
Abstract
Transient cerebral ischemia leads to increased expression of ornithine decarboxylase (ODC). Contradicting studies attributed neuroprotective and neurotoxic roles to ODC after ischemia. Using antisense oligonucleotides (ODNs), the current study evaluated the functional role of ODC in the process of neuronal damage after transient focal cerebral ischemia induced by middle cerebral artery occlusion (MCAO) in spontaneously hypertensive rats. Transient MCAO significantly increased the ODC immunoreactive protein levels and catalytic activity in the ipsilateral cortex, which were completely prevented by the infusion of antisense ODN specific for ODC. Transient MCAO in rats infused with ODC antisense ODN increased the infarct volume, motor deficits, and mortality compared with the sense or random ODN-infused controls. Results of the current study support a neuroprotective or recovery role, or both, for ODC after transient focal ischemia.
Keywords
Ornithine decarboxylase (ODC) converts L-ornithine to putrescine, which is the obligatory precursor for the polyamines, spermidine and spermine. Transient cerebral ischemia leads to increased expression of ODC (Dienel et al., 1985; Paschen et al., 1988; Dempsey et al., 1988, 1991; Rohn et al., 1992; Muszynski et al., 1993; Rao et al., 1995; Keinanen et al., 1997). The functional significance of ODC after ischemia is controversial with equivocal evidence for both the neurotoxic and neuroprotective roles.
In support of the neurotoxic role, significant neuroprotection by the ODC inhibitor α-difluoromethylornithine (DFMO) was shown in animal models of forebrain and focal ischemia (Paschen et al., 1988; Schmitz et al., 1993; Muszynski et al., 1993; Kindly et al., 1994). Ornithine decarboxylase activation and the resulting putrescine accumulation promote the blood–brain barrier breakdown and edema formation after forebrain ischemia (Dempsey et al., 1988; Rao et al., 1995). However, induction of forebrain ischemia in ODC overexpressing transgenic and healthy syngenic mice led to comparable changes in cerebral energy metabolism, immediate early gene expression, and hippocampal neuronal numbers (Lukkarainen et al., 1995). Transgenic rats overexpressing ODC showed smaller infarcts than healthy rats after permanent or transient focal ischemia (Lukkarinen et al., 1998, 1999). These studies suggest a neuroprotective role for ODC and polyamines after an ischemic insult.
The availability of ODC knockout mice would clarify the role of ODC induction after ischemia. However, as polyamines are essential for controlling the DNA, RNA, and protein synthesis during cell growth and differentiation (Bernstein and Muller, 1999), gene knockout of ODC was considered untenable (Pietila et al., 1997). Although the possibility of a conditional knockout of ODC could not be ruled out, none are currently available. Antisense oligodeoxynucleotides (ODNs) bind to a specific mRNA to prevent its translation and can effectively knockdown a protein locally (Rao et al., 2001a, 2001b). To understand the role of ischemia-induced ODC activation, transient focal cerebral ischemia was induced in rats in which the cerebral ODC protein was knocked down by continuous intracerebroventricular infusion of antisense ODNs specific for the translation initiation codon of ODC mRNA. The effect of antisense knockdown of ODC on ischemia-induced infarct volume, neuronal deficit, and mortality were evaluated.
MATERIALS AND METHODS
Animals
Adult male, spontaneously hypertensive rats (SHR; 250 to 300 g; Charles River, Wilmington, MA, U.S.A.) were used in these studies. Rats were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services Publication number 86–23 (revised 1986). All surgical procedures were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison.
Antisense knockdown
Ornithine decarboxylase antisense ODNs were designed based on rat ODC mRNA (Wen et al., 1989). Initial studies showed that the antisense ODN (5′-TCC TTA GTA AAG CTG CCC AT-3′) that targeted the translation initiation codon (nucleotides 434–453) was more effective than the antisense ODN (5′-TAA GAG CTA CAA GAA TGG CA-3′) that targeted the translation termination codon (nucleotides 1821–1840) in knocking-down ODC immunoreactive protein and catalytic activity. For routine studies, the translation initiation codon targeting antisense ODN was used. Control animals were infused with the sense (5-TCC TTA GTA AAG CTG CCC AT-3′) and the random (5′-TCA TTA CCG GGA TCC TCA AT-3′) ODNs, in which the proportion of each nucleotide is identical to that of the antisense ODN. None of the ODN sequences was related to any other nucleotide sequence in Genebank. The ODNs were synthesized with a phosphorothioate backbone and purified to analytical grade on high pressure liquid chromatography by Oligos Etc. (Wilsonville, OR, U.S.A.). The ODNs were infused as described earlier (Rothstein et al., 1996; Rao et al., 2001a, 2001b). The lyophilized ODNs were reconstituted (3.125 mg/mL) in artificial cerebrospinal fluid (aCSF; 119 mmol/L NaCl, 3.1 mmol/L KCl, 1.2 mmol/L CaCl2, 1 mmol/L MgSO4, 0.50 mmol/L KH2 PO4, 25 mmol/L NaHCO3, 5 mmol/L D-glucose, 2.2 mmol/L urea, pH 7.4), dialyzed overnight (SpectraPor cellulose ester, MW cutoff 2,000; Thomas Scientific) at 4°C, filtered (0.22 μm), and filled into osmotic minipumps. Each pump was connected to a stainless steel cannula by peristaltic tubing and primed overnight at 37°C. The cannula was implanted into the lateral ventricle (coordinates −1.5 mm, lateral; −0.8 mm, anterior-posterior; −4.8 mm, dorsal-ventral; based on the rat brain atlas of Paxinos and Watson, 1998) and secured to the skull with dental cement. The pump was placed in the skin fold on the neck of the rat, under halothane anesthesia. The osmotic minipumps used were Alzet Model 2001 (Alza Pharmaceuticals, Palo Alto, CA, U.S.A.), which will pump at a rate of 1 μL/h for 7 days. Thus, each rat received 78 μg of ODN/day (3.125 μg/μL · hr). After 5 days of ODN infusion, rats were subjected to either transient middle cerebral artery occlusion (MCAO; 1 hour) or sham operation. After 1 day of reperfusion, rats were neurologically evaluated and perfused transcardially with buffered paraformaldehyde. The brains were then removed, postfixed and cryoprotected. The effect of antisense, sense, and random ODN infusion on the levels of ODC protein was evaluated by Western blotting. Proper functioning of the osmotic minipumps was confirmed by weighing the filled pumps before implantation and immediately after killing the rats. The average total volume of pumping was observed to be 136 ± 13 μL in 6 days (0.94 ± 0.09 μL/hr; n of 73). Correct placement of the cannula into the lateral ventricle was confirmed by examining the thionine stained brain slices.
Transient middle cerebral artery occlusion
Rats were anesthetized with halothane (induction: 2%; maintenance: 1.2%) in an oxygen and nitrous oxide (50:50) mixture. Rats were ventilated mechanically with a rodent ventilator (Model 683; Harvard Apparatus, South Natick, MA, U.S.A.) through an endotracheal tube (PE-240 polyethylene tubing; Harvard Apparatus). The left femoral artery was cannulated for continuous monitoring of arterial blood pressure and to obtain blood samples for the measurements of pH, PaO2, PaCO2, hemoglobin, and blood glucose concentration (I-STAT; Sensor Devices, Waukesha, WI, U.S.A.). PaO2 and PaCO2 were maintained between 100 to 200 mm Hg and 30 to 40 mm Hg, respectively.
Middle cerebral artery occlusion was conducted by an intraluminal suture technique as described previously (Zea Longa et al., 1989; Dogan et al., 1999; Rao et al., 2001b). The rat was placed in a stereotaxic frame, a craniectomy (4 mm in diameter, 2 to 4 mm lateral and 1 to 2 mm caudal to bregma) was performed with extreme care over the MCA territory using a trephine. The dura was left intact. A laser–Doppler flowmeter probe (model PD-434; Vasamedics, St. Paul, MN, U.S.A.) was placed on the surface of the ipsilateral cortex (ischemic area) and fixed to the periosteum with a 4–0 silk suture. The probe was connected to a laser flowmeter device (Laserflo blood perfusion monitor BPM 403A; TSI, St. Paul, MN, U.S.A.) for continuous monitoring of regional cerebral blood flow (rCBF). The left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed through a ventral midline incision. A 3–0 monofilament nylon suture with a rounded tip was introduced into the ECA lumen and gently advanced to the ICA until slight resistance was felt and a reduction in rCBF was seen. The rCBF dropped to 12% to 18% of the baseline in 40 to 50 seconds and remained at that level throughout the occlusion period. After 1 hour of occlusion, the suture was withdrawn to restore the CCA-ICA-MCA blood flow (confirmed by laser–Doppler flowmeter). In less than 5 minutes after the withdrawal of the suture, the rCBF returned to the baseline level and remained unchanged through 90 minutes of reperfusion. Body and cranial temperatures were maintained with a heating blanket and a lamp at 37°C to 38°C and 36°C to 37°C, respectively, during the occlusion (1 hour) and 90 minutes of reperfusion. After recovering from anesthesia, rats were returned to their cages with free access to food and water. After one day, rats were perfused transcardially with buffered paraformaldehyde, and the brains were postfixed and cryoprotected.
Endpoint regional cerebral blood flow measurements
To confirm that ODC antisense ODN treatment had not changed the rCBF during ischemia, end ischemic rCBF was measured in additional cohorts by 4-iodo-[N-methyl- 14C]antipyrine ([14C]AIP) autoradiography as described earlier (Rusa et al., 1999; Rao et al., 2001b). For this, in a separate set of sense and antisense ODN-infused rats, the laser–Doppler flowmeter probes were attached, arterial and venous femoral catheters were inserted, and the MCA was occluded. At 60 minutes of occlusion, arterial blood pressure, PO2, PCO2, and pH were measured, and 40 μCi of [14C]AIP (specific activity, 54 mCi/mmol; Amersham Pharmacia Biotech, Arlington Heights, IL, U.S.A.) in 0.8 mL of isotonic saline was infused intravenously for 45 seconds. Simultaneously, the arterial catheter was opened and 15 free-flowing 20-μL samples were collected into heparin-coated tubes. With the filament still in place and the laser–Doppler indicating the occlusion, the rat was decapitated and the brain was snap-frozen by dripping in isopentane cooled to −30°C and stored at −80°C. Each brain was sectioned (20-μm-thick) on a cryostat and the sections from 5 coronal levels (+2.2, +0.5, −0.9, −2.1, and −3.8 from Bregma) were collected and exposed for 1 week to hyperfilm βmax (Amersham) together with 14C standards. Blood samples were decolorized with tissue solubilizer and the radioactivity was estimated by liquid scintillation spectrometry. Autoradiographic images were digitized using MCID image analysis system (Imaging Research, St. Catherine's, Ontario, Canada) and the rates of rCBF were determined as previously described (Alkayed et al., 1998).
Histopathology
Each brain was sectioned coronally (40-μm-thick at an interval of 320 μm), stained with thionine, and scanned using the NIH Image program. The volume of the ischemic lesion was computed by the numeric integration of data from 16 to 19 serial sections in respect to sectional interval. To account for the cerebral edema and differential shrinkage resulting from tissue processing, the injury volumes were corrected using the following formula: corrected injury volume = contralateral hemisphere volume − (ipsilateral hemisphere volume − measured injury volume) (Swanson et al., 1990).
Neurologic evaluation
Neurologic deficits were evaluated on a 6-point scale (Zea Longa et al., 1989) before transient MCAO and at 1 day of reperfusion (before killing the animals) by an investigator blinded to the study groups. A score of 0 suggests no neurologic deficit (healthy); 1 suggests mild neurologic deficit (failure to extend right forepaw fully); 2 suggests moderate neurologic deficit (circling to the right), 3 suggests severe neurologic deficit (falling to the right), and 4 suggests very severe neurologic deficit (the rat did not walk spontaneously and had a depressed level of consciousness).
Western blotting
Immunoblotting was performed as described earlier (Rao et al., 1999). Brain tissue was homogenized in ice-cold 25 mmol/L Tris-HCl (pH 7.4) buffer containing 2 mmol/L EDTA and protease inhibitors (aprotinin, pepstatin-A, leupeptin, bestatin, 4-(2-aminoethyl) benzenesulfonyl fluoride, and trans-epoxysuccinyl-L-leucylamido(4-guanidino) butane). Protein content was estimated by the method of Lowry et al. (1951). Equal volume of Laemmli electrophoresis sample buffer (5% sodium dodecyl sulfate, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue and 0.125 mol/L Tris-HCl buffer, pH 6.8; Sigma, St. Louis, MO, U.S.A.) was added to 15 μg protein equivalent of each sample, and the proteins were solubilized by heating at 94°C for 3 minutes. Samples were electrophoresed on polyacrylamide gels, transferred to polyvinylidene difluoride, and probed with monoclonal anti-ODC antibody (Sigma) and horse radish peroxidase-coupled anti-mouse IgG. The blots were stripped and reprobed with anti-β-tubulin antibody (Sigma). The protein bands recognized by antibodies were visualized by enhanced chemiluminescence (ECL Western blotting kit; Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) and the band intensities were quantified by densitometric scanning using the NIH Image program. Before immunodetection, blots were stained with Ponceau-S to confirm the protein loading and transfer efficiency.
Ornithine decarboxylase enzyme assay
Ornithine decarboxylase activity was assayed as described earlier (Rao et al., 1998). Brain tissue was homogenized in 50 mmol/L Tris-HCl buffer (pH 7.8) containing 40 μm pyridoxal-5-phosphate, 7.5 mmol/L dithiothreitol, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), and 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF). The homogenate was centrifuged at 4°C (43,000 g for 20 minutes) and the supernatant was incubated in the buffer containing 0.5 μCi L-[14C]ornithine (specific activity, 47.7 Ci/mmol; NEN Life Sciences Products, Boston, MA, U.S.A.) at 37°C for 1 hour. Incubations were performed in airtight tubes with center wells containing filter paper strips soaked in sodium hydroxide. The 14CO2 formed during the reaction trapped in the center wells was estimated by liquid scintillation spectrometry.
Statistical analysis
Results are presented as mean ± SD. Data from multiple groups were analyzed statistically by one-way analysis of variance followed by Tukey–Kramer multiple comparisons posttest. P < 0.05 was considered statistically significant.
RESULTS
In the Results section, antisense refers to antisense ODN-infused group, sense refers to sense ODN-infused control group, random refers to random ODN-infused control group, aCSF refers to aCSF-infused control group, sham refers to sham-operated control group, and MCAO refers to transient MCAO/reperfusion group.
Antisense infusion knocked down ornithine decarboxylase protein levels and activity
In sham-operated rats, cerebral cortex showed low ODC catalytic activity (5.8 ± 2.4 pmol/mg protein · h) and protein levels (Western blot analysis showed a faint ODC immunoreactive protein band similar to the contralateral cortex samples in Fig. 1). Transient MCAO induces ODC activity and protein levels in the ipsilateral cortex compared with the sham cortex, between 3 to 24 hours (with a peak at 6 hours) of reperfusion. Catalytic activity increased by 5-fold (at 3 hours), 9.9-fold (at 6 hours), and 4.7-fold (at 24 hours) in the ipsilateral cortex compared with the sham cortex. The ODC immunoreactive protein levels increased by 4.1-fold (at 3 hours), 7.4-fold (at 6 hours), and 3.7-fold (at 24 hours) in the ipsilateral cortex compared with the sham cortex. In comparison with the sham cortex, the contralateral cortex showed no significant changes in either the ODC activity or protein levels at any reperfusion period. Hence, in the current study, the efficiency of ODC antisense infusion on ODC protein knockdown was verified at 6 hours of reperfusion after transient MCAO. The ipsilateral cortex and striatum of the sense and MCAO group showed significantly greater ODC protein levels (by 5-to 6-fold, P < 0.01) compared with the respective contralateral cortex and striatum at 6 hours of reperfusion (Fig. 1). Infusion of ODC antisense significantly blocked the transient MCAO-induced increase in ODC protein level (by 84% ± 12%, P < 0.01) in ipsilateral cortex and striatum (Fig. 1). At 6 hours of reperfusion in the sense and MCAO group, there was a significant increase in the ODC catalytic activity in the ipsilateral cortex (by 3.5-fold, P < 0.01) and striatum (by 6.4-fold, P < 0.01), compared with the contralateral cortex and striatum (Fig. 2). Antisense infusion completely prevented the transient MCAO-induced increase in ODC activity in ipsilateral cortex and striatum (Fig. 2). Sham-operated rats infused with either sense or antisense showed little ODC activity (3.5 ± 0.6 and 2.6 ± 0.4 pmol/mg protein · h in cortex and striatum, respectively).
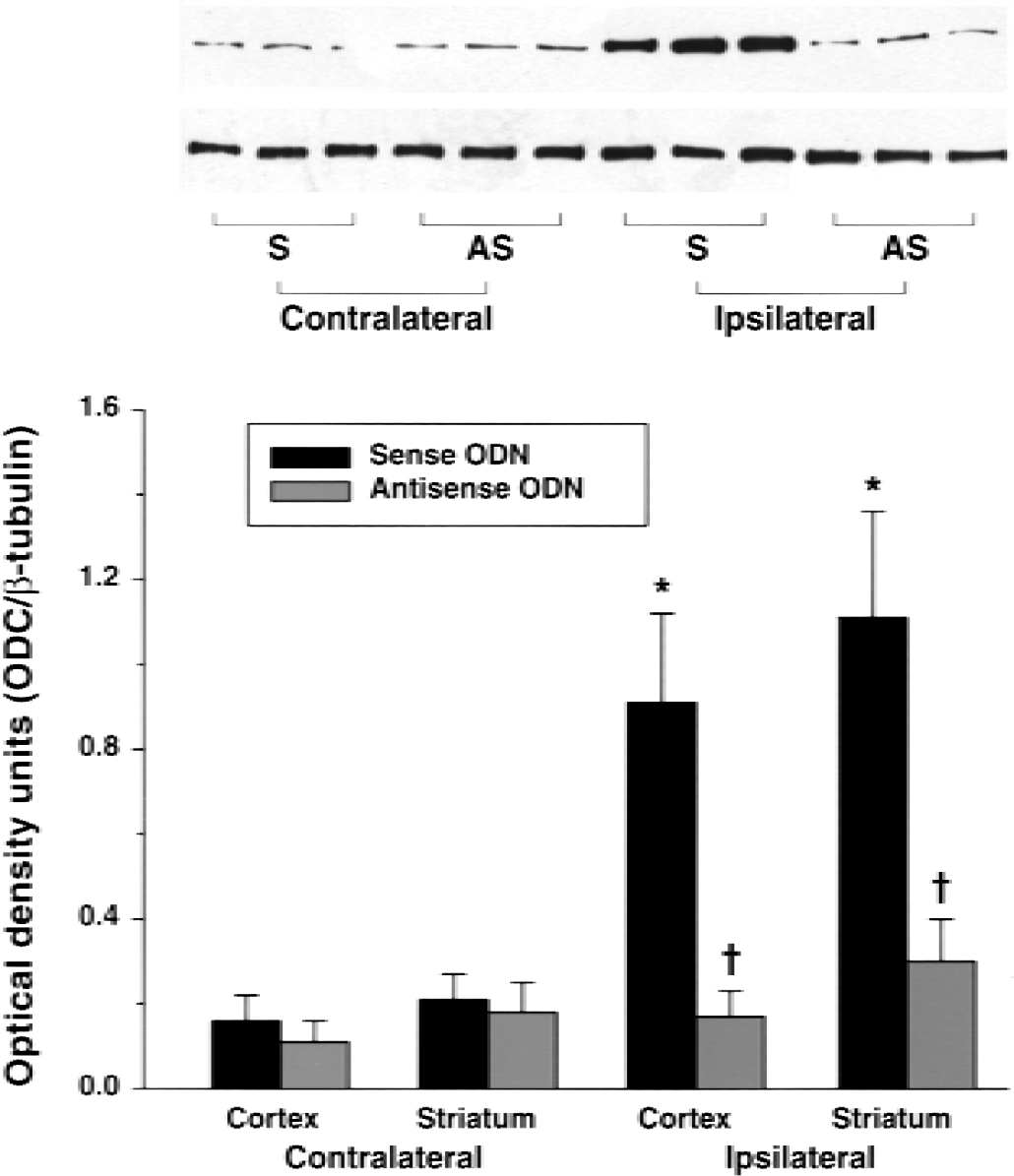
Western blot analysis of ornithine decarboxylase (ODC) immunoreactive protein in the brains of rats undergoing transient middle cerebral artery occlusion (MCAO) after the infusion of sense and antisense oligonucleotides (ODNs). In the sense-infused rats, transient MCAO resulted in a significant increase in the ODC protein levels in the ipsilateral cortex and striatum compared with the contralateral cortex and striatum. Antisense infusion significantly prevented ischemia-induced increase in the ODC protein levels in the ipsilateral cortex and striatum. Cerebral cortex and striatum from the sham-operated controls showed little ODC protein levels bilaterally. Values in the histogram are mean ± SD of four animals in each group. For each animal, Western blotting was conducted twice. * P < 0.05 (sham vs. MCAO); †P < 0.05 (sense vs. antisense) by one-way analysis of variance followed by Tukey-Kramer multiple comparisons posttest.
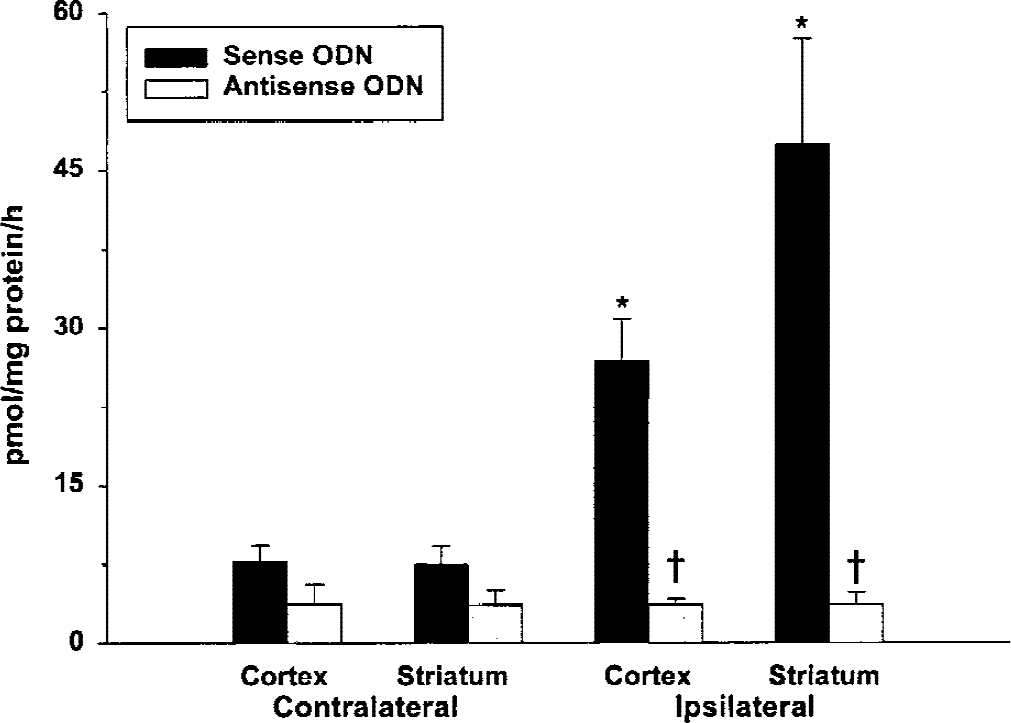
Antisense infusion significantly prevented the transient middle cerebral artery occlusion–induced increase in ornithine decarboxylase (ODC) activity. ODC activity was estimated in triplicate with <10% variation. Sham-operated rats infused with sense and antisense showed little ODC activity in cortex and striatum (3.5 ± 0.6 and 2.6 ± 0.4 pmol/mg protein/h, respectively). * P < 0.05 (contralateral vs. ipsilateral); †P < 0.05 (sense vs. antisense) by one-way analysis of variance followed by Tukey-Kramer multiple comparisons posttest.
Ornithine decarboxylase knockdown increased the ischemic infarct volume
In a group of rats not subjected to intracerebroventricular infusion, transient MCAO (1 hour) and reperfusion (24 hours) resulted in an infarct in the ipsilateral side of the brain with a total volume of 203 ± 28 mm3(cerebral cortex: 168 ± 31 mm3, striatum: 31 ± 5 mm3) (Table 1). Infusion of aCSF, sense, or random had no significant effect on the transient MCAO-induced infarct volume, whereas in the antisense/MCAO group, there was a significant increase in the total (by 33%;P < 0.05), cortical (by 35%, P < 0.05), and striatal (by 32%, P < 0.05) infarct volume compared with sense/MCAO or random/MCAO controls (Table 1). Figure 3 shows representative thionine-stained coronal sections from the brains of sense/MCAO, antisense/MCAO, sense/sham, and antisense/sham groups. There were no significant differences between the antisense/MCAO, sense/MCAO, and random/MCAO groups in any of the physiologic parameters (MABP, pH, PCO2, PO2, hemoglobin, and blood glucose) (Table 2) and the rCBF measured by laser–Doppler flowmetry (Fig. 4). The endpoint rCBF measured at 1 hour of MCAO, using [14C]AIP autoradiography, also was not significantly different between the sense and antisense-infused rats (Fig. 5).
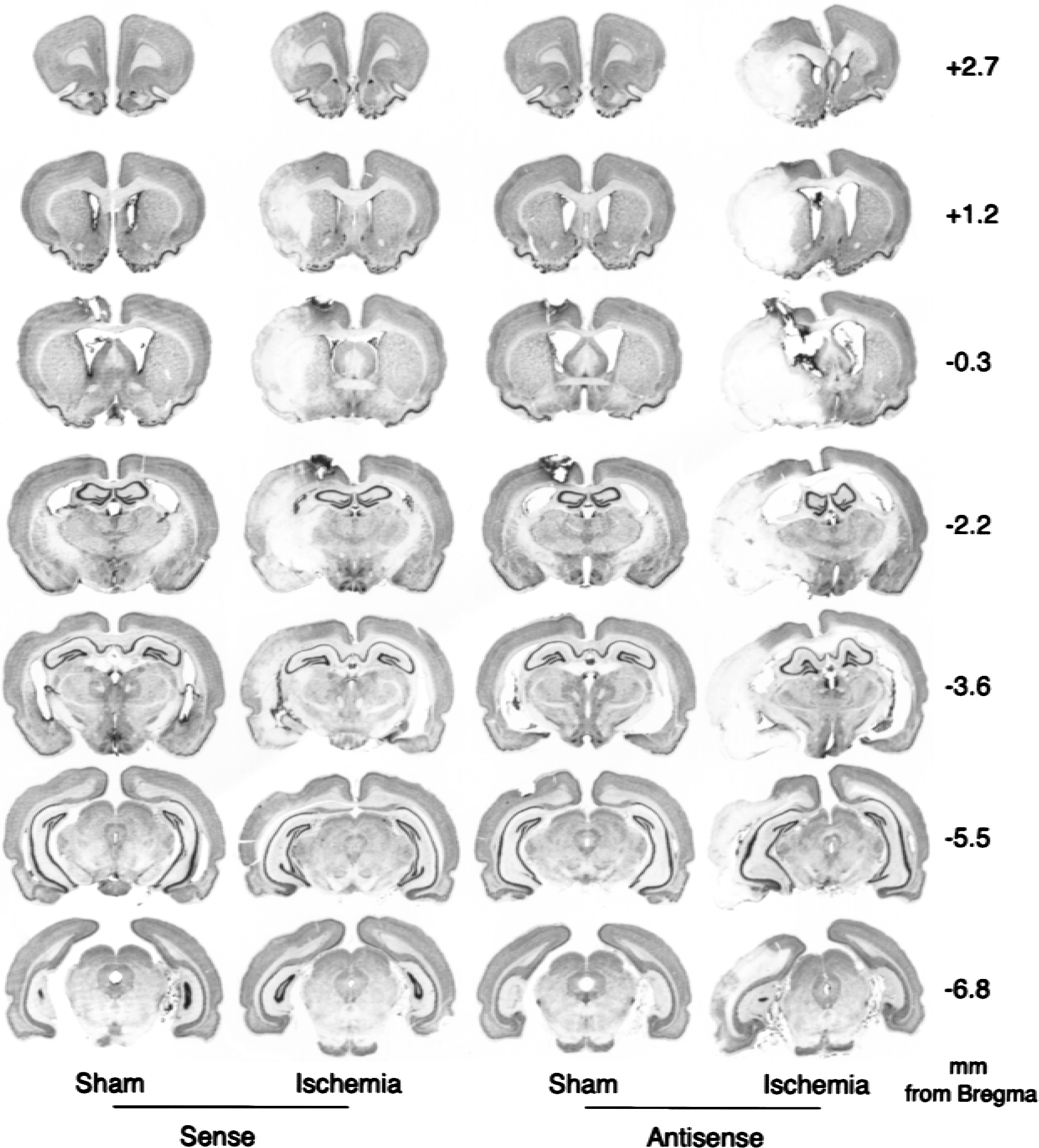
Thionine-stained serial coronal sections from the brains of representative rats infused with sense and antisense, which underwent transient middle cerebral artery occlusion (ischemia) or sham operation (sham). Figure shows only the sense-infused controls, as there were no observable differences between the sense-and random-infused groups. Mean ± SD of the total, cortical, and striatal infarct volume in each group is given in Table 1.
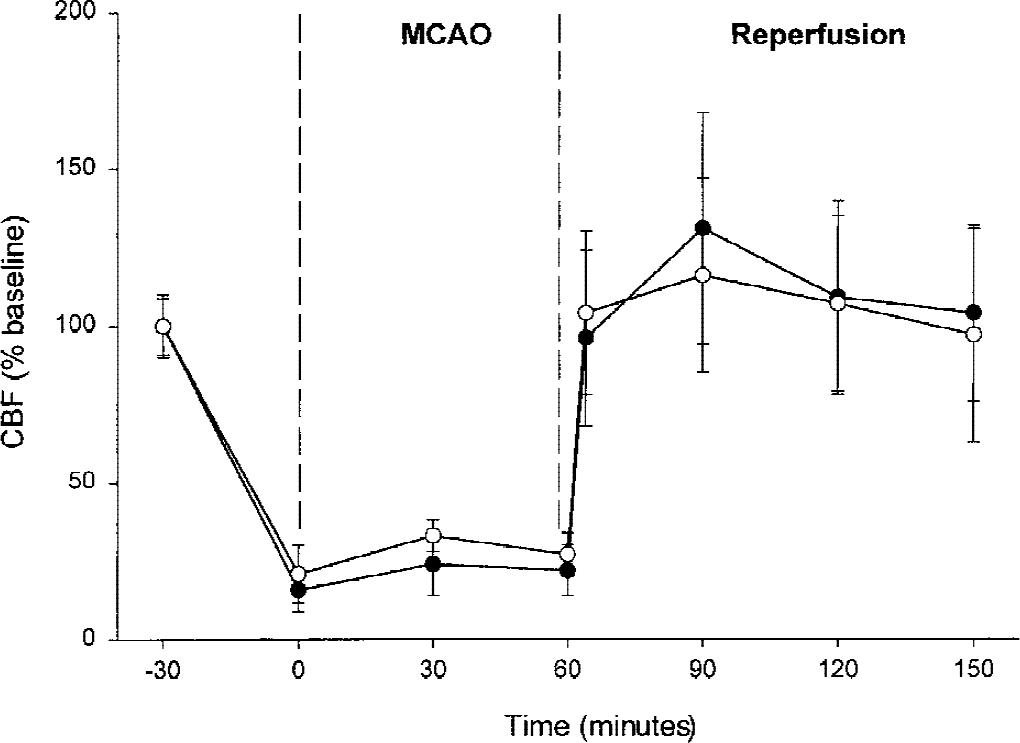
Effect of sense (open circles) and antisense (filled circles) infusion on regional cerebral blood flow (rCBF) after transient middle cerebral artery occlusion (MCAO). Changes are expressed as percentage of the baseline. Values are mean ± SD. The rCBF was measured using a laser–Doppler flowmeter probe placed on the surface of the ipsilateral cortex (ischemic area) through a craniectomy over the MCA territory. There were no statistically significant differences between the two groups.
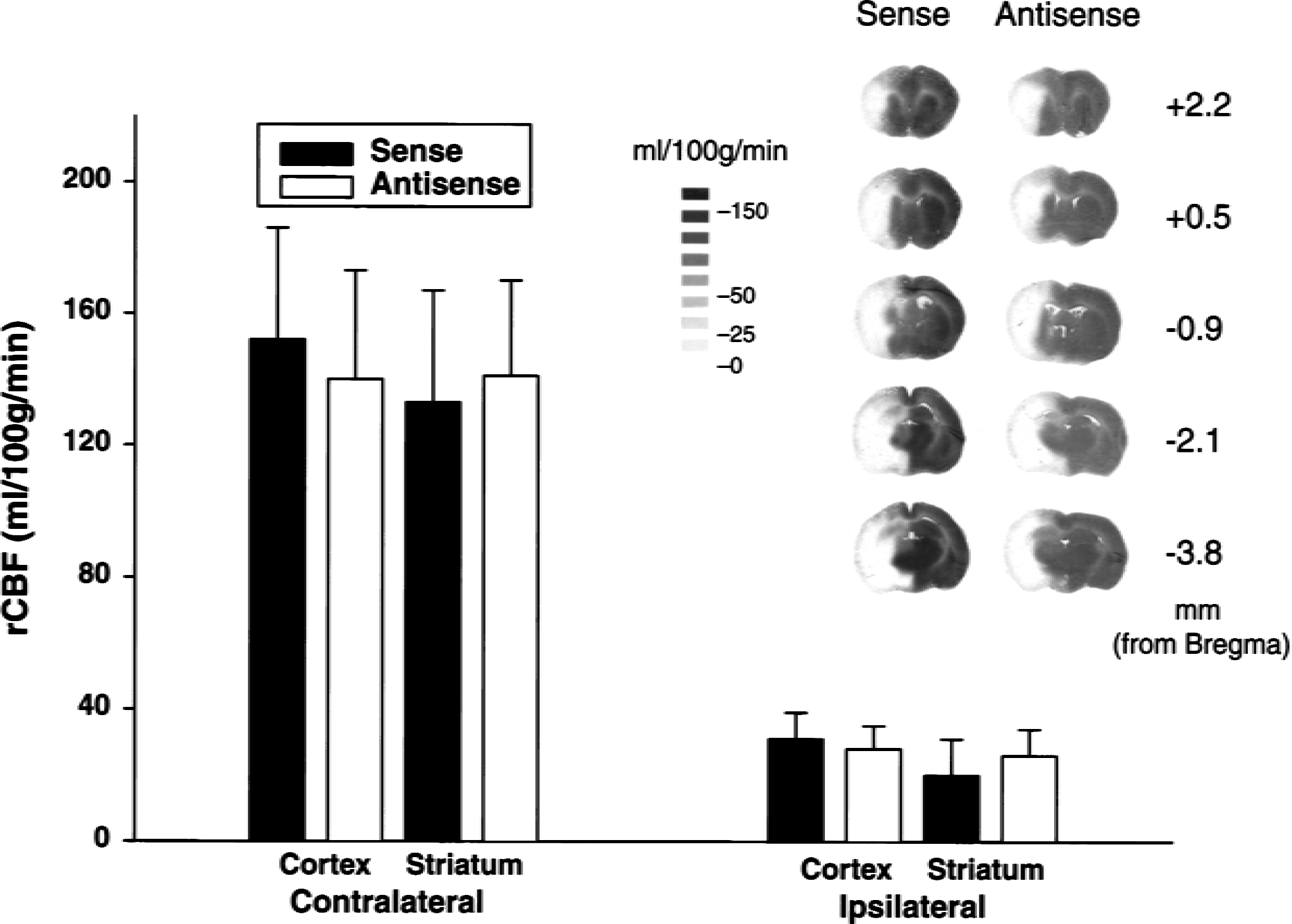
Endpoint regional cerebral blood flow (rCBF) rates of 1 hour of middle cerebral artery occlusion as measured by autoradiography using [14C]AIP in the ornithine decarboxylase (ODC) sense- (n of 5) and antisense- (n of 5) infused rats. Flow rates in the representative areas of cerebral cortex and striatum were averaged across the contralateral and ipsilateral sides of the brain. No significant differences were observed between sense-and antisense-infused groups. The inset shows autoradiographs generated using the coronal brain sections (+2.2, +0.5, −0.9, −2.1, and −3.8 from Bregma) of [14C]AIP administered sense-and antisense-infused groups.
Ischemic injury volumes in ODC sense, random, and antisense ODN-infused rats after transient MCAO
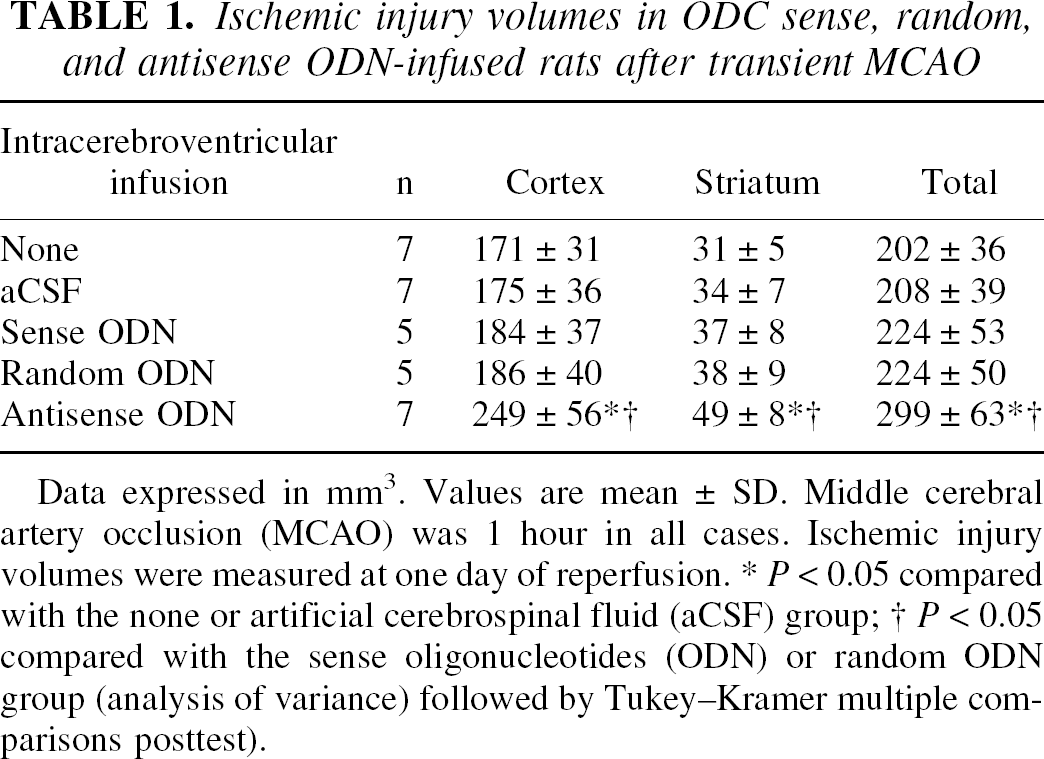
Data expressed in mm3. Values are mean ± SD. Middle cerebral artery occlusion (MCAO) was 1 hour in all cases. Ischemic injury volumes were measured at one day of reperfusion.
P < 0.05 compared with the none or artificial cerebrospinal fluid (aCSF) group
P < 0.05 compared with the sense oligonucleotides (ODN) or random ODN group (analysis of variance) followed by Tukey–Kramer multiple comparisons posttest).
Physiologic parameters in ODC sense, random, and antisense ODN-infused rats before, during, and after MCAO
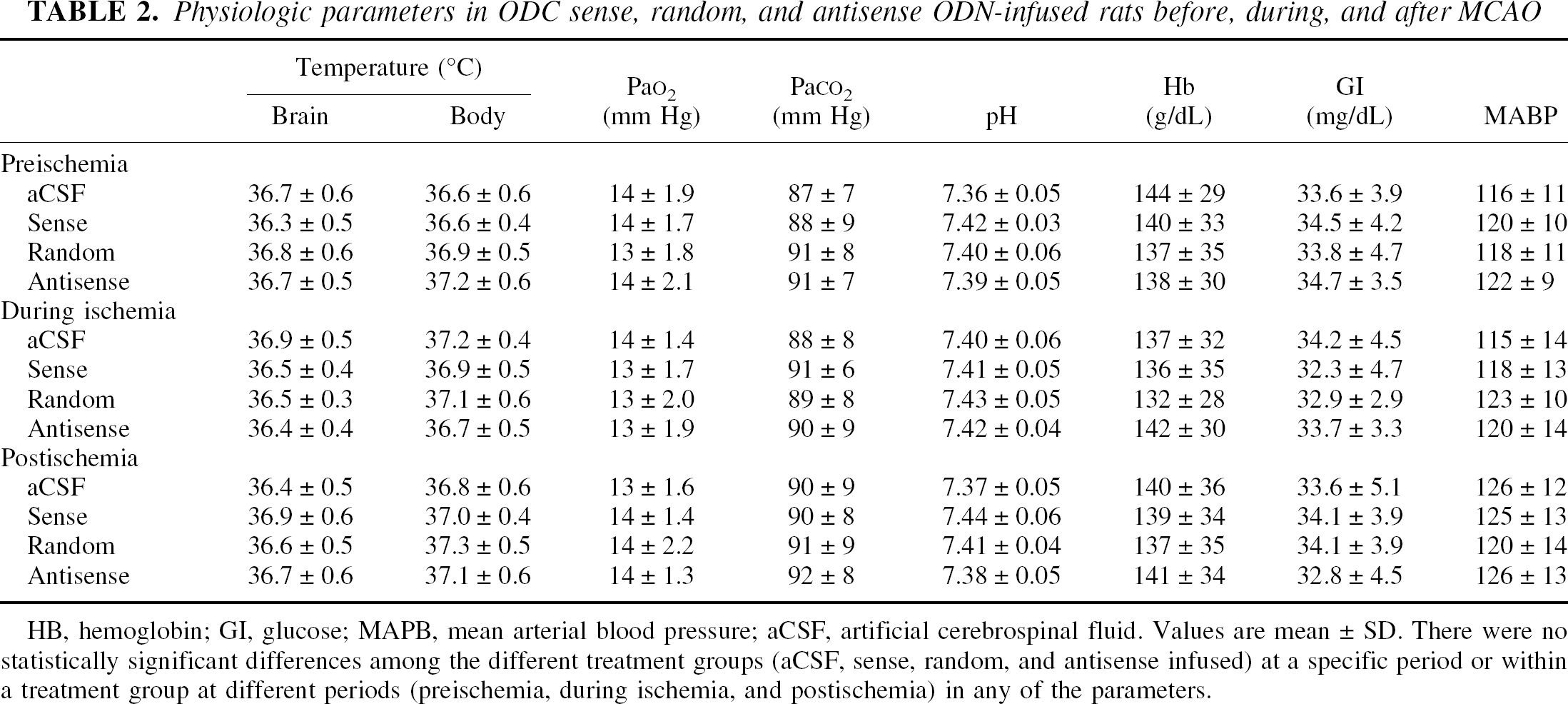
HB, hemoglobin; GI, glucose; MAPB, mean arterial blood pressure; aCSF, artificial cerebrospinal fluid. Values are mean ± SD. There were no statistically significant differences among the different treatment groups (aCSF, sense, random, and antisense infused) at a specific period or within a treatment group at different periods (preischemia, during ischemia, and postischemia) in any of the parameters.
Ornithine decarboxylase knockout increased the transient MCAO-induced mortality and motor deficits
Transient MCAO (1 hour) resulted in 14% mortality and mild-to-moderate neurologic deficit at 24 hours of reperfusion in the rats not subjected to intracerebroventricular infusion (Table 3). There was no significant change in the mortality or neurologic deficit in the aCSF/MCAO, sense/MCAO, and random/MCAO groups (Table 3). Whereas, the antisense/MCAO group showed a significant increase in the mortality rate to 58% (P < 0.05) with a severe to very severe neurologic deficit (Table 3). There was no mortality or observable neurologic deficit in any of the sham groups (aCSF/sham, sense/sham, random/sham, and antisense/sham) (Table 3).
ODC knockdown worsened the MCAO-induced neurologic deficit and mortality
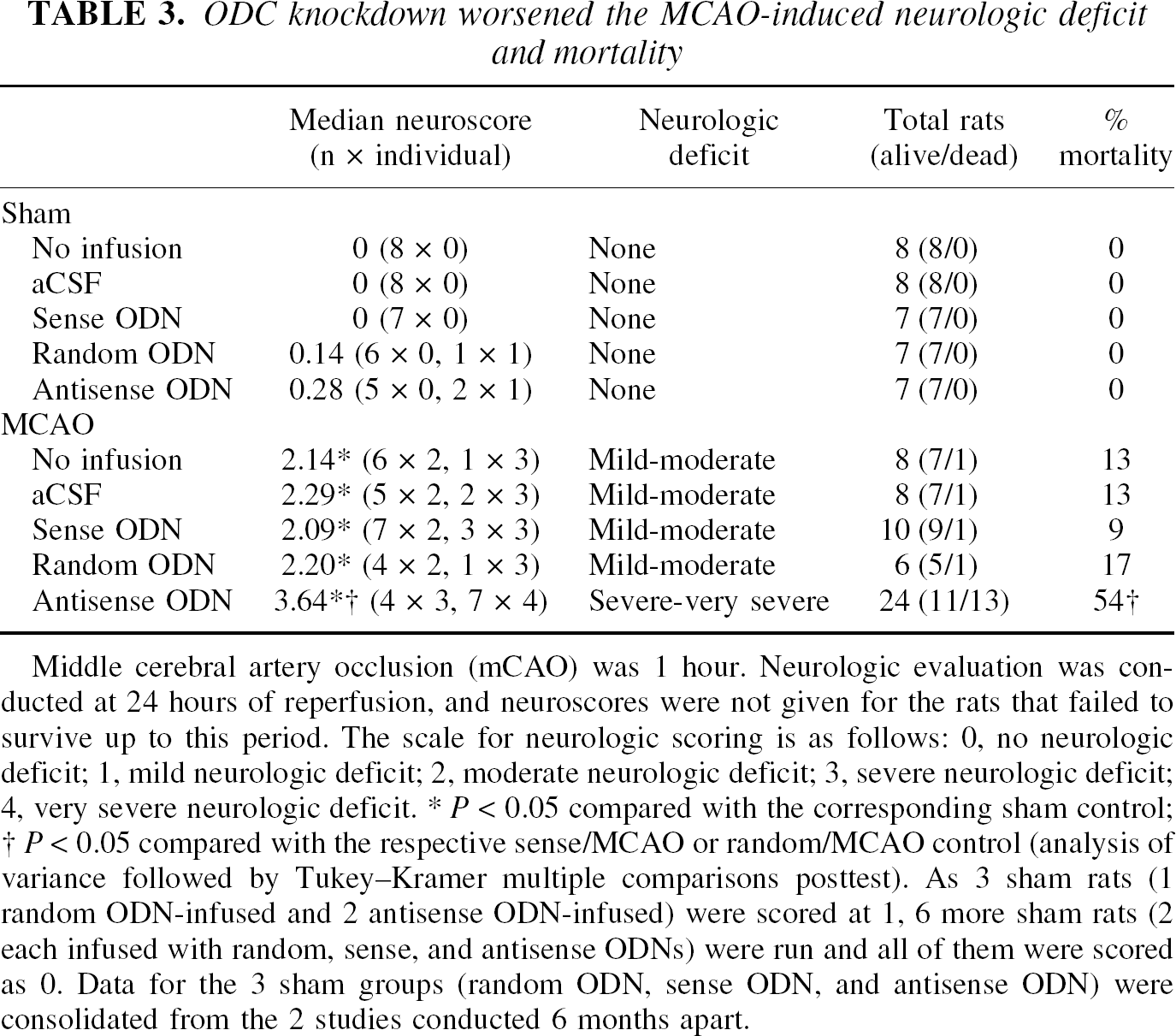
Middle cerebral artery occlusion (mCAO) was 1 hour. Neurologic evaluation was con-ducted at 24 hours of reperfusion, and neuroscores were not given for the rats that failed to survive up to this period. The scale for neurologic scoring is as follows: 0, no neurologic deficit; 1, mild neurologic deficit; 2, moderate neurologic deficit; 3, severe neurologic deficit; 4, very severe neurologic deficit.
P < 0.05 compared with the corresponding sham control
P < 0.05 compared with the respective sense/MCAO or random/MCAO control (analysis of variance followed by Tukey–Kramer multiple comparisons posttest). As 3 sham rats (1 random ODN-infused and 2 antisense ODN-infused) were scored at 1, 6 more sham rats (2 each infused with random, sense, and antisense ODNs) were run and all of them were scored as 0. Data for the 3 sham groups (random ODN, sense ODN, and antisense ODN) were consolidated from the 2 studies conducted 6 months apart.
DISCUSSION
Briefly, the current study shows that transient MCAO after ODC knockdown results in bigger infarcts, exacerbated neurologic deficit, and higher mortality. Increased infarct volume was evident in cerebral cortex and striatum. These results suggest that it is detrimental to prevent the ODC induction after transient focal cerebral ischemia and support a neuroprotective role for ODC.
Although there is a consensus showing increased ODC expression in all models of ischemia, there is no agreement regarding its physiologic significance. Previous studies indicated that inhibition of ischemia-induced ODC activity and putrescine levels by the ODC inhibitor DFMO protects the hippocampal neurons after transient forebrain ischemia in gerbil (Kindy et al., 1994). A recent report also showed that DFMO decreases the infarct volume after transient MCAO in rats (Dogan et al., 1998). Notably, despite DFMO being a potent, irreversible, substrate binding site inhibitor of ODC, it is neuroprotective in both of the models only at high concentrations (Kindy et al., 1994; Dogan et al., 1998). These studies led the authors to question the mechanism of neuroprotection afforded by DFMO. At high concentrations, DFMO may be neuroprotective because of its nonspecific actions on other proteins in addition to ODC. Furthermore, Paschen et al. (1988) showed that DFMO inhibits the ODC activation, but not formation of polyamine putrescine at 8 hours of recirculation after transient forebrain ischemia.
As antisense ODNs are very specific and inhibit a protein at its translational step, they were used in this study to prevent ODC induction after transient focal ischemia. Based on earlier reports, the hypothesis was that ODC induction after ischemia is neurotoxic and the increased ODC activity is responsible for edema, blood–brain barrier breakdown, and neuronal death after ischemia. If this is true, knockdown of ODC should afford neuroprotection. However, the current results showed exacerbated ischemic neuronal damage in the brains of rats infused with ODC antisense. The increase in infarct size cannot be simply attributed to hemodynamic-vascular factors, as the rCBF decrease produced by MCAO did not differ between the ODC sense and antisense infused rats. The authors also confirmed the effectiveness and specificity of ODC antisense in the cortical and striatal tissue by analyzing the ODC catalytic activity and immunoreactive protein levels. It is reasonable to hypothesize that under certain conditions the polyamine system may modulate repair or cell growth in light of its role in embryogenesis and neoplasia (Nishioka et al., 1995; Mehrotra et al., 1998). The authors conclude that a toxic response from the polyamine system after transient ischemia is not mediated by ODC activation.
Intracortical and intrastriatal administration of exogenous putrescine do not promote neuronal damage (Bourdoil et al., 1992; Ivanova et al., 1998). Gilad and Gilad (1991) showed that exogenous polyamines protect the hippocampal CA1 neurons from ischemic death. Later studies with the ODC overexpressing rodents showed that high levels of putrescine do not induce neurotoxicity (Lukkarinen et al., 1997, 1998, 1999). In fact, ODC overexpressing mice showed increased cerebral expression of neurotrophins and no histopathologic damage. These mice also showed no changes in the intracellular pH, free magnesium concentration, and hippocampal long-term potentiation (Kauppinen et al., 1992; Alhonen et al., 1995; Lukkarainen et al., 1995; Reeben et al., 1996; Pussinen et al., 1998). High concentrations of polyamines are also essential for the growth and development of neurons during healthy brain development and under cell culture conditions (Seiler et al., 1984; Slotkin and Bartolome, 1986). Furthermore, neurons are regularly cultured in medium containing serum with high levels of putrescine and cultured cerebellar neurons grown in the absence of serum undergo apoptosis, which can be prevented by the addition of polyamines (Harada and Sugimoto, 1997).
Based on the observation that metabolic, mechanical, thermal, and chemical injury induces cerebral ODC activity, ODC was suggested to be a heat shock and trauma protein (Dienel and Cruz, 1984; Johnson, 1998). The current study extends this concept and suggests that if ODC is a stress protein, indeed it is a necessary one, which might be neuroprotective after ischemia.
The only known biologic function of ODC is to form putrescine, and ODC is the entry point for l-ornithine into polyamine biosynthesis (Johnson, 1998). Putrescine also is formed from the endogenous polyamines by the interconversion pathway (Paschen, 1992; Seiler, 2000).
In this pathway, spermine will be converted to spermidine and the spermidine will be converted to putrescine by the coordinated action of spermine/spermidine N1 -acetyltransferase (SSAT) and polyamine oxidase (PAO). SSAT converts spermine to N1 -acetylspermine and spermidine to N1 -acetylspermidine. PAO converts N1 -acetylspermine to spermidine and N1 -acetylspermidine to putrescine (Bernstein and Muller, 1999).
Although Seiler (2000) suggested that in healthy brain 30% of putrescine will be formed by ODC and 70% by SSAT/PAO, this ratio may not be the same after a neurologic insult like ischemia. Previous studies showed the PAO-specific inhibitor MDL 72527 treatment reduces transient MCAO-induced putrescine formulation by only 40%, arguing against the possibility of 70% putrescine coming from interconversion pathway under ischemic conditions (Dogan et al., 1999). Polyamine metabolism is a dynamic process and the relative contribution of ODC and interconversion toward putrescine formation under ischemic conditions is not investigated properly. In addition to ODC, cerebral ischemia also activates SSAT and PAO. SSAT mRNA levels increase 8-to 10-fold in the early phases after transient forebrain ischemia (Zoli et al., 1996). The product of SSAT, N1− acetylspermidine, accumulates in the brain if PAO is blocked after transient focal and forebrain ischemia (Rao et al., 2000). Permanent MCAO results in a significant elevation of PAO activity within 2 hours after the onset of ischemia (Ivanova et al., 1998). However, the consequences of putrescine formation by ODC and SSAT/PAO may be different. Ornithine decarboxylase is the entry point of ornithine into polyamine synthesis, whereas SSAT/PAO uses the existing polyamines to form putrescine. In addition to putrescine, SSAT/PAO reaction also yields neurotoxic H2 O2, 3-acetamidopropanal and 3-aminopropanal as byproducts that contribute to the ischemic infarct development (Ivanova et al., 1998; Chan et al., 1998). These findings raise the possibility of a dual role for the polyamine system after ischemia (a protective/repair role mediated by putrescine, spermine, and spermidine and a toxic role mediated by polyamine interconversion pathway byproducts). The current antisense studies show that after transient ischemia, ODC induction is an essential neuroprotective event. Knockdown of ischemia-induced ODC activity and the ensuing prevention of putrescine formation may render the neurons electrically unstable as putrescine is known to block the inwardly rectifying potassium channels (Shyng et al., 1996).
Ornithine decarboxylase overexpressing rats manifesting 30 times more putrescine than healthy rats show smaller infarcts and a slower rate of infarct maturation after MCAO (Lukkarinen et al., 1998, 1999). Putrescine formation may be an essential and unavoidable consequence of transient cerebral ischemia. The ischemic brain will form putrescine, by both ODC activity and the polyamine interconversion pathway. Of the two, ODC activation may be less deleterious as it will produce only CO2 as a byproduct, which is less toxic than H2 O2 and 3-acetamidopropanal produced by the interconversion pathway. Ischemia-induced ODC activation may be a response to increased tissue putrescine levels that may act as a negative feedback on the polyamine interconversion to decrease the formation of toxic byproducts. Transient MCAO after ODC knockdown may disturb the balance between ODC and interconversion pathway leading to more putrescine formation by interconversion, and the toxic byproducts formed during this process may have increased the neuronal damage.
Recent studies showed the MDL72527 significantly decreases the neuronal damage after transient MCAO, suggesting PAO activation as a possible promoter of ischemic neuronal damage (Dogan et al., 1999). However, SSAT overexpressing transgenic mice with significantly elevated putrescine levels are resistant to kainate neurotoxicity, supporting the concept that increased cerebral putrescine levels are neuroprotective, regardless of whether derived from ODC or SSAT overexpression (Kaasinen et al., 2000). However, the effect of ischemia in SSAT transgenic rodents is not yet investigated. Furthermore, the consequences of coinduction of SSAT/PAO (like in ischemia) may be different from overexpression of only SSAT.
Footnotes
Acknowledgments:
The authors thank Dr. B-T. Kim, Dr. A.M. Rao, and J. Hatcher for helping with 14 C-AIP autoradiography and ODC assays.