
Abstract
Infarct size and vascular hemodynamics were measured 24 h after middle cerebral artery (MCA) occlusion in mice genetically deficient in the endothelial nitric oxide synthase (eNOS) isoform. eNOS mutant mice developed larger infarcts (21%) than the wild-type strain when assessed 24 h after intraluminal filament occlusion. Moreover, regional CBF values recorded in the MCA territory by laser-Doppler flowmetry were more severely reduced after occlusion and were disproportionately reduced during controlled hemorrhagic hypotension in autoregulation experiments. Unlike the situation in wild-type mice, nitro-L-arginine superfusion (1 mM) dilated pial arterioles of eNOS knockout mice in a closed cranial window preparation. As noted previously, eNOS mutant mice were hypertensive. However, infarct size remained increased despite lowering blood pressure to normotensive levels by hydralazine treatment. Systemic administration of nitro-L-arginine decreased infarct size in eNOS mutant mice (24%) but not in the wild-type strain. This finding complements published data showing that nitro-L-arginine increases infarct size in knockout mice expressing the eNOS but not the neuronal NOS isoform (i.e., neuronal NOS knockout mice). We conclude that NO production within endothelium may protect brain tissue, perhaps by hemodynamic mechanisms, whereas neuronal NO overproduction may lead to neurotoxicity.
Keywords
Nitric oxide (NO) is synthesized from L-arginine by the action of one of the following NO synthase (NOS) isoforms: neuronal (Type I; nNOS), endothelial (Type III; eNOS), and inducible (Type II; iNOS) NOS (Nathan and Xie, 1994; Snyder, 1995). NOS is found in neurons [nNOS (Bredt and Snyder, 1990)], endothelial cells [eNOS (Palmer et al., 1988)], astrocytes [iNOS (Murphy et al., 1993)], and perivascular nerve fibers [nNOS (Nozaki et al., 1993)]. NO has been implicated in many normal and abnormal functions such as regulation of vascular tone (Moncada et al., 1991; Knowles and Moncada, 1992), platelet aggregation (Radomski et al., 1990), N-methyl-D-aspartate-mediated cytotoxicity (Dawson et al., 1991), and neurotransmission (Snyder and Bredt, 1991; Dawson et al., 1992; O'Dell et al., 1994).
Recently, an important, albeit complex role for NO has been proposed in the pathophysiology of cerebral ischemia (Dalkara and Moskowitz, 1994; Iadecola et al., 1994). Whether NO is beneficial or detrimental to brain, however, remains controversial. Since NO is a diffusible, short-lived, and reactive free radical gas that is difficult to measure in vivo (Archer, 1993), most studies examining ischemic outcomes have based their conclusions on results following NOS inhibition by arginine analogues such as nitro-L-arginine or nitro-L-arginine methyl ester. These inhibitors, however, lack enzyme selectivity and block multiple isoforms (Rees et al., 1990). This nonselectivity might account in part for the discrepant outcomes after administration of NOS inhibitors following middle cerebral artery (MCA) occlusion.
Targeted gene disruption of the e- or nNOS isoforms provides a novel approach to dissect the relevance of NO in brain ischemia. We previously reported that mice deficient in nNOS gene expression were relatively resistant to brain injury after permanent focal cerebral ischemia (Huang et al., 1994). In this report, we document the consequences of MCA occlusion in mice lacking eNOS gene expression.
MATERIALS AND METHODS
Wild-type (SV-129 and C57 Black/6; Taconic Farms, Germantown, NY, U.S.A.) and eNOS mutant (Huang et al., 1995) male and female mice weighing 20–26 g were housed under diurnal lighting conditions and allowed free access to food and water ad libitum. Nitro-L-arginine, nitro-D-arginine, hydralazine hydrochloride, and 2,3,5-triphenyltetrazolium chloride (TTC) were purchased from Sigma.
Focal ischemia study
Mice were anesthetized with 2% halothane for induction and maintained on 1% halothane in 70/30% nitrous oxide/oxygen by mask. The right femoral artery was cannulated with PE-10 polyethylene tubing for arterial blood pressure measurement (Gould, Valley View, OH, U.S.A.) and blood gas determination (Corning 178; Ciba Corning Diag., Medford, MA, U.S.A.). Rectal temperature was maintained between 36.5 and 37.5°C with a homeothermic blanket system (YSI, Yellow Springs, OH, U.S.A.).
Focal cerebral ischemia was induced by occlusion of MCA using the intraluminal filament technique (Zea-Longa et al., 1989; Huang et al., 1994). Through a ventral midline incision, the right common and external carotid arteries were isolated and ligated. A microvascular clip (Zen temporary clip; Ohwa Tsusho, Tokyo, Japan) was temporarily placed on the internal carotid artery and the pterygopalatine artery. An 8–0 nylon monofilament (Ethicon, Somerville, NJ, U.S.A.) coated with silicone was introduced through a small incision in the common carotid artery and advanced 10 mm distal to the carotid bifurcation so as to occlude the MCA and posterior communicating artery. The wound was sutured and the animal returned to its cage and allowed free access to water and food. Twenty-four hours later, animals were killed with an overdose of pentobarbital and the brains were removed and sectioned coronally into five 2-mm slices in a mouse brain matrix. Slices were placed in 2% TTC solution, followed by 10% formalin overnight (Morikawa et al., 1994a). The infarction area, outlined in white, was measured (Bioquant IV image analysis system) on the posterior surface of each section, and infarction volume was calculated by summing the infarct volume of sequential 2-mm-thick sections.
In randomly selected mice, regional CBF (rCBF) was determined by laser–Doppler flowmetry (PF2B; Perimed, Stockholm, Sweden) and recorded on a Mac Lab/8 data acquisition system (AD Instruments, Milford, MA, U.S.A.). Two fiberoptic probe tips (Perimed PF 319:2, diameter = 0.5 mm) were fixed 2 mm posterior and 3 mm lateral to bregma and 2 mm posterior and 6 mm lateral to bregma on the ipsilateral hemisphere. These two coordinates identify sites on the convex brain surface within the vascular territory supplied by distal and proximal segments of the MCA, respectively, and they correspond to periinfarct zone and deeply ischemic territory, respectively (Huang et al., 1994; Yang et al., 1994). Steady-state baseline values were recorded before MCA occlusion. rCBF was recorded continuously during and after ischemia and expressed as percentage relative to the baseline value.
In protocol 1, MCA occlusion was produced in SV-129 (n = 12), C57 Black (n = 11), eNOS mutant mice (n = 14), and eNOS mutant mice injected with hydralazine (1 mg/kg i.p. 1 h before and 5 and 17 h after MCA occlusion; n = 10) to match the arterial blood pressure of wild-type mice (as determined in preliminary experiments).
In protocol 2, eNOS mutant and wild-type animals were injected with nitro-L-arginine (6 mg/kg, i.p. 5 min and 3 and 6 h after ischemia) or an equivalent volume of saline vehicle to test the hypothesis that inhibition of nNOS activity alleviated ischemic brain injury. The investigator was blinded to the treatment group in this protocol.
Autoregulation study
Mice were anesthetized with urethane (1.5 g/kg, i.p.) and ventilated (SAR-830 ventilator; CWE, Ardmore, PA, U.S.A.) with 70/30% nitrous oxide/oxygen after tracheotomy. Both femoral arteries were cannulated for arterial blood pressure measurement, blood gas determination, and blood withdrawal. Respiratory parameters were adjusted to keep the Paco2 in normal ranges (30–40 mm Hg). The core temperature was kept normothermic as described. The level of rCBF was monitored by laser-Doppler flowmetry (Dalkara et al., 1995).
Following reflection of the skin and subcutaneous tissue, an rCBF probe tip was secured directly over the parietal skull with glue (Borden, Columbus, OH, U.S.A.), away from pial vessels. An initial rCBF recording was taken as 100% and subsequent flow changes were expressed relative to this value. After heparin (10 units i.v.) administration, arterial blood pressure was lowered — 10 mm Hg every 5 min by withdrawing femoral artery blood (0.05–0.15 ml). Corresponding rCBF readings were averaged for each 10-mm Hg stepwise reduction. The duration of total experiment was ∼2–2.5 h. The upper limit of autoregulation was not tested in these mice.
Closed cranial window preparation and pial vessel diameter measurement
The mouse head was fixed in a stereotaxic frame and the skull exposed by a longitudinal skin incision. A stainless-steel cranial window ring (8.0 mm in inner diameter, 2.0 mm in height) containing three ports was embedded into a loop of bone wax over the skull. Dental acrylic was then applied. A craniotomy (2 × 1.5 mm) was made in the left parietal bone within the ring of the window. After the dura was opened and the brain surface superfused with artificial CSF (aCSF), a cover glass was placed to close the window. The volume under the window was ∼0.1 ml. The composition of aCSF was as follows (in mmol/L): Na+ 156.5, K+ 2.95, Ca2+ 1.25, Mg2+ 0.67, CP 138.7, HCO3− 24.6, dextrose 3.7, and urea 0.67. The pH value of aCSF was kept at 7.35–7.45 and monitored continuously with a pH meter (Corning, Corning, NY, U.S.A.). The aCSF was superfused by an infusion pump (0.4 ml/min) via PE-100 tubing connected to a window port. Intracranial pressure was maintained at 5–8 mm Hg. The temperature of aCSF within the windows was maintained at 36.5–37.0°C.
Pial vessels were visualized through a cranial window by an intravital microscope (Leitz, Germany) equipped with a video camera (C2400; Hamamatsu Photonics, Hamamatsu, Japan). The diameter of a single pial arteriole (20–30 μm) was continuously measured by a video width analyzer (C3161; Hamamatsu) and recorded using the MacLab data acquisition and analysis system. After baseline stabilization, nitro-L-arginine or nitro-D-arginine solution (1 mM) was superfused into the window and the diameter of pial arteriole measured continuously 40 min thereafter.
Statistical analysis
Data are expressed as means ± SD. Statistical evaluation was performed by analysis of variance (ANOVA) followed by t test to compare the data among groups in protocol 1. Unpaired Student t test was used to test the significance between two groups in protocol 2 and rCBF measurement. ANOVA with repeated measures and ANOVA followed by t test were used to evaluate significance within group differences and individual points between groups in the autoregulation experiment. Probability values of <0.05 were considered of statistical significance.
RESULTS
As reported previously (Huang et al., 1995), the mean arterial blood pressures in eNOS mutant mice were higher (115 ± 8 mm Hg) than in wild-type animals (98 ± 7 and 94 ± 7 mm Hg in SV-129 and C57 Black/6, respectively). After hydralazine administration, however, MABP did not differ between groups (Table 1).
Physiological data 10 min before MCA occlusion and 24 h after ischemia
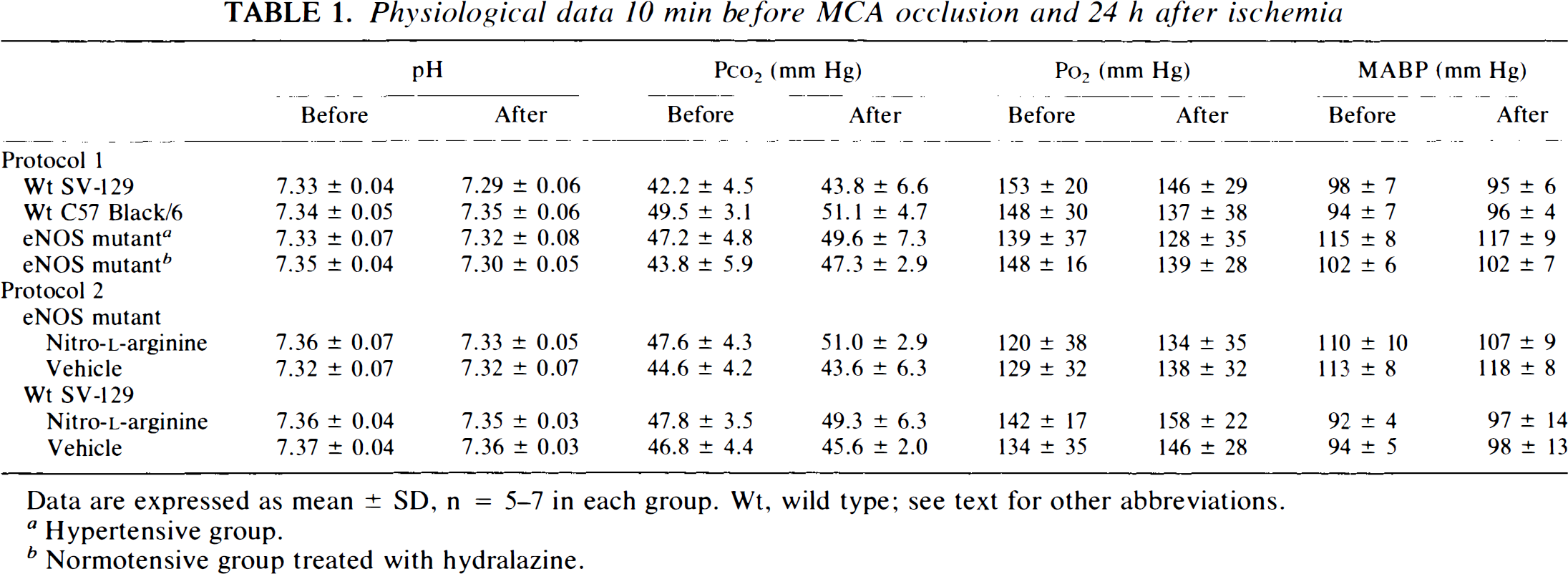
Data are expressed as mean ± SD, n = 5–7 in each group. Wt, wild type; see text for other abbreviations.
Hypertensive group.
Normotensive group treated with hydralazine.
In protocol 1, wild-type SV-129 and C57 Black/6 developed infarcts that were 37 ± 7% (n = 12) and 38 ± 15% (n = 11) of their respective hemispheres. Larger infarct volumes (46 ± 9% of hemisphere, n = 14, p < 0.05 as compared with wild-type SV-129 and C57 Black/6) were measured in eNOS mutant mice. Larger infarct volumes were also recorded in eNOS mutant mice made normotensive by hydralazine treatment (48 ± 7% of hemisphere, n = 10, p < 0.05 vs. wild-type SV-129 or C57 B/6) (Fig. 1). There were no significant group differences in physiology or blood gases before and 24 h after MCA occlusion to explain these differences (Table 1).
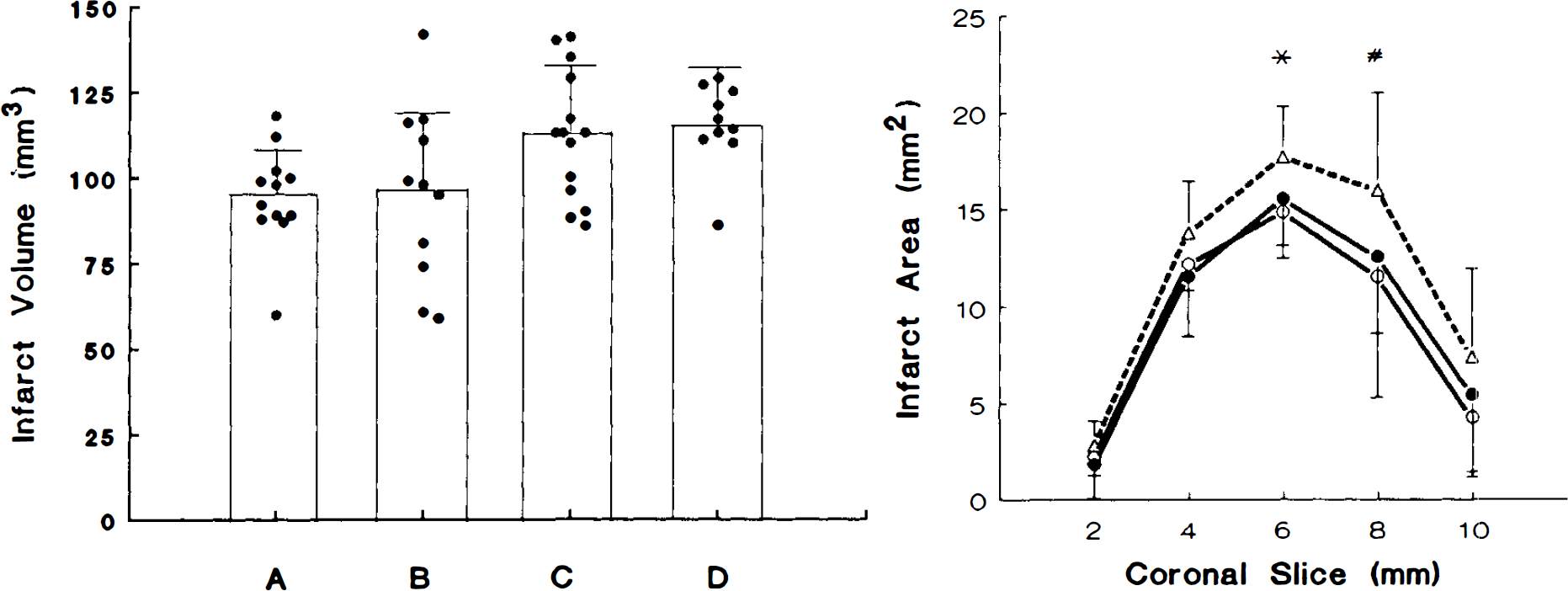
rCBF reduction was greater in the zone corresponding to the periinfarct area in eNOS mutant mice (30 ± 16% of baseline, n = 11, p < 0.05) than in SV-129 (40 ± 13% of baseline, n = 10) (Fig. 2), although there was no significant difference in the MCA core territory (data not shown).
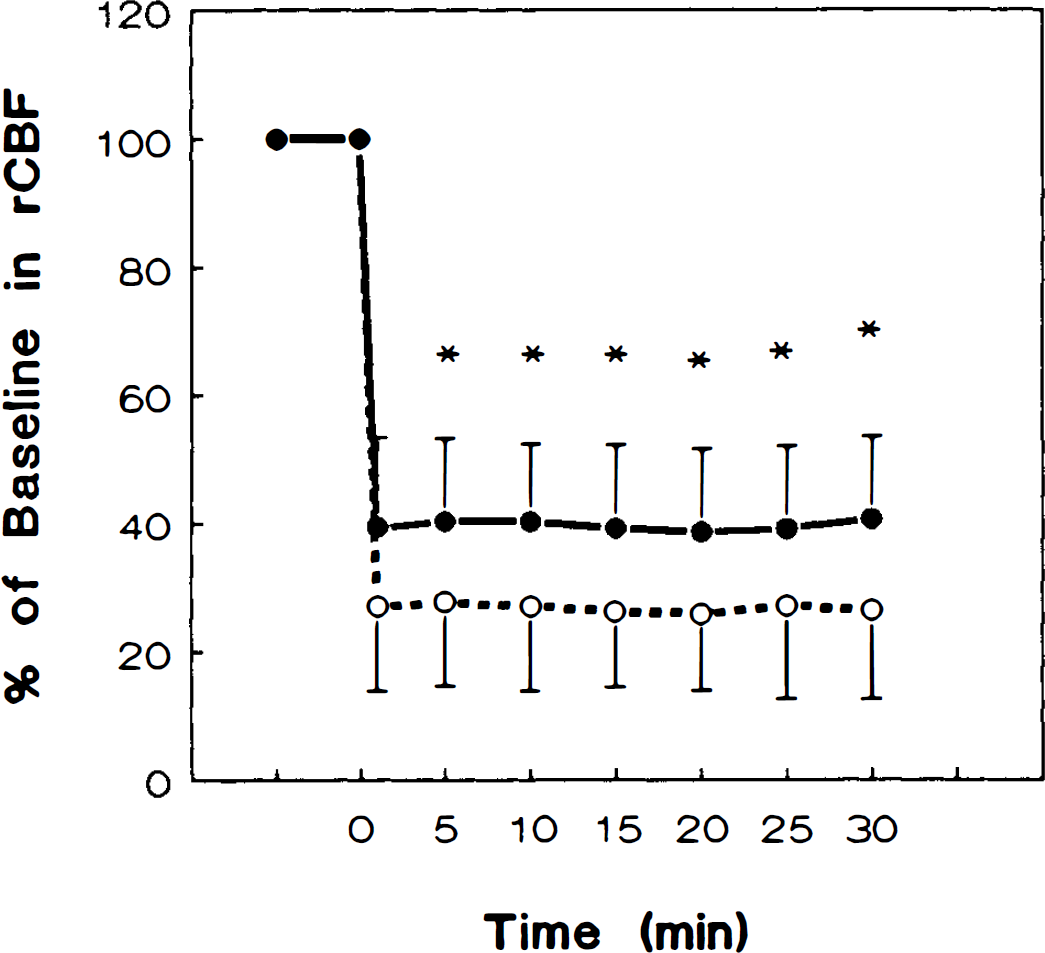
A greater rCBF reduction was observed by laser-Doppler flowmetry after MCA occlusion in the periinfarct area in eNOS mutant mice (dashed line; n = 11) than in wild-type SV-129 animals (solid line; n = 10, p < 0.05). A flexible optic probe tip (diameter = 0.5 mm) was secured 2 mm posterior and 3 mm lateral to bregma on the ipsilateral hemisphere. Steady-state rCBF values prior to MCA occlusion were taken as baseline (100%), and the subsequent changes after the onset of ischemia were shown as the percentage relative to the baseline. Time zero represents the point of MCA occlusion. There were no significant differences in rCBF reduction between SV-129 and C57 B/6 mice (data not shown). See text for abbreviations.
When arterial blood pressure was lowered stepwise by controlled hemorrhagic hypotension, CBF stayed relatively constant until <40 mm Hg. However, at low blood pressures, the autoregulation curve was shifted slightly to the right in eNOS mutant animals, suggesting higher cerebrovascular resistance than in wild type at lower perfusion pressures (Fig. 3).
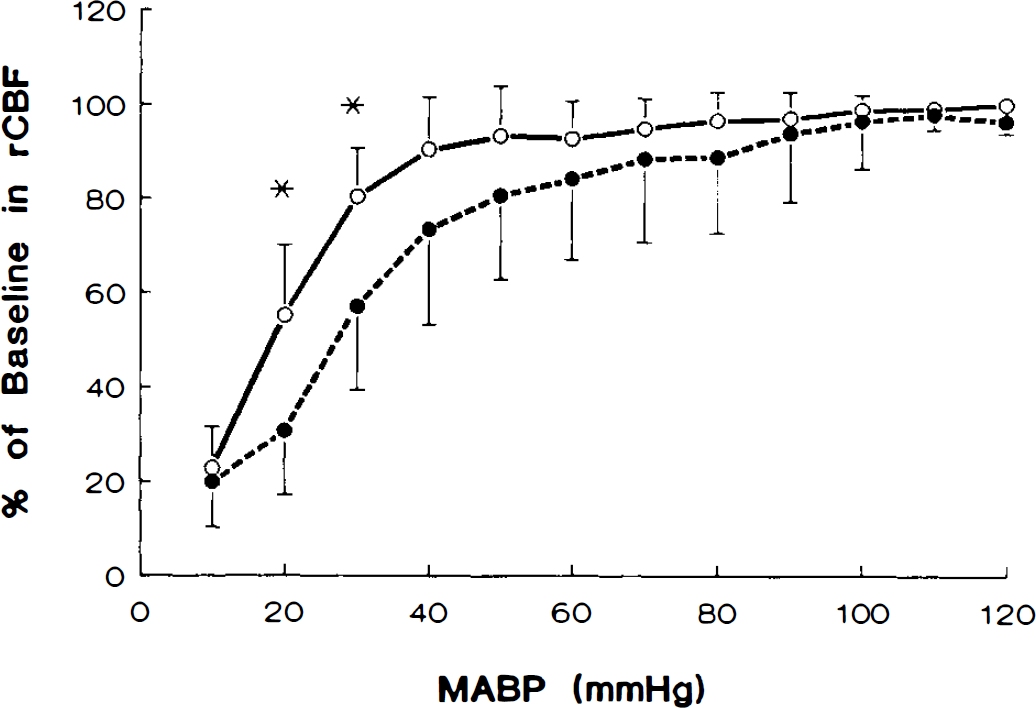
Urethane-anesthetized eNOS mutant mice (dashed line) showed greater decreases in rCBF during hemorrhagic hypotension than wild-type SV-129 animals (solid line). Hypotension was induced by gradually withdrawing arterial blood (see Materials and Methods), and rCBF was measured by laser-Doppler flowmetry. The initial rCBF values were taken as 100% and the corresponding changes thereafter calculated as percentage relative to the initial value. The baseline MABPs in wild-type and mutant were 104 ± 12 (n = 7) and 117 ± 13 (n = 7) mm Hg, respectively. There was a greater tendency toward hypoperfusion at higher levels of MABP in the mutant animals. Data are expressed as means ± SD. * p < 0.05 as compared with wild-type animals. See text for abbreviations.
In protocol 2, administration of nitro-L-arginine decreased infarct volume in the eNOS mutant mice by 24% and injury volumes became equivalent to those in wild type. Nitro-L-arginine treatment, however, did not change lesion size after MCA occlusion in wild-type mice (Table 2).
Effects of nitro-L-arginine on infarct size 24 h after MCA occlusion in mice

Data are expressed as means ± SD, n = 8–12 in each group. See text for abbreviations.
p < 0.05 vs. wild type.
p < 0.05 vs. vehicle.
Unlike in wild-type mice, nitro-L-arginine superfusion alone increased vessel diameter in eNOS mutant animals, reaching maximum at 30 min (p < 0.05 vs. wild-type) (Fig. 4). Similar changes in rCBF were recorded in preliminary experiments by laser-Doppler flowmetry using the closed cranial window technique (data not shown). There was no change in MABP during nitro-L-arginine superfusion. No change was found in pial diameter after nitro-D-arginine superfusion in eNOS mutant mice (data not shown).
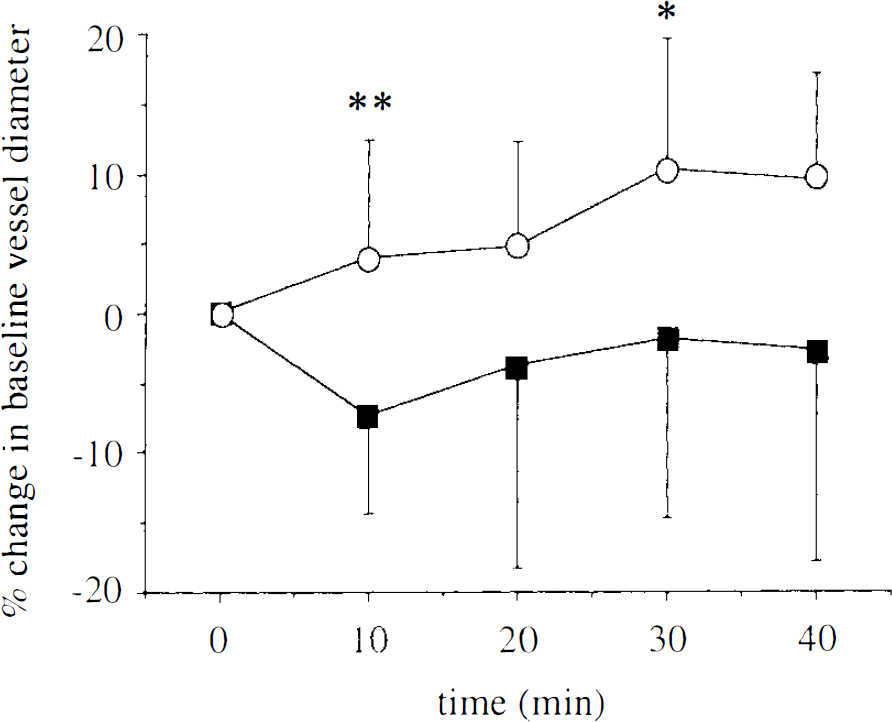
The diameter of pial arterioles was increased in eNOS mutant (circles; n = 7) but not wild-type (squares; n = 8) mice after nitro-L-arginine superfusion in a closed cranial window. Nitro-L-arginine (1 mM) was topically superfused for 40 min. Superfusion of nitro-D-arginine (1 mM; n = 3) did not have any effect on diameter of pial arterioles in eNOS mutant mice (data not shown). The data are calculated as percentage from baseline and expressed as means ± SD. *p < 0.05, **p < 0.01 vs. wild-type. See text for abbreviations.
DISCUSSION
Deletion of the mouse eNOS gene is associated with larger brain infarcts after MCA occlusion. Based upon inspection (dissecting microscope) of the circle of Willis after intracardiac Evans blue injection in SV-129, C57/Black, and eNOS mutant mice (n = 5/group), this result could not be accounted for by vascular anatomical differences. eNOS mutants exhibit more pronounced rCBF reductions in corresponding brain regions after MCA occlusion and exhibit proportionally lower rCBFs at reduced perfusion pressures during controlled hemorrhagic hypotension (Figs. 2 and 3). The latter may be due to hypertension, although no hypertensive changes were noted in the vessel wall on preliminary histopathological analyses (P. Huang et al., unpublished data). The contribution of high blood pressure to infarct enlargement was probably minor in this instance because infarct size did not change in eNOS mutants made normotensive by hydralazine administration.
Second, nitro-L-arginine decreased infarct volume in eNOS mutant mice, and we presume this decrease was caused by nNOS inhibition, the only constitutively expressed isoform in the eNOS mutant. However, it is possible that nitro-L-arginine-induced vasodilation and possible rCBF increases provided a second mechanism, and additional blood flow studies are needed to clarify this point. Infarct sparing after nitro-L-arginine was not as robust as expected (24% decrease), possibly because nNOS inhibition was subtotal. In the eNOS mutant, nitro-L-arginine superfusion inhibits nNOS within subjacent brain parenchyma (W. Meng et al., unpublished data), and there is published evidence of a contribution from parenchymally derived NO to cerebrovascular tone that could negatively impact on ischemic outcome during enzyme inhibition (Iadecola et al., 1994).
NO or a closely related chemical is proposed as endothelium-derived relaxing factor (Ignarro et al., 1987; Palmer et al., 1988). There is evidence to support the hypothesis that endothelium-derived NO or reaction products may be beneficial to stroke by augmenting rCBF in the ischemic territory. We previously reported that infusion of L-arginine, a substrate for NOS, caused NO-dependent vasodilation and increased rCBF distal to MCA occlusion in rats (Morikawa et al., 1994a). Dynamic susceptibility contrast magnetic resonance imaging also suggests that L-arginine infusion increases CBF and blood volume (Hamberg et al., 1993). Zhang et al. (1994) reported that NO donors improved rCBF in the ischemic area and reduced infarct size as well. In addition, sectioning of NOS-containing parasympathetic cerebrovascular fibers in sphenopalatine ganglia (Nozaki et al., 1993) increased infarct size after focal ischemia (Kano et al., 1991) and reduced cerebral perfusion during hemorrhagic hypotension (Koketsu et al., 1992). This evidence coupled with our observation that arterial blood pressures are less stable in eNOS mutant animals (e.g., during hypercapnic challenge) (unpublished data) speak to the importance of eNOS in regulation of vascular hemodynamics and its potential importance to stroke outcome.
eNOS mutant mice may be more susceptible to ischemic injury because NO modulates the microcirculation. Rosenblum and colleagues (1992) showed that NO protects the endothelium of mouse pial arterioles from platelet aggregation after vessel wall injury. NO may also block leukocyte adhesion and decrease microvascular stasis often seen following MCA occlusion (Garcia et al., 1993). Kubes and colleagues (1991) reported that superfusing mesenteric vessels with NOS inhibitors increased leukocyte adhesion. Kurose et al. (1994) found that L-arginine attenuated ischemia-induced platelet-leukocyte aggregation, mast cell degranulation, and albumin extravasation. We also found that rCBF was more severely reduced in homologous brain areas after MCA occlusion in eNOS but not nNOS (Huang et al., 1994) mutant mice. These findings confirm that NO plays a role in the modulation of the microcirculation that may contribute to the outcome of ischemia.
We have speculated that eNOS inhibition accounts for the increases in infarct size in some studies after nitro-L-arginine, particularly after large doses (Yamamoto et al., 1992; Zhang and Iadecola, 1993; Morikawa et al., 1994b). By contrast, neuroprotection was reported after selective nNOS inhibition with 7-nitroindazole (Yoshida et al., 1994) or FPL 17477 (Zhang et al., 1995). Consistent with the present findings, infarction size increased (Huang et al., 1994) when nitro-L-arginine was administered to mutant mice expressing only the eNOS isoform (i.e., nNOS knockout mice). Importantly, iNOS enzyme activity is not measurable in mouse (SV-129 strain) brain for at least 4 days after permanent MCA occlusion (Yoshida et al., 1995).
We do not believe that the protective effect of the eNOS gene is due to eNOS in neurons. In fact, neuronal eNOS expression appears to be more focal, site specific (hippocampus) (Dinerman et al., 1994), and at lower levels of activity than nNOS expression. For example, we presume that the small amount of residual NOS in nNOS mutants is due to eNOS activity. In nNOS mutants, brain NADPH diaphorase staining disappears almost completely, enzymatic NOS activity is reduced by 95% in brain (Huang et al., 1993), and specific [3H]nitro-L-arginine binding approaches background levels (H. Hara et al., 1996). For these reasons, we believe that infarct sparing after nitro-L-arginine is most likely secondary to nNOS inhibition in eNOS mutants.
We conclude that NO possesses a dual role in focal cerebral ischemia. Depending upon its source, NO may be toxic or protective to brain under ischemic conditions. Parenchymal NO overproduction may lead to neurotoxicity, whereas endothelial NO may protect brain tissue by increasing rCBF or some other hemodynamic mechanism. Our findings emphasize the need to develop selective inhibitors of the neuronal isoform to protect brain from injury. Our results also emphasize the importance of knockout mice to dissect the role of individual proteins in complex pathophysiological events.
Footnotes
Acknowledgment:
Studies were supported by the Massachusetts General Hospital Interdepartmental Stroke Program Project NS10828 (M.A.M.) and by a Sponsored Research Agreement with Bristol-Myers Squibb (M.C.F.) We thank Drs. H. Hara and C. Waeber for their advice.