
Abstract
The role of intracerebral complement activation after traumatic brain injury remains unclear. In this study, the authors demonstrate that transgenic mice with astrocyte-targeted expression of the soluble complement inhibitor sCrry have a significantly reduced neurologic impairment and improved blood–brain barrier function after closed head injury compared with wild-type C57BL/6 littermates. This work further implicates the complement system as a participant in secondary progression of brain damage after head trauma and provides a strong rationale for future studies of posttraumatic pharmacologic complement inhibition.
Traumatic brain injury still represents the leading cause of death and residual neurologic impairment in persons younger than 40 years of age in industrialized countries, despite substantial recent progress in basic research and clinical therapeutic concepts for amelioration of secondary brain damage (Graham et al., 2000; Kossmann and Stahel, 2002; Marshall, 2000). Clinical and experimental studies suggest a pathophysiologic role for intracerebral complement activation in contributing to inflammation, blood–brain barrier (BBB) dysfunction, intracranial leukocyte recruitment, and neuronal cell death after traumatic brain injury (Bellander et al., 2001; Kaczorowski et al., 1995; Keeling et al., 2000; Morganti-Kossmann et al., 2002; Nataf et al., 1999; Stahel et al., 1998, 2001; Van Beek et al., 2003). A significant limitation for experimental therapeutic approaches to inhibit complement activation has been the lack of speciesspecific reagents for the mouse. Complement receptorrelated protein y (Crry) is a functional homologue of human decay-accelerating factor (CD55) and membranecofactor protein (CD46) and inhibits both the classical and alternative pathways of complement activation in mice (Foley et al., 1993). Transgenic mice with astrocyte-targeted overexpression of soluble Crry (GFAP-sCrry mice) provide a model system to assess the role of complement in CNS inflammatory disease and injury. These mice were protected against neuropathology and demyelination in a model of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis, compared with nontransgenic littermates (Davoust et al., 1999). In this study, we used sCrry transgenic mice to assess the pathophysiologic role of intracerebral complement activation in traumatic brain injury.
MATERIALS AND METHODS
The GFAP-sCrry transgenic mice used in this study have been previously characterized (Davoust et al., 1999) and are in the C57BL/6 background. These mice express a soluble form of Crry, a murine complement regulatory protein, with expression limited to the CNS under the control of an astrocyte-specific promoter (GFAP). The GFAP-sCrry transgenic mice were initially found to be 80% to 90% pure C57BL/6 when assessed by DNA marker analysis. They were then back-crossed for more than 10 generations, whereby each subsequent cross was selected as pure C57BL/6 × sCrry until all mice were >99% C57BL/6 background, as detected by DNA marker analysis by Charles River Therion International (Saratoga Springs, NY, U.S.A.). Wild-type C57BL/6 littermates were used as control. All mice were male, 10 to 16 weeks of age, and weighed 30 to 35 g. Mice were bred in a specific pathogen-free environment under standard conditions of temperature and light (four to six animals per cage), and provided with food and water ad libitum.
Experimental closed head injury was performed using a standardized weight-drop model, as previously described (Chen et al., 1996; Stahel et al., 2000; Yatsiv et al., 2002). After trauma, the mice received supporting oxygenation with 100% oxygen until they awakened. Sham-operated animals underwent anesthesia and scalp incision, but were not subjected to experimental trauma. The mice in the sham-operated control group were killed at identical corresponding time-points as the trauma group. The animal experiments were performed in compliance with the guidelines of the Federation of European Laboratory Animal Science Association and approval was granted by the Institutional Animal Care Committee (Tierschutzkommission des Kantonalen Veterinäramtes Zürich, permission No. 143/200, Int. 2124).
Posttraumatic neurologic impairment was analyzed using a neurologic severity score (NSS), as previously described (Beni-Adani et al., 2001; Stahel et al., 2000). The score consists of 10 individual clinical tasks on motor function, alertness, and physiologic behavior in which one point is given for failure of task performance and no points for successful task performance (Table 1). A maximal NSS of 10 points indicates severe neurologic dysfunction, with failure of all tasks, whereas a score of zero is achieved by healthy uninjured mice. Two independent, blinded investigators evaluated task performance at 1, 24, and 72 hours, 7 days, and 4 weeks after trauma.
Neurologic severity score for mice

The BBB integrity in injured brains was assessed 4 hours after head injury, which corresponds to the peak of posttraumatic BBB dysfunction in this model (Chen et al., 1996). Quantitative analysis of the BBB function was performed by quantification of Evans blue extravasation in the injured hemisphere (n = 4 mice for each group), as previously described (Chen et al., 1996; Stahel et al., 2000; Uyama et al., 1988). Briefly, 20 μL of a 2% solution of Evans blue (Sigma, St. Louis, MO, U.S.A.) in saline was injected into the tail vein of mice at 3 hours after trauma and allowed to circulate for 60 minutes. After removal of the intravascular dye by saline perfusion, the brain hemispheres were separated and weighed before homogenization in 1 mL 50% trichloroacetic acid in dH2O. Thereafter, the samples were centrifuged for 20 minutes at 10,000 rpm and the supernatants were diluted 1:4 in 100% ethanol. After vigorous mixing, an aliquot of each sample was diluted threefold in a solution of 50% trichloroacetic acid/100% ethanol. Evans blue fluorescence was then measured by fluorospectrophotometry at an extinction wavelength of 620 nm and an emission wavelength of 680 nm. A serial dilution of 2% stock Evans blue was used to establish a standard curve. The concentrations were normalized for the brain tissue weight and calculated as nanograms Evans blue per gram brain tissue.
For qualitative characterization of the BBB integrity, 10-μm-thick cryosections of murine brains (n = 5 mice for each group) were analyzed by immunohistochemistry for intracerebral albumin deposition 4 hours after trauma, using a polyclonal rabbit anti–mouse albumin antibody diluted 1:500 (Accurate Chemical and Scientific Corporation, Westbury, NY, U.S.A.) and detection by a biotin/avidin/peroxidase technique (Vector, Burlingame, CA, U.S.A.). Isotype-matched control immunoglobulin G from nonimmunized rabbits (Vector) was used as a control for the primary antibody.
The analysis of complement activation in injured wild-type and transgenic mouse brains was performed by immunohistochemistry for complement C3 deposition in injured brain tissue, using the above-mentioned technique. Complement C3 represents the central component of complement activation of all three activation pathways. A polyclonal goat anti-mouse C3c antibody (Accurate Chemical and Scientific Corporation) was used as primary antibody, because C3c is the major fragment resulting from C3 cleavage by C3 convertases and factor I, and antisera to C3c have been shown to react with the C3-derived fragments C3b, C3bi, and smaller fragments, all of which carry antigenic determinants of the C3c domain. This antibody does not to react with any other protein component of mouse serum or plasma, as determined by the manufacturer's instructions (Accurate Chemical and Scientific Corporation). Isotype-matched nonspecific goat immunoglobulin G (Vector) was used as a control for the C3c antibody.
Statistical analysis was performed using the SPSS 9.0 for Windows statistical software package. The Mann-Whitney U test was used for analysis of neurologic impairment (NSS) and the unpaired Student's t-test for analysis of BBB function (Evans blue extravasation). Differences in posttraumatic mortality were evaluated on a contingency table using the Fisher exact test. A P level < 0.05 was considered statistically significant.
RESULTS
In accordance with previous studies (Bellander et al., 2001; Keeling et al., 2000, Kyrkanides et al., 2001), experimental brain injury induced a massive intracranial complement activation in wild-type mice within 24 hours after trauma, as determined by immunohistochemical assessment of complement C3 deposition in injured brains, using a polyclonal anti–mouse C3c antibody. In contrast, complement C3 deposition was clearly attenuated in injured brains of transgenic GFAP-sCrry mice at 24 hours, indicating an attenuated extent of complement activation in the transgenic mouse brains (data not shown).
The GFAP-sCrry mice (n = 36) had a significantly reduced NSS at all time-points from 1 hour to 4 weeks after closed head injury, compared with nontransgenic littermates (n = 31; P<0.05, Mann-Whitney U test; Fig. 1A). In addition, the transgenic mice had a reduced posttraumatic mortality within the first 24h after trauma compared with nontransgenic littermates (n = 2 / 5.5% vs. n = 5 / 16.1%), however, the difference was not statistically significant (P = 0.157, Fisher exact test; Fig. 1B). No animal in either group died at time-points after 24 hours. Sham-operated animals of both groups (n = 3 per group) had normal NSS and no mortality.
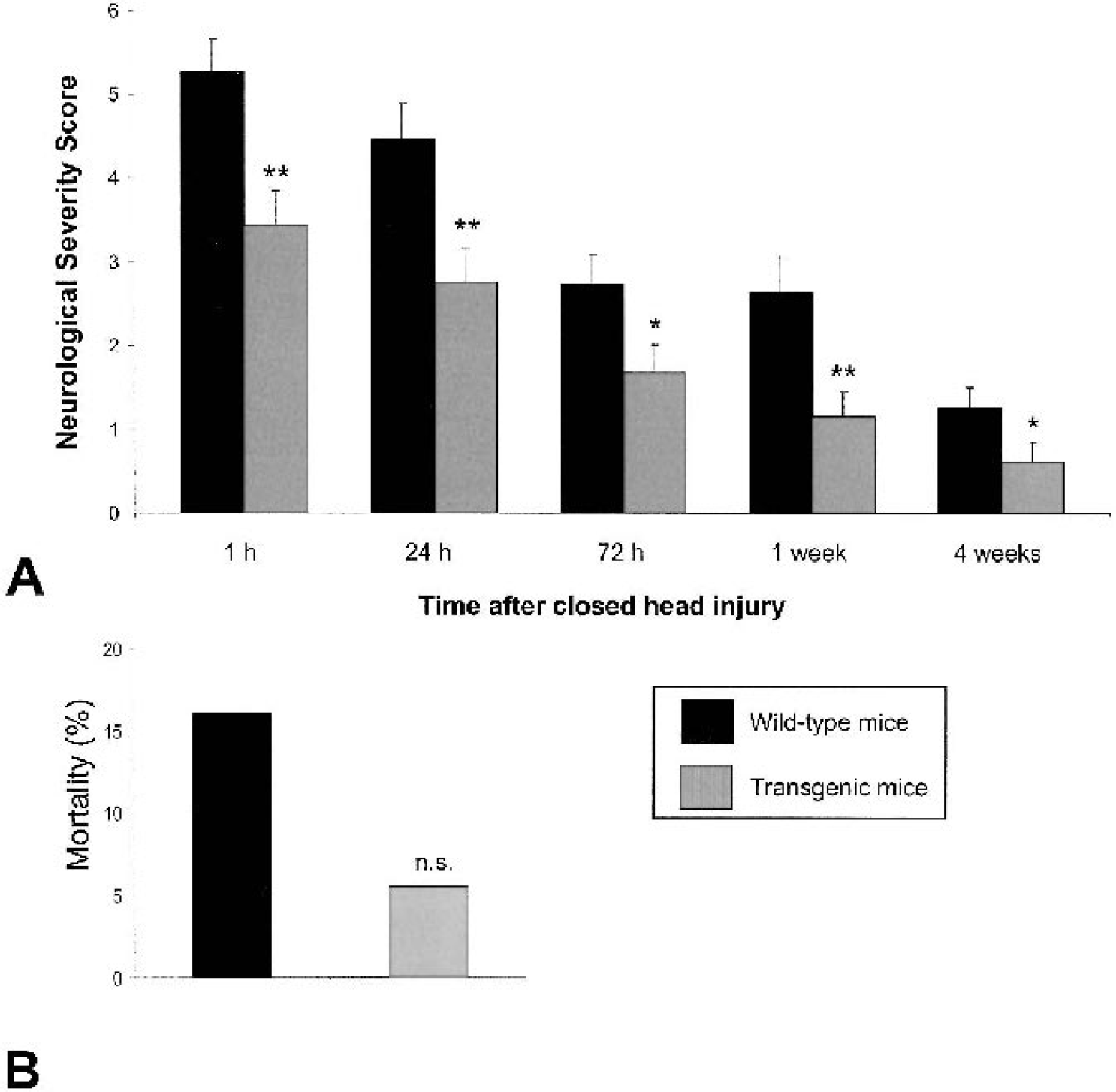
Neurologic impairment
In this closed head injury model, it has been shown that BBB disruption peaks at 4 hours after trauma, as assessed by quantitative Evans blue extravasation (Chen et al., 1996). Transgenic GFAP-sCrry mice had significantly lower intracerebral Evans blue extravasation in the injured hemisphere compared with nontransgenic littermates (1,140 ± 480 vs. 495 ± 188 ng/mg brain tissue, mean ± SD; P < 0.05, unpaired Student's t-test), indicating reduced posttraumatic BBB dysfunction (Fig. 2A). In accordance with previous studies (Chen et al., 1996; Stahel et al., 2000), Evans blue concentrations were significantly higher in the ipsilateral (injured) hemispheres compared with the contralateral (control) hemispheres in both animal groups (P < 0.05, unpaired Student's t-test, Fig. 2A). Because Evans blue binds to serum albumin before leaking across a dysfunctional BBB (Uyama et al., 1988), we also determined the qualitative pattern of albumin deposition in injured brains by immunohistochemistry. As shown in Fig. 2, albumin deposition was detectable in the subarachnoid space of injured brains in control mice (Figs. 2B and 2C), but not in GFAP-sCrry mice (Fig. 2D). Staining of adjacent tissue sections with an isotype-matched control immunoglobulin G demonstrated low or undetectable nonspecific binding (data not shown).
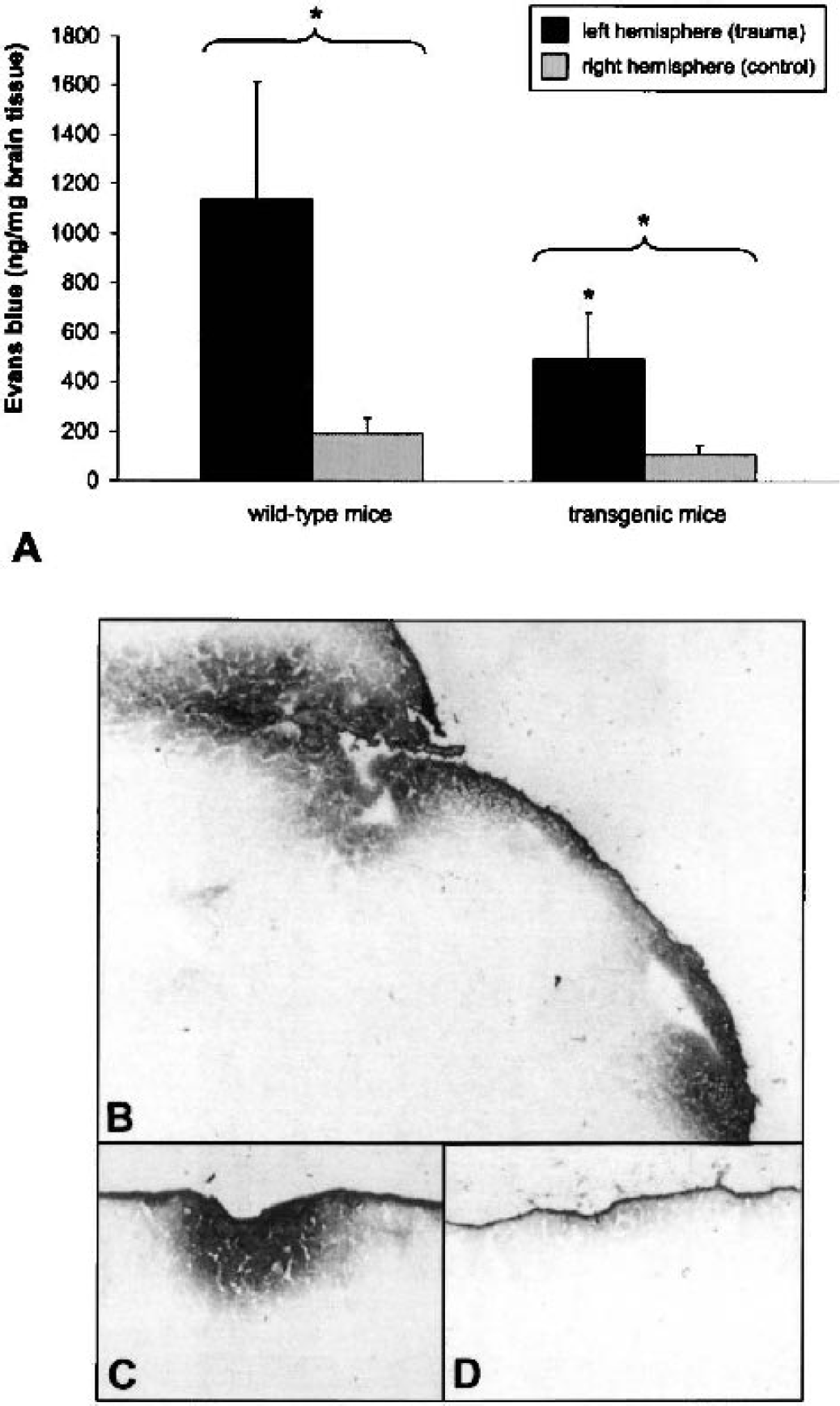
Posttraumatic blood–brain barrier dysfunction in wild-type C57BL/6 and transgenic GFAP-sCrry mice at 4 hours after experimental closed head injury. Quantitative assessment was performed by systemic Evans blue injection and analysis of extravasation into brain tissue
DISCUSSION
These are the first data to demonstrate that endogenous, intracerebral inhibition of complement activation mediates neuroprotection in mice after closed head injury. Using a standardized weight-drop model, we observed improvement of neurologic deficit and BBB function in sCrry transgenic mice compared with nontransgenic controls. The observation of strongly enhanced GFAP protein expression in the injured, but not in the contralateral hemisphere (data not shown), implies that the posttraumatic activation of the GFAP promoter induces increased sCrry synthesis via the activated transgene. Davoust et al. (1999) have previously shown that activation of the transgene in these mice leads to increased sCrry levels in murine cerebrospinal fluid, thus mediating intrathecal inhibition of complement activation. Our results are in sharp contrast to those of Hicks et al. (2002), who used a recombinant Vaccinia virus complement control protein (VCP) in a fluid-percussion model of head injury. In their study, VCP and VCPt (the truncated version lacking complement inhibitory activity) protected against impairment in spatial memory but not against neuropathologic damage. The fact that VCPt also provided protection against spatial memory impairments indicates that VCP-mediated protective effects are independent of complement inhibition. Given that VCP binds heparin and heparin sulfate proteoglycans (Hicks et al., 2002), it is possible that VCP and its derivative are sequestered and unable to effectively inhibit complement activation in the setting of head injury. In addition, VCP and VCPt were injected intracranially, thus raising the concern that this model system induces further brain damage in terms of a cortical stab wound injury (Hicks et al., 2002). This fact significantly complicates the interpretation of their results, particularly because cortical stab injury induces the expression of many inflammatory genes, including cytokines and complement components (Kyrkanides et al., 2001). This bias can be avoided in our transgenic model, because sCrry is locally produced, murine specific, and freely diffusible in the CNS (Davoust et al., 1999).
Some potential limits of the transgenic model system used in this study should be addressed. The kinetics of BBB opening after closed head injury have been extensively studied in wild-type mice (Chen et al. 1996), but not in the transgenic GFAP-sCrry mice used in this study. Thus, a delayed opening of the BBB in the transgenic mice beyond the peak seen in wild-type mice cannot be excluded. Furthermore, the finding of neuroprotection by complement inhibition in transgenic animals cannot be automatically extrapolated to a pharmacologic concept of “postinjury” complement inhibition as a basis for a new therapeutic strategy in neurotrauma.
This study raises the question of which complement activation fragments and pathways contribute the most to complement-mediated neuropathology in the setting of closed head injury. Increased levels and activation of C3 and other complement proteins are readily detected in head injury (Bellander et al., 2001; Keeling et al., 2000; Kossmann et al., 1997; Kyrkanides et al., 2001; Stahel et al., 1998), implicating both the classical and alternative pathways in the pathophysiology of brain injury. Using a selected group of complement mutant mice, we are currently examining which effector molecules in the complement system are crucial to inflammation and cell death in head injury. Our study, based on the GFAP-sCrry transgenic mice, suggests that inhibition of complement at the level of the C3 and C5 convertases provides an exciting new therapeutic avenue for treatment of traumatic brain injury. The observation of early significant improvement of the NSS in sCrry mice implies that inhibition of complement activation as early as possible after injury may be of clinical value. Given that there are numerous human-specific complement inhibitors currently in clinical use or in development (Barnum, 1999), this therapeutic strategy has the potential for further implementation in the clinic.
Footnotes
Acknowledgments:
The authors thank Dr. Shabaan Mousa and Dr. Michael Schäfer (Department of Anesthesiology, University Hospital Benjamin Franklin, Berlin) for their continuing support.