle-treated groups at 2 days after MCAO. The same tendency of gene transfer effects on infarct volume was confirmed at 35 days after MCAO with hematoxylin/eosin staining. Enzyme-linked immunosorbent assay revealed that FGF-2 concentration was increased significantly at 2 days after MCAO, not only in cerebrospinal fluid but also in cerebral substance in the lesioned and treated animals. These results suggested that FGF-2 gene transfer using these adenoviral vectors might be a useful modality for the treatment of occlusive cerebrovascular disease even after the onset of stroke.
Keywords
Recently, treatment for occlusive cerebrovascular disease has progressed (Bogousslavsky et al., 2002;Longa et al., 1989). However, according to an announcement of the Ministry of Health, Labor and Welfare in 2002, stroke is still the third cause of the death in Japan and can result in various sequelae, including motor weakness, sensory disturbance, visual disturbance, and speech disturbance. Moreover, recovery is often limited in cases of severe ischemia. Fibroblast growth factor (FGF)-2 is an 18 kDa protein with potent trophic effects on brain neurons, glia, and endothelial cells (Barid, 1994). In particular, FGF-2 supports the survival and outgrowth of a wide variety of brain neurons both in vitro and in vivo (Barid, 1994;Patel and McNamara, 1995;Walicke, 1988). To date, studies of FGF-2 protein in ischemic animal models have shown reduction in infarct volume or neurologic recovery by intraventricular (Koketsu et al., 1994) or intravenous administration of FGF-2 protein (Fisher et al., 1995;Jiang et al., 1996). However, it may be unfortunately difficult for FGF-2 protein to pass through the blood brain barrier because of its high molecular weight. Furthermore, systemic administration of FGF-2 protein may cause digestive system complications, leukocytosis, and severe hypotension (Boussairi and Sassard, 1994), among other complications. In a previous report, we developed a method by which to transfer the FGF-2 gene, using a replication defective recombinant adenoviral vector, directly into the paraventricular region in healthy rat to augment the angiogenesis (Yukawa et al., 2000). In the present report, we examine possible protective effects of FGF-2 gene expression against transient focal cerebral ischemia in rats.
MATERIALS AND METHODS
Adenoviral vector
We prepared the replication-deficient adenoviral vector expressing human FGF-2 with an artificially fused interleukin (IL)-2 secretory signal sequence (AxCAMAssbFGF) (Yukawa et al., 2000). This vector was constructed according to the COS/TPC method (Akimoto et al., 1999;Matsuoka et al., 2000). The control vector was AxCALacZ carrying the Escherichia coli lacZ cDNA in the same expression unit as FGF-2. All virus vectors were propagated in 293 cells; then the viral seeds were purified by centrifugation in CsCl at a discontinuous gradient and dialyzed against 10% glycerol in PBS and then stored at −80°C until use.
Experimental protocol
Each animal was assigned to one of four experimental protocols, and each protocol was applied to three groups in random fashion. In protocol 1, we examined whether intraventricular administration of AxCAMAssbFGF affects neurologic function. Experimental groups consisted of the following: group 1, rats given middle cerebral artery occlusion (MCAO) alone (n = 6) (nontreated); group 2, intraventricular administration of AxCALacZ (used as a control vector) at the time of reperfusion MCAO (n = 4); and group 3, intraventricular administration of AxCAMAssbFGF at the time of reperfusion (n = 8). Neurologic evaluations were performed before ischemia and at 1, 4, 7, 14, 21, 28, and 35 days after MCAO. In protocol 2, we checked the infarct volume both in acute phase and chronic phase independently. To examine an effect of AxCAMAssbFGF upon infarct volume, brains of each group were removed, and we evaluated their infarct volumes at 2 or 35 days after MCAO. The numbers of animals used for each experiment are shown in the legend of Fig. 1. In protocol 3, we killed the animals at 2 days after MCAO to quantify the FGF-2 protein content in the cerebral cortex and in the cerebrospinal fluid in the brains (n = 4 in each group). In protocol 4, immunohistochemical study for FGF-2 was performed to examine the FGF-2 immunoreactivity at 2 days after MCAO. Experimental groups consisted of group 1, rats given MCAO alone (n = 3) (non-treated), and group 2, intraventricular administration of AxCAMAssbFGF at the time of reperfusion MCAO (n = 3).
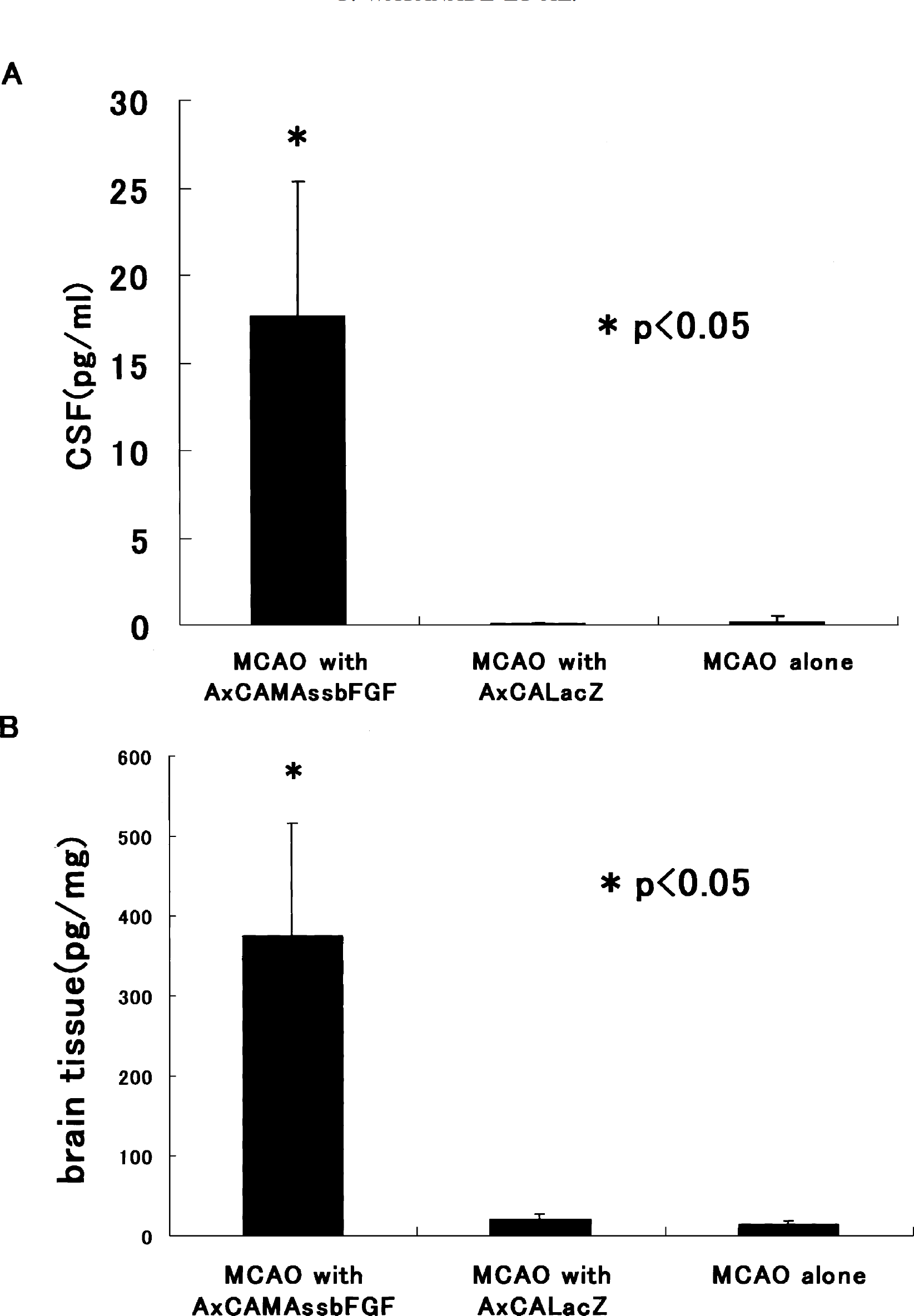
(A) ELISA for FGF-2 in the CSF of rats at 48 hours after MCAO. The Y-axis is the concentration of FGF-2. (B) ELISA for FGF-2 in the S-4 coronal slice. The FGF-2 protein content in the CSF and cortex was significantly higher in the AxCAMAssbFGF-treated group (n = 4; *P < 0.05) than in the MCAO-alone group (n = 4) or in the AxCALacZ-treated group (n = 4). Data represent mean ± SD.
Transient middle cerebral artery occlusion model
All procedures and virus inocula were approved by the Committee of Recombinant DNA and Animal Experiments, Osaka Medical College. Male Wistar rats (body weight, 270–310 g) were anesthetized with 1.0 to 2.0% halothane in 70% NO2/30% O2. Body temperature was maintained at 37° ± 0.5°C by use of a heating pad and rectal probe (Shibaura Electronics Co., Ltd., City, Japan) from the beginning of the surgical procedure through recovery from anesthesia. We induced the transient (2 hours) MCAO model by using the intraluminal suture method (Longa et al., 1989). In brief, the right common carotid artery, external carotid artery (ECA), and internal carotid artery were exposed through a midline neck incision, and the ECA and its branches were isolated and coagulated. A 4-0 nylon suture (18.5–19.5 mm, determined by the animal's weight), its tip, rounded by heating near a flame, was advanced from ECA into the lumen of the internal carotid artery until it blocked the origin of the MCA. After 2 hours of MCAO, the suture was carefully removed to restore blood flow, and then the skin was sutured.
In vivo gene transfer
Two hours after onset of MCAO (i.e., upon reperfusion), the animals were placed in a stereotactic head-holder (DAVID KOPF, model 900) under inhalation anesthesia described in this section. The dorsal surface of the skull was exposed by a mid-line skin incision. The location of the burr hole was 2 mm lateral to the right of the bregma. A small burr hole (1.5 mm in diameter) was drilled by an electric drill to avoid traumatic brain injury. A needle (31G) attached to a Hamilton syringe was inserted into the right lateral ventricle (3.5 mm in depth) (Paxinos and Watson, 1986), and 50 μL of AxCAMAssbFGF or AxCALacZ (containing 1 × 108 p.f.u) was slowly injected into the right lateral ventricle over the course of 15 minutes. After the needle was kept in place for 5 minutes and then withdrawn, the wounds were sutured. The injected rats were kept in a prone position for 20 minutes after administration to avoid variation of the infected region (Ooboshi et al., 1995). Immunosuppressive drug were not used in any animals.
Neurologic evaluation
Each animal's neurologic function was evaluated by a set of modified Neurological Severity Scores (NSS) (Borlongan et al., 1995;Chen et al., 1996;Schallert et al., 1997;Shohami et al., 1995) (Table 1) before MCAO and at 1, 4, 7, 14, 21, 28, and 35 days after MCAO by an investigator who was blinded to the experimental groups. Neurologic function was graded on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18). NSS is a composite of motor, sensory, reflex, and balance tests. In the severity scores of injury, one score point is awarded for the inability to perform the test or for the lack of a tested reflex; thus, the higher the score, the more severe the injury.
Neurologic Severity Score

One point is awarded for the inability to perform the tasks or for the lack of a tested reflex. 13 to 18 indicates severe injury; 7 to 12, moderate injury; 1 to 6, mild injury.
Determination of infarct volume
Infarct volumes after MCAO with or without gene transfer was determined by 2,3,5-triphenyltetrazolium chloride (TTC) or hematoxylin/eosin (H&E) staining. In brief, 2 days after the MCAO, the rats were deeply anesthetized with sodium pentobarbital at a dose of 100 mg/kg, and the brains were rapidly removed and cut into seven equally spaced (2 mm) coronal sections (designed S-1 to S-7 from the frontal pole to the midbrain). These slices were reacted with 2% solution of TTC at 37°C for 30 minutes to reveal the infarct area. TTC reacts with intact mitochondrial respiratory enzyme to generate a bright red color that contrasts with the pale color of the infarct (Chen and Cheung, 2002). The digitized images of the TTC-stained brain slice were analyzed using a software package (Scion Image, Version Beta 4.0.2, Scion Corporation, Frederick, MD, U.S.A.). An investigator blinded to the experimental groups then outlined the zones of infarction as well as the outlines of the left and right hemispheres on each section on the computer screen. Infarct volume (mm3) was determined by multiplying the appropriate area by the section's interval thickness. The infarct volume is presented as the percentage of infarct volume of the contralateral hemisphere (indirect volume calculation) (Paxinos and Watson, 1986). Additionally, 35 days after the MCAO, coronal sections of the brain were prepared as described above. A series of adjacent 10-μm thick sections were cut from each block in the coronal plane and then stained with H&E. Infarct volume was measured by the same method as described previously in this article.
Enzyme-linked immunosorbent assay for FGF-2
We quantified the concentrations of FGF-2 protein in the brain tissue and cerebrospinal fluid (CSF) (n = 4 in each group). The rats were anesthetized with sodium pentobarbital at a dose of 100 mg/kg at 2 days after MCAO, 50 to 100 μL of CSF were sampled from cisterna magna (Yukawa et al., 2000), and their brains were removed. The brains were cut as described above, and the S-4 coronal section (injection side) was used. The right half of the S-4 was homogenized with cell lysis buffer, and then centrifuged at 20,000 g for 45 minutes at 4°C. The supernatant was collected as a sample. The protein concentration of each supernatant or CSF was determined by the Bradford assay. Then we used a FGF-2 enzyme-linked immunosorbent assay (ELISA) kit (Wako Pure Chemical, Osaka, Japan), and 450 nm absorbance was determined on a microplate reader (Bio-Rad, CA, U.S.A.). Using these assays, FGF-2 protein concentrations of each sample were calculated.
Fibroblast growth factor-2 immunohistochemistry
For the immunohistochemical staining of FGF-2, the rats were anesthetized as described previously and perfused with the 4% buffered formalin. Then the brains were removed and embedded in paraffin, and 5-μm thick sections were cut from the block and placed on slides. After being deparaffined and rehydrated, the sections were microwaved in the Target Retrieval Solution (DAKO, Carpinteria, CA, U.S.A.) and then incubated for 30 minutes in 0.3% H2O2 in methanol to quench endogenous peroxidase activity. The sections were incubated with 1% normal blocking serum in phosphate-buffered saline (PBS) to prevent nonspecific binding of antibodies, 10 μg/mL of anti-FGF-2 (#05-117 Upstate, Lake Placid, NY, U.S.A.) in 1% albumin in PBS for 2 hours, and biotinylated anti-mouse IgG (Funakoshi, Tokyo, Japan) at a 1:200 dilution for 1 hour. This primary antibody can cross-react with FGF-2 from bovine, human, rat, and mouse sources. Reaction products were visualized with the VECSTAIN Elite ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.) and DAB substrate kit (Funakoshi, Tokyo, Japan). The sections were counterstained with hematoxylin and examined with light microscope.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc analysis for modified NSS, infarct volume, and quantification of FGF-2 amount. All data are expressed as mean ± SD. The differences were considered to be statistically significant at P < 0.05.
RESULTS
Neurologic outcome
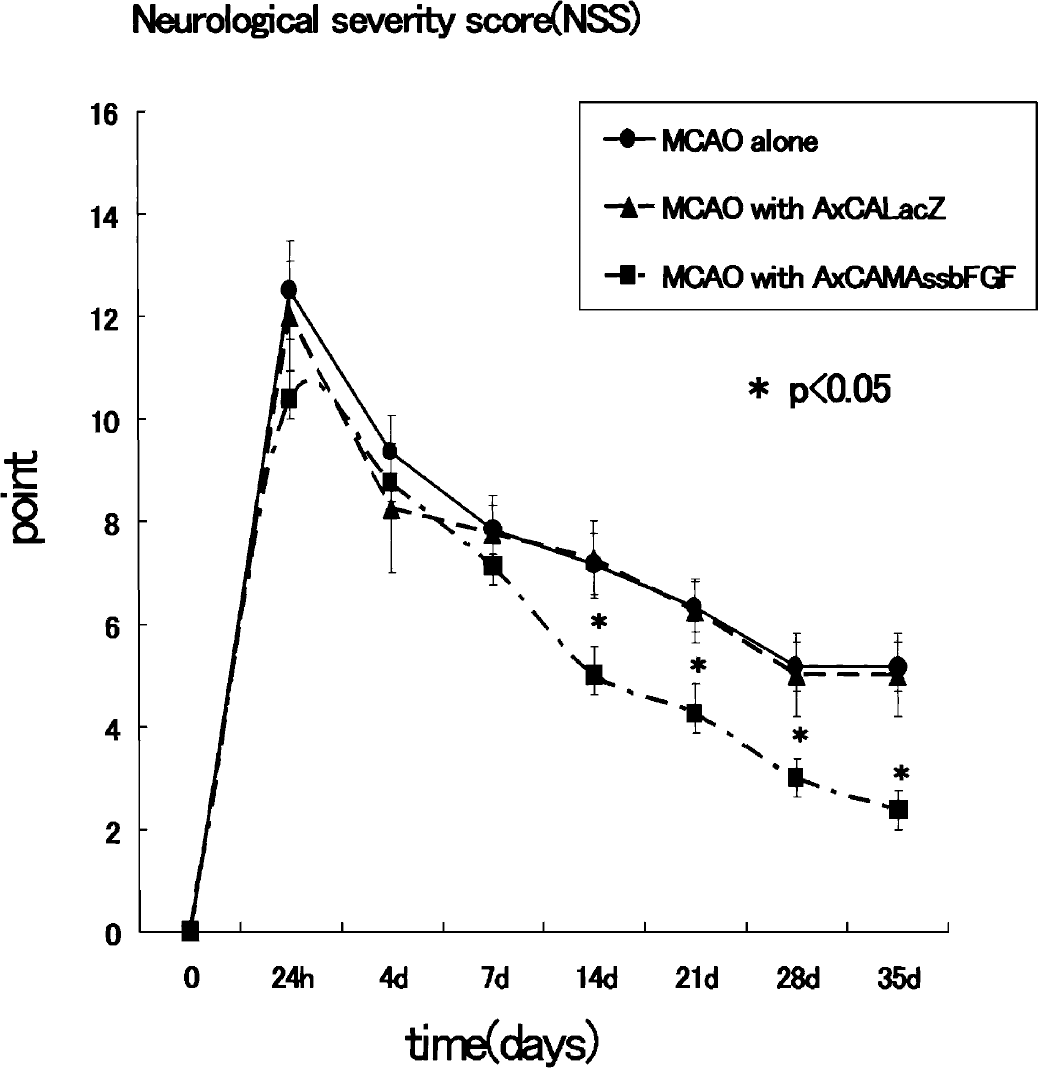
The changes in NSS in different groups are shown in Fig. 2. No significant difference in NSS was found between AxCALacZ-treated groups and MCAO-alone groups. However, AxCAMAssbFGF-treated rats had significantly lower NSS at days 14, 21, 28, and 35 as compared with the nontreated group and AxCALacZ-treated rats (P < 0.05). We confirmed that intraventricular administration of AxCAMAssbFGF contributed to neurologic improvement even after MCAO.
NSS before and after MCAO. Group1, MCAO alone (n = 6); group 2, MCAO with AxCALacZ (n = 4); group 3, MCAO with AxCAMAssbFGF (n = 8). *P < 0.05. Data represent mean ± SD. NSS, Neurological Severity Scores; MCAO, middle cerebral artery occlusion.
Infarct volume
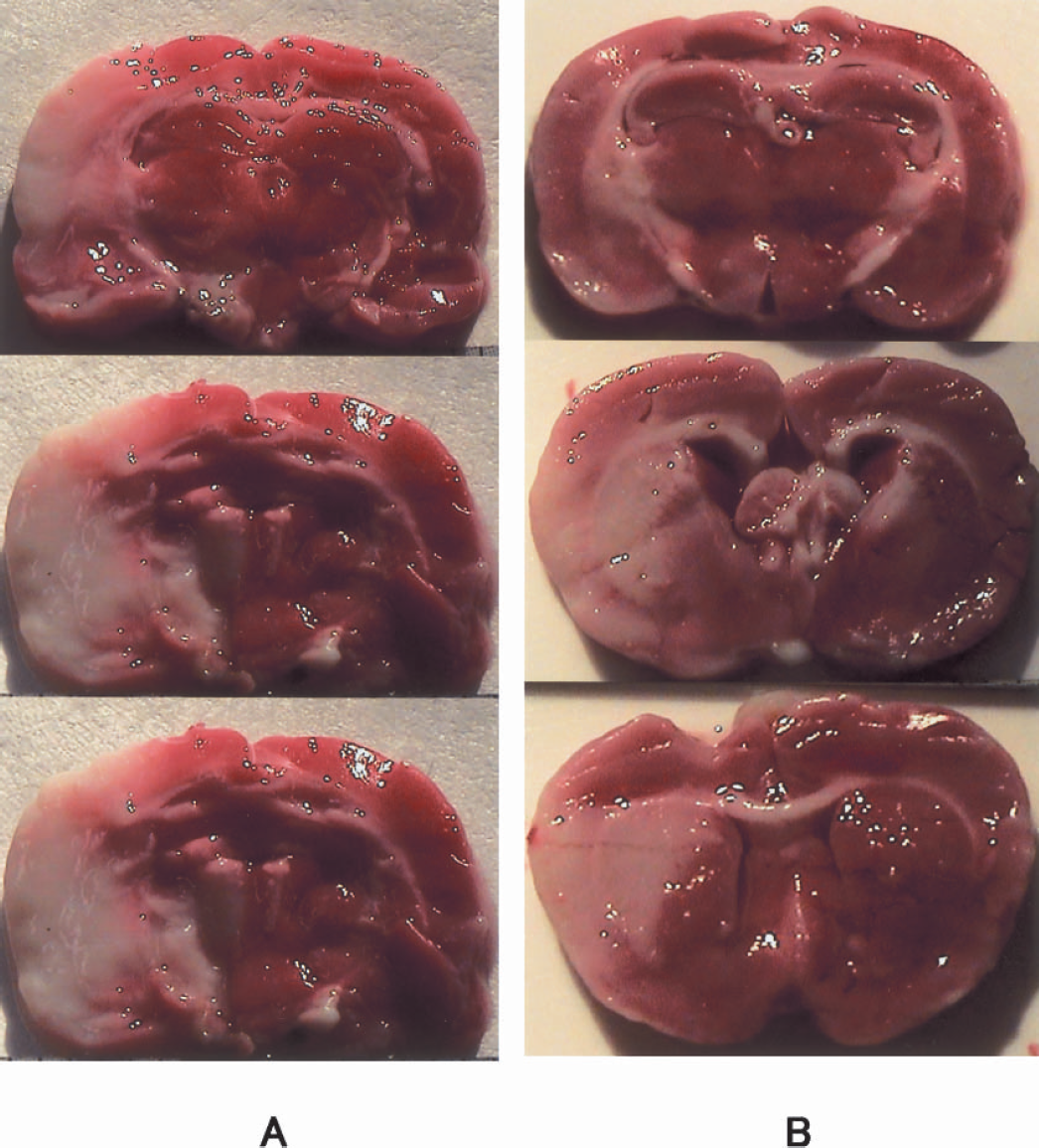
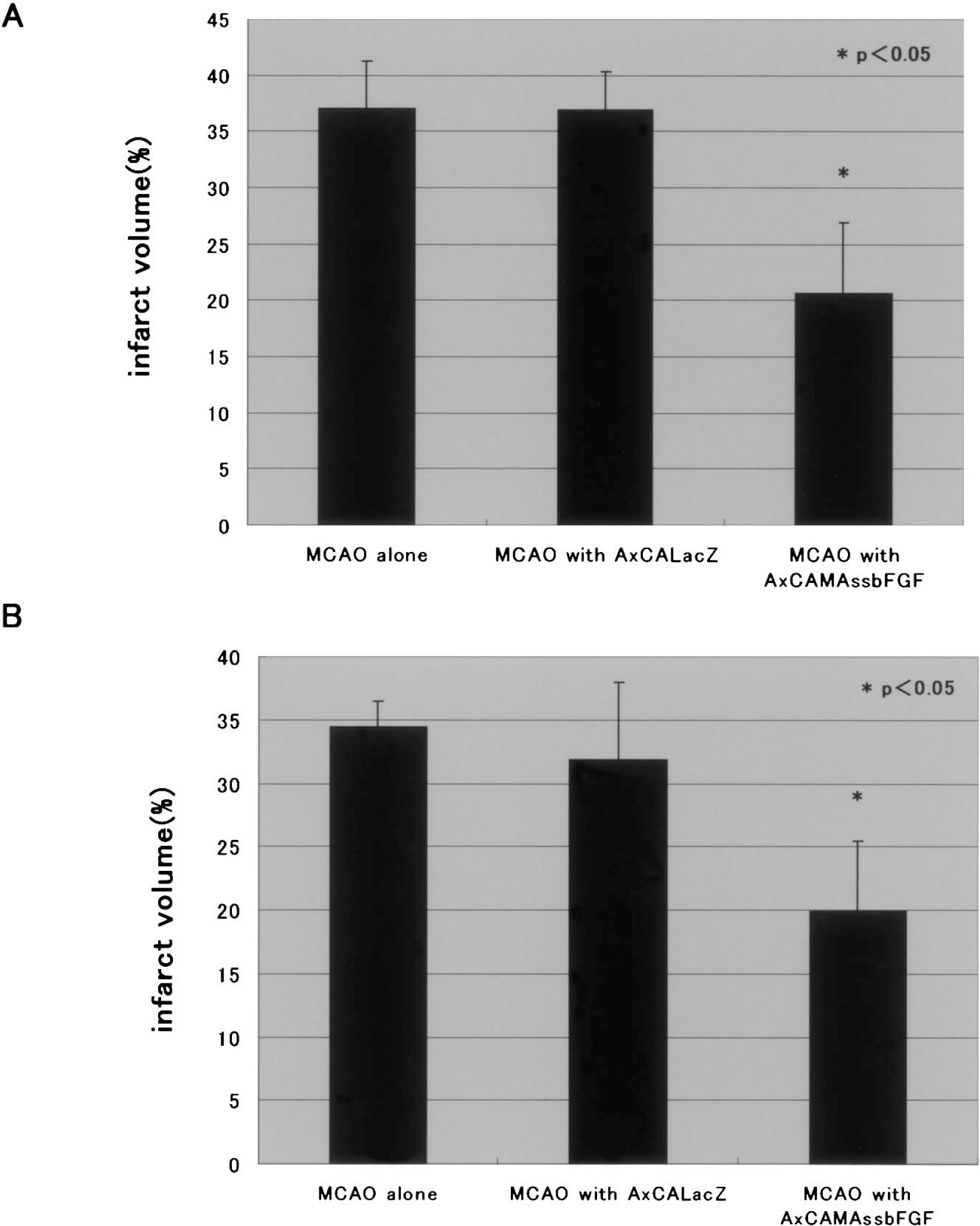
The intraluminal suture method resulted in an infarct in the right MCA perfused region. Representative photographs stained with TTC at 2 days after MCAO are shown in Fig. 3. The infarct volume values of the non-treated and AxCALacZ-treated groups at day 2 after MCAO were 37.0 ± 4.3% (n = 5) and 36.9 ± 3.4% (n = 4), respectively (Fig. 4A), but the difference between the two groups was not significant (one-way ANOVA). On the other hand, the infarct volume of the AxCAMAssbFGF-treated group was 20.6 ± 6.6% (n = 5). Postischemic intraventricular administration of AxCAMAssbFGF significantly reduced the infarct volume compared with that in control groups (P <0.05 versus nontreated and AxCALacZ-treated groups) (Fig. 4B). Quantitative assessment of infarct size revealed that the infarct size was significantly reduced by 44.3%via the postischemic intraventricular administration of AxCAMAssbFGF, compared with that in the nontreated group. The similar tendency was also confirmed at 35 days after MCAO with H&E staining, as shown in Fig. 4B. The infarct volume of the nontreated and AxCALacZ-treated groups at day 35 after MCAO were 34.4 ± 2.1% (n = 6) and 31.9 ± 6.1% (n = 4), respectively, and the infarct volume of the AxCAMAssbFGF-treated group was 19.9 ± 5.5% (n = 8).
Infarct areas are observed at 2 days after MCAO as pale regions by 2,3,5-triphenyltetrazolium chloride(TTC) staining. (A) Nontreated ischemic brain section shows infarct of MCA perfused area. (B) AxCAMAssbFGF-treatment reduced infarct volume in the cortical and subcortical areas.
(A) Infarct volume determined with TTC staining at 2 days after MCAO; volume was significantly reduced with intraventricular administration of AxCAMAssbFGF (n = 5; *P < 0.05) in comparison with that of MCAO alone (n = 5) or that of AxCALacZ (n = 4). (B) Infarct volume determined with H&E staining at 35 days after MCAO, which was also significantly reduced with intraventricular administration of AxCAMAssbFGF (n = 8; *P < 0.05) as compared with that of MCAO alone (n = 6) or that of AxCALacZ (n = 4). Data represent mean ± SD.
Quantification of immunoreactive fibroblast growth factor-2
To determine FGF-2 protein content in the brain tissue and cerebrospinal fluid in each group at 2 days after MCAO (n = 4 in each group), ELISA assays for FGF-2 were performed. A large amount of FGF-2 protein in the CSF (17.7 ± 7.7 ng/mL) was detected in the AxCAMAssbFGF-treated group (Fig. 1A). In the non-treated group and the AxCALacZ-treated group, however, only a slight amount of FGF-2 protein was detected in CSF. Furthermore, the level of FGF-2 protein in the brain tissue (S-4 coronal section) of the AxCAMAssbFGF-treated group (374.4 ± 140.8 pg/mg) was significantly higher (P <0.05) than that in the non-treated (20.5 ± 3.6 pg/mg) or the AxCALacZ-treated group (14.4 ± 2.1 pg/mg) (Fig. 1B).
Immunohistochemistry
Some cells of paraventricular brain tissue of the non-treated (MCAO alone) brain showed the immunoreactivity of FGF-2, but the number of the positive cells was limited and the immunoreactivity was not so strong (Fig. 5A). On the other hand, FGF-2 immunoreactivity was intensely observed not only in ependymal cells but also in three cell layers that consisted of ventricular wall of the rat treated with AxCAMAssbFGF (Fig. 5B). It is interesting to note that some staining patterns showed with a gradient fashion in the brain parenchyma.
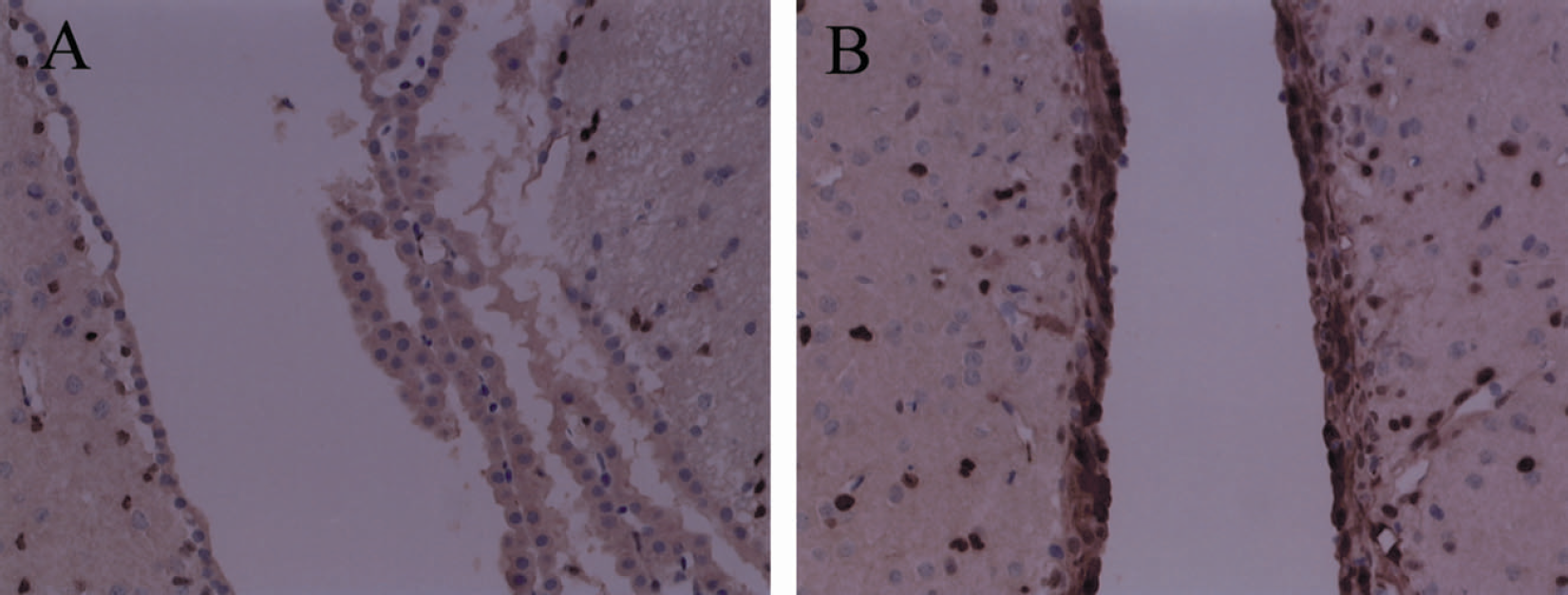
FGF-2 immunohistochemical study of the brain 2 days after MCAO. FGF-2 positive cells were stained with DAB (brown). (A) The right paraventricular region of MCAO alone (nontreated); (B) right paraventricular region of MCAO with treatment of AxCAMAssbFGF. Original magnification, ×200.
DISCUSSION
Thus far, there have been several reports of exogenous application of trophic factors, which can induce angiogenesis, neurologic recovery, and infarct volume reduction in animal stroke models (Bao et al., 1999;Fisher et al., 1995;Jiang et al., 1996;Kawamata et al., 1997;Kitagawa et al., 1998a;Koketsu et al., 1994;Yamashita et al., 1997). However, if these trophic factors, including FGF-2, are given intravenously as protein, it might be difficult for them to appear effectively in the ischemic brain because of the blood brain barrier. In addition, their half-life in vivo is too limited (Hawker and Granger, 1992;Moscatelli, 1992). In light of these considerations, we adopted the use of a replication defective virus vector, which has some advantages compared with protein administration, and showed that FGF-2 gene transfer using these adenoviral vectors might be a useful modality for the treatment of occlusive cerebrovascular disease even after the onset of stroke.
Adenovirus vectors have some advantages in comparison with other vectors, such as those of retrovirus or herpes simplex virus. Adenovirus vectors can be produced relatively easily at high titers. However, the gene of interest delivered by adenovirus vector is not integrated into the genome of the host cell, so that transgene expression is transient. It has been shown that the adenovirus vector can transfer the gene of interest with a high efficiency in lung, muscle, and brain tissue (Akli et al., 1993;Davidson et al., 1993;Quentin et al., 1992;Rosenfeld et al., 1991). Thus the adenoviral vector is one of the most suitable vectors for the treatment of stroke. We previously showed that intraventricular gene transfer with AxCAMAssbFGF can promote angiogenesis in the healthy rat brain (Yukawa et al., 2000) and that the same treatment can also promote neurogenesis in the gerbil forebrain ischemic model (Matsuoka et al., 2003). These results suggested that intraventricular administration of AxCAMAssbFGF might be useful for the treatment of ischemic cerebrovascular disease. In the present study we demonstrated that postischemic intraventricular administration of AxCAMAssbFGF can contribute to neurologic improvement and reduction of infarct volume after MCAO.
It was reported that the adenoviral vector itself could induce several proteins such as inflammatory cytokines, adhesion molecules and stress proteins in vivo (Cartmell et al., 1999;Kitagawa et al., 1998b;Schachtner et al., 1995). Therefore, there is a possibility that the adenoviral vector itself could influence or protect against ischemic brain injury. However, we found that intraventricular administration of AxCALacZ had no effect upon either neurologic recovery or infarct volume after transient MCAO (Figs. 2 and 4). Thus we concluded that these improvements were caused by the FGF-2 protein, which was produced by intraventricularly administered AxCAMAssbFGF.
In immunohistochemical analysis, some paraventricular cells of the rat treated with the vector showed intense immunoreactivity for FGF-2 with some gradient pattern, which is consistent with our previous study using other models (Matsuoka et al., 2003;Yukawa et al., 2000). However, the numbers of FGF-2 positive cells in the border zone between infarction and healthy brain were not apparently different in immunohistochemical analysis in both groups, that is, virus vector-treated and untreated groups (data not shown). Some investigators had already proven that even focal ischemia alone could increase FGF-2 immunoreactive positive cells in the borderzone, as discussed previously (Kumon et al., 1993, Speliotes et al., 1996). This coincides with our results. AxCAMAssbFGF has the transgene containing human FGF-2 cDNA, with an artificially fused IL-2 secretory signal sequence. This recombinant vector causes secretion of the protein into extracellular space that is approximately 10-fold greater than that in native FGF-2 cDNA, which lacks a secretory signal sequence (Yukawa et al., 2000). The data of ELISA show that a large amount of FGF-2 protein is detected not only in the CSF but also in the brain substance in response to only a single intraventricular administration of the virus vector. Therefore, we speculate that the increased FGF-2 protein both in CSF and brain substance could contribute to the reduction of infarction size and the improvement of the symptoms.
The neuroprotective mechanism of FGF-2 remains unclear. However, because high affinity FGF-2 receptors are widely distributed in neurons, glia, and some endothelial cells in the rodent brain (Wanaka et al., 1990), there are many possible mechanisms, and hypotheses have been demonstrated. First, FGF-2 could show antiapoptotic effects. This might be the main reason of the reduction of infarction volume in the present study. We demonstrated this protective effect of neuronal cell death by FGF-2-expressing adenoviral vector in vivo elsewhere (Akimoto et al., 1999). It has also been reported that FGF-2 prevented the downregulation of an antiapoptotic protein Bcl-2 and the DNA fragmentation in ischemic brain tissue (Ay et al., 2001). It was especially prominent in the cortex at the borders (“penumbra”) of infarcts, which had been rescued by the FGF-2 treatment. Such antiapoptotic effects may contribute the neuroprotective action of FGF-2. Second, FGF-2 could show the neuro-protection by promoting the angiogenesis. We have already shown that the gene transfer of FGF-2 using this vector induced angiogenesis (Yukawa et al., 2000). Also, some reports set forth that intracisternal administration of FGF-2 protein could improve neurologic behavior, while the infarction size did not alter if the treatment started 24 hours after the stroke (Kawamata et al., 1996, 1997). These studies suggest another mechanism of FGF-2 for functional recovery. Finklestein and his group demonstrated that this functional recovery by FGF-2 without reduction of infaction volume might be caused by upregulation of markers of neuronal plasticity, such as synaptophysin and GAP43, and stimulation of the new neuronal sprouting in the uninjured brain (Kawamata et al., 1996, 1997, 1999). Other mechanisms could include promoting the neurogenesis and the synaptic plasticity. Wagner et al. (1999) reported that subcutaneous injection of FGF-2 protein stimulated the neurogensis even in adult rat brain. In addition, Wada et al. (2003) reported ischemic insult and FGF-2 administration augmented neurogensis in rat brain. Recently, we also have shown the possible neurogenesis by the gene transfer of FGF-2 using this vector and ischemic insult (Matsuoka et al., 2003). These mechanisms might work in combination also in our study; however, it is still unclear, and it should be elucidated in the future which one was predominant.
Anyway, our preliminary results suggest that postischemic intraventricular administration of AxCAMAssbFGF improved neurologic score and satisfactory reduction of infarct volume in a permanent MCAO model (Watanabe and Miyatake, unpublished data, August 2003). These results suggest that FGF-2 gene transfer using these adenoviral vectors might be useful for the treatment of occlusive cerebrovascular disease with or without reperfusion. Moreover, we previously demonstrated that ex vivo gene transfer by this virus vector to effector cells could result in good angiogenesis in the rabbit hind limb ischemia model (Ohara et al., 2001) and in good regeneration of damaged heart tissue in swine (Ninomiya et al., 2003). In the next step, these fusion modalities using cell therapy and gene therapy may be applicable for occlusive cerebrovascular disease.