
Abstract
Increased transport of Na+ across an intact blood-brain barrier (BBB) participates in edema formation during the early hours of cerebral ischemia. In previous studies, the authors showed that the BBB Na-K-Cl cotransporter is stimulated by factors present during ischemia, suggesting that the cotransporter may contribute to the increased brain Na+ uptake in edema. The present study was conducted to determine (1) whether the Na-K-Cl cotransporter is located in the luminal membrane of the BBB, and (2) whether inhibition of the BBB cotransporter reduces brain edema formation. Perfusion-fixed rat brains were examined for cotransporter distribution by immunoelectron microscopy. Cerebral edema was evaluated in rats subjected to permanent middle cerebral artery occlusion (MCAO) by magnetic resonance diffusion-weighted imaging and calculation of apparent diffusion coefficients (ADC). The immunoelectron microscopy studies revealed a predominant (80%) luminal membrane distribution of the cotransporter. Magnetic resonance imaging studies showed ADC ratios (ipsilateral MCAO/contralateral control) ranging from 0.577 to 0.637 in cortex and striatum, indicating substantial edema formation. Intravenous bumetanide (7.6–30.4 mg/kg) given immediately before occlusion attenuated the decrease in ADC ratios for both cortex and striatum (by 40–67%), indicating reduced edema formation. Bumetanide also reduced infarct size, determined by TTC staining. These findings suggest that a luminal BBB Na-K-Cl cotransporter contributes to edema formation during cerebral ischemia.
Brain edema that forms during the early hours of ischemic stroke involves a net uptake of Na+ and water from blood into brain across an intact blood-brain barrier (BBB) (Betz, 1996; Betz et al., 1994, 1995; Menzies et al., 1990, 1993; Schielke et al., 1991) along with swelling of astrocytes (Bourke et al., 1980; Gotoh et al., 1985; Iadecola, 1999; Kempski et al., 1991; Kimelberg, 1995, 1999). Previous studies have shown that BBB breakdown, allowing paracellular flux of solute and water into the brain, generally occurs after 3 to 6 hours of continuous reduction in blood flow, and that the majority of edema formation is accounted for by the net uptake of brain cations and water that occurs well before BBB breakdown (Betz, 1996). Previous studies have also indicated that a luminally located BBB Na+ transport pathway plays a major role in this phenomenon, although the transporter(s) involved have not been identified (Betz, 1996; Ennis et al., 1996; Schielke et al., 1991). Under healthy, normoxic conditions, the BBB endothelium normally generates up to 30% of the volume of brain interstitial fluid by mechanisms that involve luminal Na+ and Cl− transporters (Cserr et al., 1989; Keep, 1993), which very likely work in conjunction with abluminal Na/K ATPase and Cl− efflux pathways. This BBB secretion of Na+, Cl−, and water into the brain appears to be greatly increased by conditions present during cerebral ischemia. During ischemia, astrocytes rapidly swell as they are stimulated to take up Na+ and Cl− from the brain interstitial fluid (Bourke et al., 1980; Gotoh et al., 1985; Iadecola, 1999; Kempski et al., 1991; Kimelberg, 1995, 1999). Thus, early edema formation appears to involve not only astrocyte swelling but also increased supply of Na+, Cl− and water to the astrocytes across an intact BBB. In this sense, increased BBB Na+ and Cl− transport makes the barrier facilitate, or become permissive to, astrocyte swelling.
A number of findings from this and other laboratories suggest a role for the Na-K-Cl cotransporter in the transport of Na+ and Cl− across the BBB from blood into brain during cerebral ischemia. The Na-K-Cl cotransport inhibitor bumetanide was found in an early study to inhibit Cl− dependent uptake of Na+ across the BBB of rats (Betz, 1983). We subsequently showed that a Na-K-Cl cotransporter is indeed present in cultured bovine cerebral microvascular endothelial cells (CMEC) and freshly isolated brain microvessels (O'Donnell et al., 1995a, b ; Yerby et al., 1997), and the cotransporter has also been demonstrated in cultured rat (Kawai et al., 1996b, 1997; Vigne et al., 1994) and human (Spatz et al., 1997) CMEC. The endothelial cotransporter is upregulated by exposure to astrocyte-conditioned medium or to astrocyte co-culture, suggesting that the cotransporter is important to BBB function (O'Donnell et al., 1995a; Sun et al., 1995, 1997; Sun and O'Donnell, 1996). Consistent with this, the tissue expression of Na-K-Cl cotransporter NKCC1 isoform mRNA (the extrarenal isoform) is 5- to 40-fold higher in brain than all other tissues examined (except salivary gland, with a comparable level) (Yerby et al., 1997). Our previous studies have also provided evidence suggesting that the cotransporter may be located in the luminal membrane of CMEC (Sun et al., 1995). Furthermore, others and we have shown that the CMEC cotransporter is stimulated by conditions present during brain ischemia including vasopressin (O'Donnell et al., 1995a), endothelin (Kawai et al., 1996b, 1997; Spatz et al., 1997), and hypoxia (Kawai et al., 1996a, b ; O'Donnell et al., 1999). Collectively, these findings have led us to hypothesize that the Na-K-Cl cotransporter, located at the luminal membrane of BBB endothelial cells, is stimulated by conditions present during ischemia to increase secretion of Na+, Cl−, and water from blood into brain, promoting edema formation.
The present studies were designed with two primary goals: (1) to determine whether the Na-K-Cl cotransporter is indeed located predominantly in luminal membranes of brain microvascular endothelial cells in situ; and (2) to determine whether inhibition of brain microvascular endothelial Na-K-Cl cotransporter activity reduces edema formation in cerebral ischemia. To this end, we first conducted immunoelectron microscopy studies of rat brain sections, using antibodies that specifically identify the Na-K-Cl cotransporter protein. Second, we used magnetic resonance diffusion-weighted imaging to follow in vivo brain edema formation in rats subjected to up to 3 hours of permanent middle cerebral artery occlusion (MCAO), and thereby to study edema formation induced by ischemia (rather than by ischemia/reperfusion) during the early hours of ischemia before BBB breakdown occurs.
MATERIALS AND METHODS
Immunoelectron microscopy
Rat brains were perfusion fixed for 40 to 60 minutes using 4% paraformaldehyde plus 0.05% glutaraldehyde in 0.1-M phosphate buffer (pH 7.4). Brains were removed and postfixed in 4% paraformaldehyde overnight, then subjected to freeze substitution, as described previously (Golshani et al., 2001; Liu et al., 2001b). Tissues were embedded in lowiacryl resin, then sectioned and placed on Butvar-coated grids. Sections were then immunolabeled using as primary antibody either T4 (monoclonal; University of Iowa Developmental Studies Hybridoma Bank) or T84 (polyclonal; gift of Dr. Christian Lytle) Na-K-Cl cotransporter antibodies. Secondary antibodies used were 15-nm gold particle-conjugated goat anti-mouse IgG (for T4) and 15-nm gold particle-conjugated goat anti-rabbit IgG (for T84) (Ted Pella, Inc, Redding, CA, U.S.A.). Sections were subsequently stained with uranyl acetate and lead citrate and observed on a Phillips 410 electron microscope. To quantitate gold particle distribution in the microvessel plasma membranes, a total of 80 immunoelectron micrographs were examined and gold particles counted using a double-blind method. Data are presented as the percentage of total plasma membrane gold particles found in the luminal membrane.
Induction of ischemia by permanent middle cerebral artery occlusion
This study was conducted in accordance with the Animal Use and Care Guidelines issued by the National Institutes of Health using a protocol approved by the Animal Use and Care Committee at University of California, Davis. Normotensive Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, U.S.A.) weighing 250 to 300 g were used in the study. Animals were anesthetized with intraperitoneal injection of sodium pentobarbital (65 mg/kg body weight). For experiments using nephrectomized rats, a dorsoventral incision was made into the abdominal cavity and renal blood vessels and ureter were ligated. Kidneys were excised and the incision was closed using a 9-mm Autoclip Applier (Vibratome Co., St. Louis, MO, U.S.A.). In all experiments, body temperature was monitored and maintained at 36.8°C to 37.0°C using an electric heating pad and rectal probe (Cole-Parmer Instruments, Vernon Hills, IL, U.S.A.) during surgery, and a water heating pad (Gaymar Inc., Orchard Park, NY, U.S.A.), and rectal probe during nuclear magnetic resonance data acquisition. The left femoral artery was cannulated with PE-50 polyethylene tubing for continuous monitoring of arterial blood pressure during nuclear magnetic resonance data acquisition. PE-50 tubing was also used to cannulate the left femoral vein for injection of bumetanide or vehicle. Blood samples were drawn from the descending aorta immediately before animal euthanasia for determination of electrolytes, pH, P
Focal cerebral ischemia was induced by occlusion of the left middle cerebral artery as described previously (Hasegawa et al., 1994; Karpiak et al., 1989; Zea Longa et al., 1989). Under a dissecting microscope, the left common carotid artery was exposed through blunt dissection and a ventral midline neck incision. The occipital and thyroid artery branches of the external carotid artery (ECA) were ligated and dissected using loop tip surgical cauterizer (Abco Inc., Nashville, TN, U.S.A.). The pterygopalatine artery was also isolated and ligated close to its origin and the ECA was ligated approximately 3 to 5 mm distal to its origin. The common and internal carotid arteries were then temporarily clamped using microvascular clips to allow insertion of a 3–0 dermalon filament (3 cm in length) with blunted tip into the internal carotid artery by way of the ECA. The ECA was dissected and the dermalon filament was loosely secured inside the ECA using 5–0 suture. This allowed gentle movement of the dermalon into the internal carotid artery and the eventual occlusion of the middle cerebral artery. Occlusion was confirmed using laser Doppler (LD) (Moor Instruments, Wilmington, DE, U.S.A.). In these measurements, CBF was determined immediately before MCAO and again immediately after occlusion. We found that CBF was reduced by MCAO to an average of 23% preocclusion CBF. Specifically, CBF was 272.1 ± 43.6 and 259.4 ± 27.2 LD units before MCAO for rats treated with and without intravenous bumetanide, respectively, and 61.5 ± 11.2 and 58.4 ± 21.9 LD units after MCAO for rats treated with and without intravenous bumetanide, respectively (mean ± SD, 11 experiments with and 16 experiments without bumetanide).
Rats were subjected to MCAO by advancing a nylon suture in the internal carotid artery to the origin of the middle cerebral artery. Alternatively, rats were subjected to MCAO surgery without occlusion (sham). For bumetanide treatment, rats were administered bumetanide intravenously (7.6–30.4 mg/kg in one to four doses of 7.6 mg/kg) 20 minutes before initiation of MCAO. Animals were then placed in the nuclear magnetic resonance magnet and diffusion-weighted imaging was conducted over a 180-minute period. For both MCAO and sham, rats were treated intravenously with either vehicle or bumetanide. Bumetanide (ICN Biomedicals, Costa Mesa, CA, U.S.A.) was prepared as a fresh stock solution for each experiment by dissolving 10 to 18 mg in 0.5 mL of 0.5 N NaOH, diluting to the desired concentration in a 0.5% saline solution containing albumin, and adjusting the pH with HCl.
Magnetic resonance imaging analysis of brain edema
Diffusion-weighted spin echo images (2-mm slices) were acquired 30 to 180 minutes after occlusion of the MCA, using a 7-T Bruker Biospec MRS/MRI system (Bruker Biospin MRI, Inc., Billerica, MA, U.S.A.). The anesthetized rats were placed supine on a homemade Plexiglas stage with water-heated pad, bite bar, and ear clamps for securing the head. The stage and rat were then placed into a 72-mm radiofrequency probe inside the 7-T magnet to acquire diffusion-weighted images. These images were 2-mm thick axial slices with a field of view of 4 cm, matrix of 256 by 128 pixels, repetition time of 2 seconds, and echo time of 40 milliseconds. Apparent diffusion coefficient (ADC) values were determined for selected brain regions using four gradient strengths (Tatlisumak et al., 1996) and ParaVision 2.1 software (Bruker BioSpin GmbH, Rheinstetten, Germany). For each region, ADC values for ipsilateral (left) and contralateral (right) hemisphere regions were compared and a ratio of left/right ADC values was calculated.
TTC assessment of infarct size
At the conclusion of diffusion-weighted imaging experiments (180–200 minutes), rats were killed and their brains were quickly removed and sectioned into 2-mm-thick slices starting at the frontal pole using a Brain Matrix Slicer (Vibratome Co.). Slices were then immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma Aldrich Corp., St. Louis, MO, U.S.A.) in a Petri dish and incubated at 37°C for 20 minutes. Slices were flipped at the 10-minute mark for consistent staining of anterior and posterior faces. Slices were then scanned using an Epson Perfection 1200U scanner (Epson America, Inc., Long Beach, CA, U.S.A.) and Adobe Photoshop software (Adobe, San Jose, CA, U.S.A.). Brain slices for each of the experimental groups were analyzed for infarct volume using Image-J analysis software (public domain software developed at NIH, and available on the Internet at http://rsb.info.nih.gov/nih-image/). Percent infarct was calculated as described by Swanson and colleagues (Swanson et al., 1990).
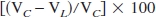
where VC is volume of control hemisphere and VL is volume of noninfarcted tissue in the lesioned hemisphere.
Gravimetric analysis of brain water content
Brain water content was determined as described by Menzies et al. (1993), with some modifications. Rats treated with either bumetanide or vehicle were subjected to 180 minutes of MCAO under continuous anesthesia and were killed by decapitation. Brains were immediately removed and separated into left and right hemispheres. After determining the wet weight of each hemisphere, the tissue was dried for 72 hours at 100°C and then reweighed to determine dry weight. Percent brain water was determined as 100 x (wet weight – dry weight)/wet weight.
Statistical analysis
All values are presented as means ± SD. Data shown were analyzed for significance using analysis of variance for repeated measures with a Scheffé post hoc test unless otherwise indicated. P values less than 0.05 were considered to indicate significant difference.
RESULTS
In situ distribution of Na-K-Cl cotransport in rat brain microvessel endothelial cells
If the Na-K-Cl cotransporter contributes to transport of Na+, Cl−, and water from blood to brain during cerebral ischemia, then the cotransporter should reside predominantly in the luminal membrane of the cerebral microvascular endothelial cells. To evaluate the in situ cellular distribution of Na-K-Cl cotransport in cerebral microvessels, we used perfusion-fixed, sectioned rat brains and immunoelectron microscopy to examine labeling by T4 (monoclonal) or T84 (polyclonal) antibodies that recognize the Na-K-Cl cotransporter (Lytle et al., 1995; O'Donnell et al., 1995b; Sun et al., 1995). Figure 1 shows representative immunoelectron micrographs generated using the T4 and T84 antibodies (panels A/B and C/D, respectively). Note that the majority of plasma membrane-associated gold particles in these images is found at the luminal membrane with a minor fraction at the abluminal membrane. In these micrographs, gold particles can also be seen at the astrocyte end feet plasma membrane (adjacent to the lower aspect of the endothelial basal lamina; best seen in Figs. 1B and 1D). Because the focus of the present study was the BBB cotransporter, we did not further evaluate cotransporter distribution in the astrocytes. To quantitate the in situ distribution of Na-K-Cl cotransporter protein in plasma membranes of brain microvessel endothelial cells, immunoelectron micrographs were generated using three different dilutions for both primary antibodies and gold particle distributions were evaluated in 80 separate micrographs. As shown in Fig. 2, we found that the majority of plasma membrane cotransporter protein is located in the luminal membrane (approximately 80%), with a minor fraction (approximately 20%) residing in the abluminal membrane. This was observed whether we used the T4 or T84 antibody to locate the cotransporter, and did not vary significantly over a wide range of primary antibody dilutions.
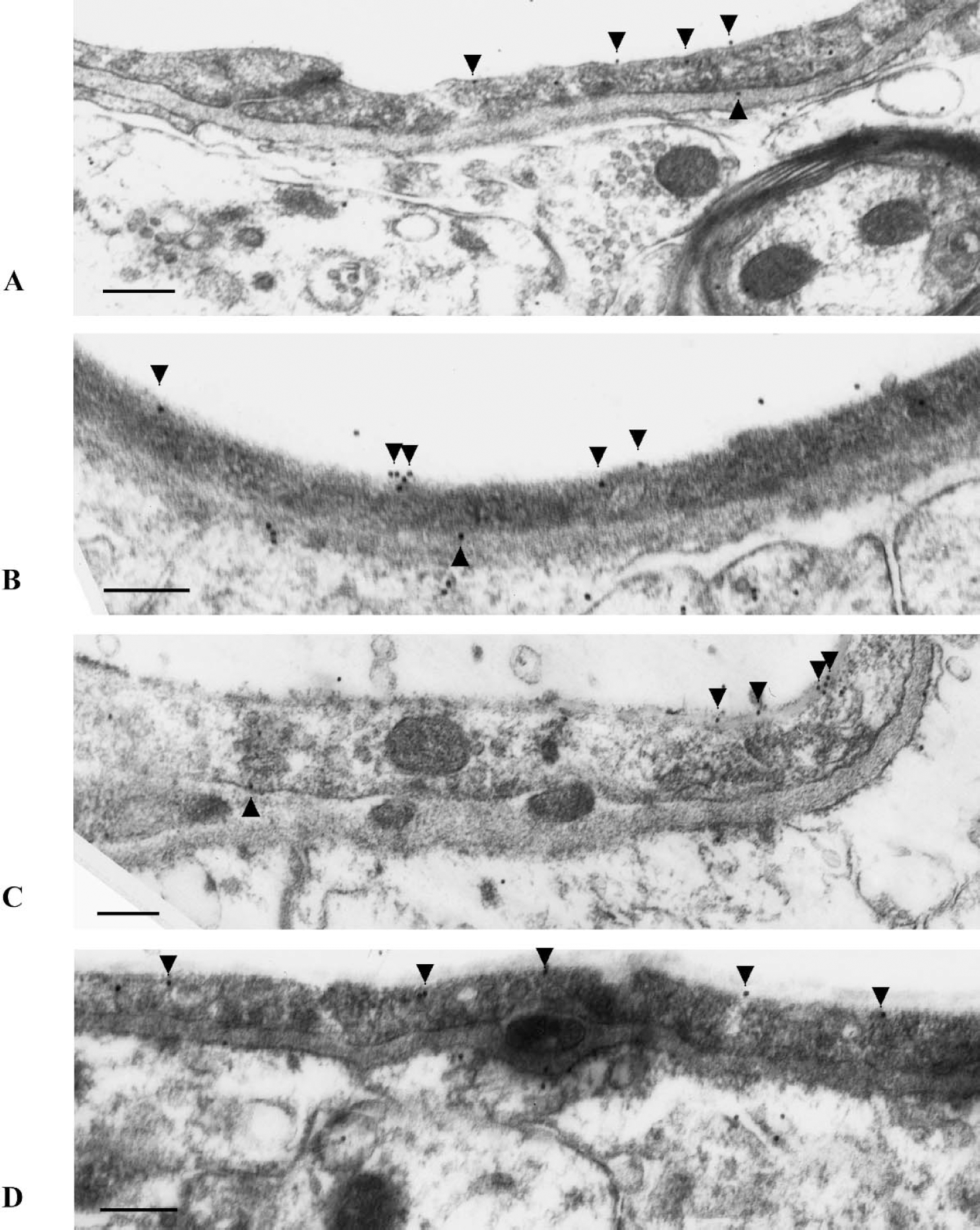
Immunoelectron microscopy localization of Na-K-Cl cotransport protein in rat brain microvascular endothelial membranes. Rat brains were perfusion-fixed as described in Materials and Methods, then labeled with primary antibody (T4 monoclonal or T84 polyclonal, panels
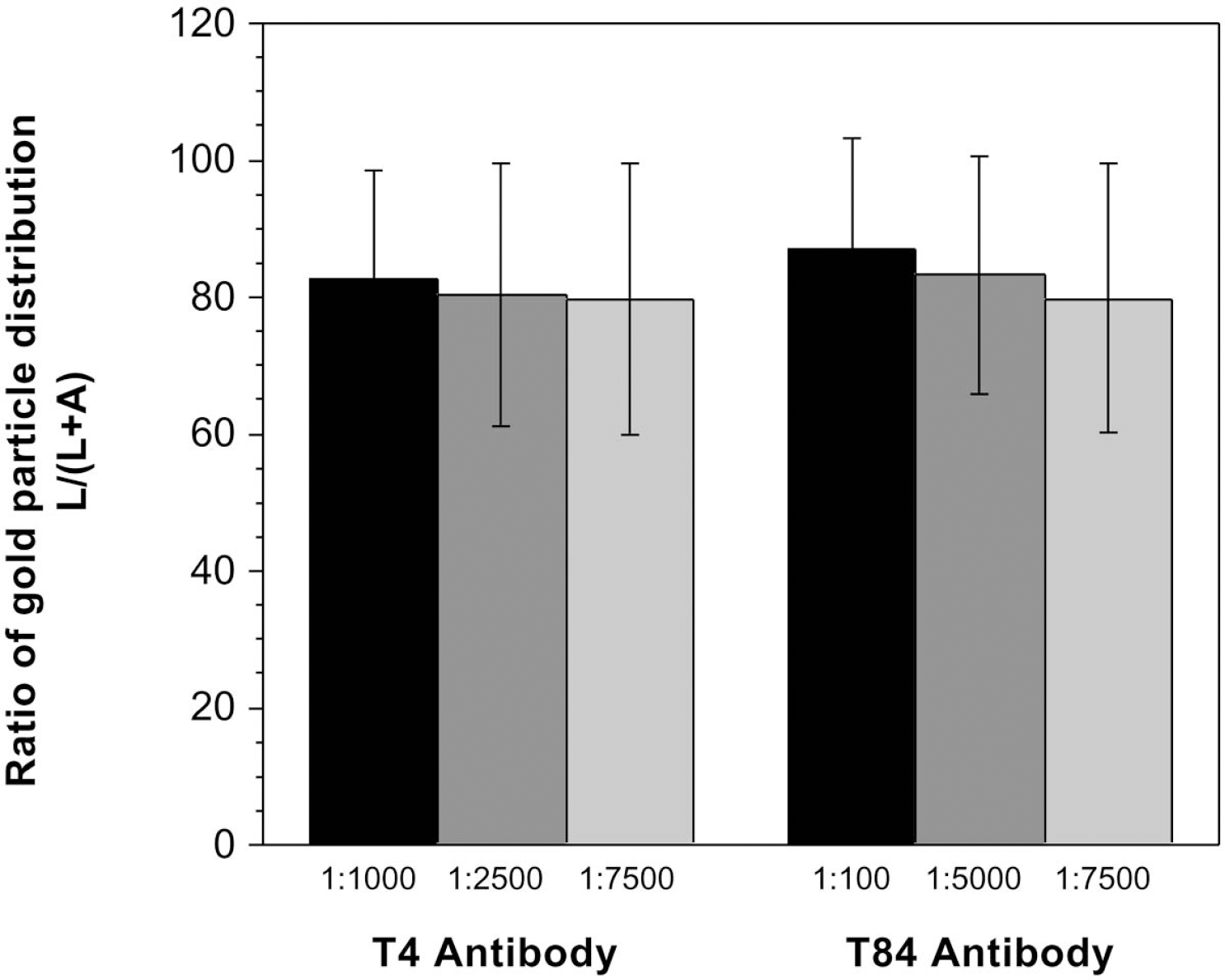
Na-K-Cl cotransporter distribution between luminal and abluminal membranes of brain microvessels as determined by quantitation of gold particles in immunoelectron micrographs. Immunoelectron micrographs generated using either T4 or T84 antibodies were evaluated for relative distribution of Na-K-Cl cotransporter protein in luminal (L) and abluminal (A) membranes of brain microvessels of perfusion-fixed rat brains. Data are presented as the means ± SD of 21, 6, and 12 microvessels for T4 antibody dilutions of 1:1,000, 1:2,500, and 1:7,500, respectively, and 6, 29, and 6 microvessels for T84 antibody dilutions of 1:100, 1:5,000, and 1:7,500, respectively. No significant differences were found among the groups by analysis of variance testing.
Effect of bumetanide on cerebral edema in permanent middle artery occlusion
If the BBB Na-K-Cl cotransporter contributes to ischemia-induced edema formation, then edema should be attenuated or slowed by Na-K-Cl cotransporter inhibition. To investigate this possibility, we evaluated the effect of the Na-K-Cl cotransport inhibitor bumetanide on edema formation occurring in response to permanent MCAO. In these studies, we first subjected rats to MCAO to cause focal ischemia, and then evaluated edema formation by diffusion-weighted magnetic resonance imaging over a 180-minute time course. The purpose of these studies was to evaluate edema formation during the early stages after induction of ischemia and thus, we used only permanent MCAO and did not evaluate rats subjected to ischemia and reperfusion. Figure 3 is a magnetic resonance proton image of a rat brain with left MCAO. This diffusion-weighted image, acquired 90 minutes after occlusion, shows a relative hyperintensity on the left side of the image, which corresponds to edema formation. From the diffusion-weighted imaging data, we calculated apparent diffusion coefficient (ADC) values for four different brain regions in both the ipsilateral and contralateral sides of the brain (regions L1–L4 and R1–R4, respectively). For each brain region, the ratio of left/right ADC values was then determined. Figure 4 shows the results of experiments in which rats were subjected to MCAO surgery or sham surgery and edema formation was measured in the cortex and striatum during a 180-minute period. To eliminate renal diuretic effects of bumetanide, rats were nephrectomized immediately before bumetanide (or vehicle) injection and MCAO or sham surgery. The values shown in Figs. 4A and B are ratios of left (occluded) to right (nonoccluded control) ADC values for regions 1 and 4 of Fig. 3. As predicted, MCAO caused a decrease in ADC values for the occluded hemisphere (increased edema formation), seen as a decrease in the left/right ADC ratios for all four regions evaluated. Previous studies have shown that permanent MCAO causes a net increase in brain water and a decrease in ADC values, and that the latter provides a very good index of brain swelling (Betz et al., 1994; Rudin et al., 2001). In these studies, we also used gravimetric methods (Menzies et al., 1993) to evaluate the effect of 180 minutes of permanent MCAO on brain water (Table 1). As predicted, MCAO caused a significant (2.5%) increase in the percent brain water of the ipsilateral occluded hemisphere compared with the contralateral, nonoccluded hemisphere.
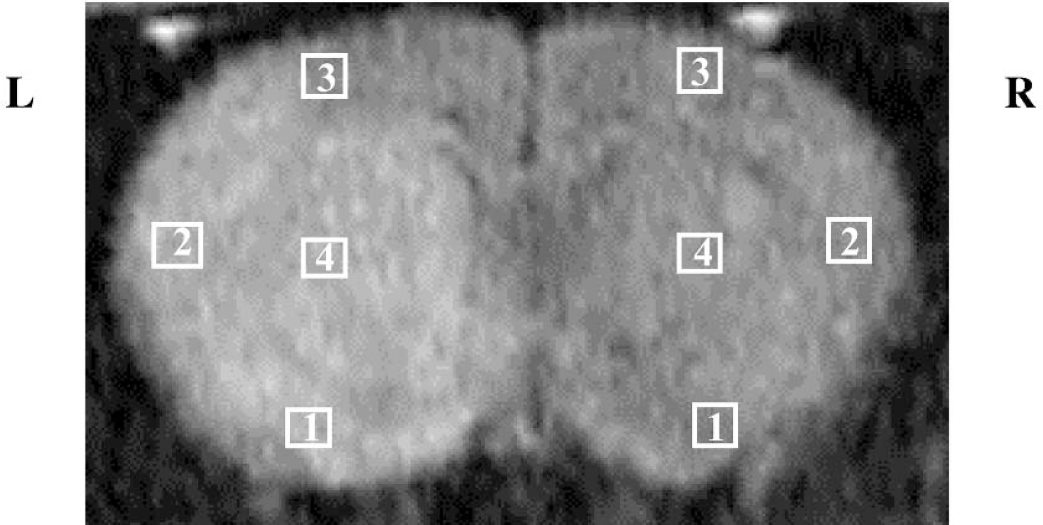
Magnetic resonance imaging assessment of edema in focal ischemia. This magnetic resonance diffusion-weighted image of a rat brain with left MCAO shows a relative hyperintensity (edema) on the left side (L) of the image. For experiments shown in Figs. 4 and 5, ADC values were determined for the regions indicated by the boxes (regions L1–L4 and R1–R4) and ratios of left/right ADC values calculated for each region.
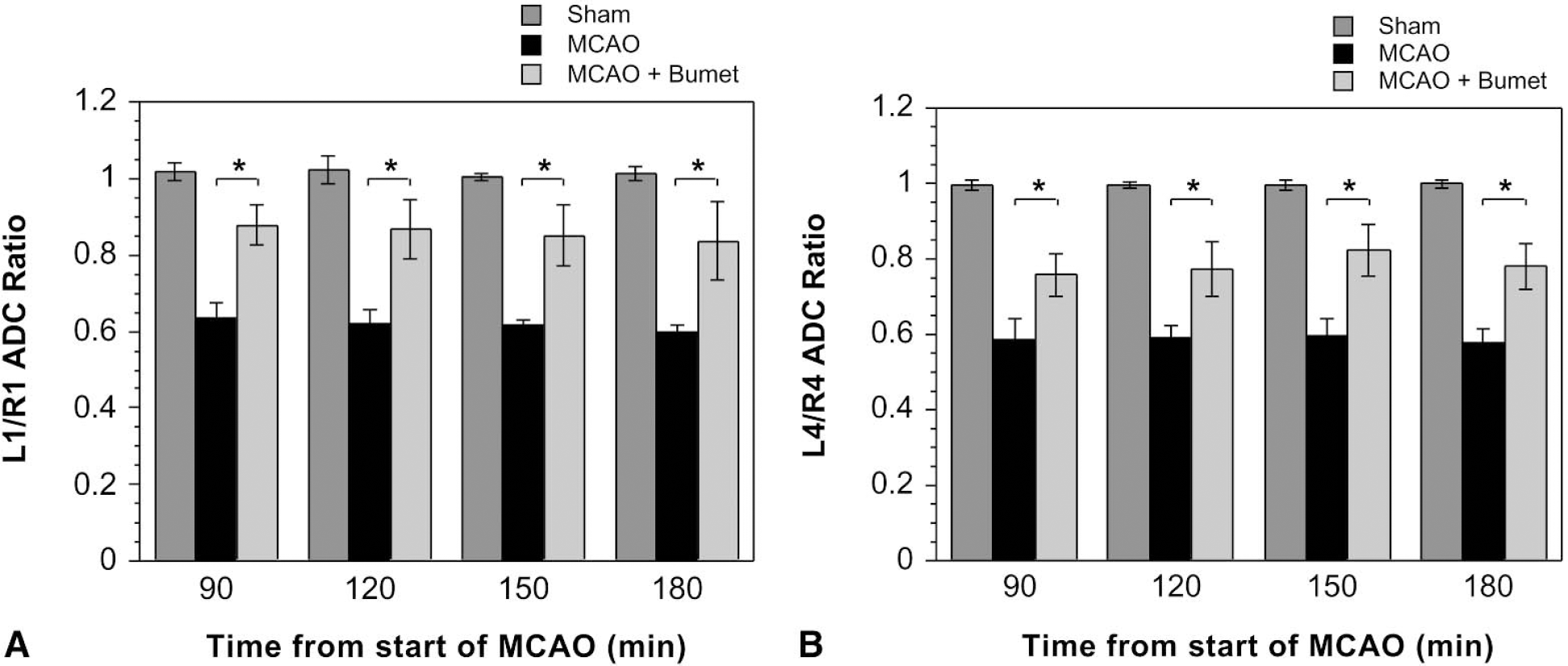
Brain edema formation in permanent MCAO: inhibition by intravenous bumetanide. ADC values were determined for the regions shown in Fig. 3 over a 180-minute period after the initiation of permanent MCAO. Rats were administered bumetanide or vehicle intravenously for 20 minutes, and then subjected to MCAO or MCAO surgery without occlusion (sham) as described in Materials and Methods. The values shown in the figure are ratios of left (occluded) to right (nonoccluded control) ADC values for cortical region 1 (
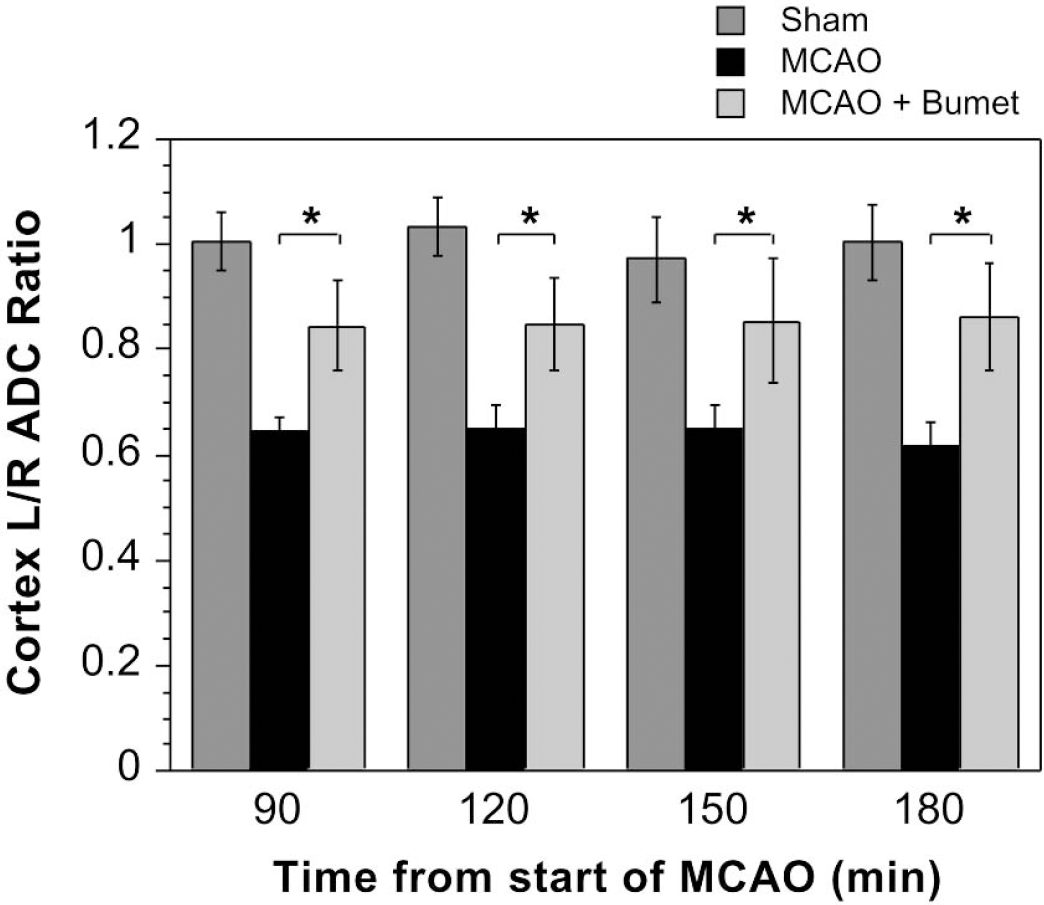
Bumetanide inhibition of cerebral edema in intact (nonnephrectomized) rats subjected to MCAO. ADC ratios were evaluated after MCAO as described in Materials and Methods. Rats underwent sham nephrectomy surgery immediately before MCAO. Bumetanide (7.6 mg/kg) or vehicle was administered intravenously in a single dose 20 minutes before the initiation of MCAO. Values shown are ADC ratios of left (occluded) to right (nonoccluded control) cortical brain regions depicted in Fig. 3 (data from regions 1, 2, and 3 combined). ADC values for sham animals were not affected by bumetanide (data not shown). ADC ratios shown represent means ± SD of six to eight, five or six, and five or six rats each for sham, MCAO, and MCAO plus bumetanide, respectively. At all time points, ADC ratios for MCAO plus bumetanide were found to be significantly different from ADC ratios for MCAO by analysis of variance with a Bonferroni-Dunn post hoc test (*P < 0.0001).
Effect of bumetanide on brain water content after 180 minutes of permanent MCAO
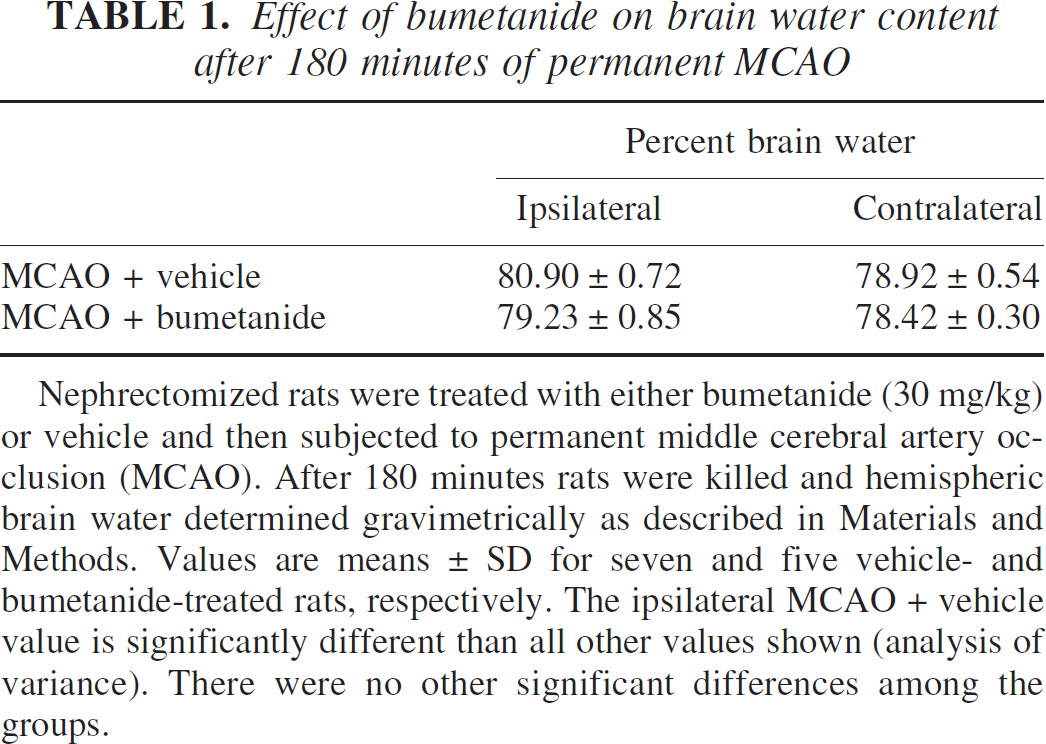
Nephrectomized rats were treated with either bumetanide (30 mg/kg) or vehicle and then subjected to permanent middle cerebral artery occlusion (MCAO). After 180 minutes rats were killed and hemispheric brain water determined gravimetrically as described in Materials and Methods. Values are means ± SD for seven and five vehicle- and bumetanide-treated rats, respectively. The ipsilateral MCAO + vehicle value is significantly different than all other values shown (analysis of variance). There were no other significant differences among the groups.
To determine whether bumetanide reduces edema formation resulting from MCAO, bumetanide (7.6, 15.2, or 30.4 mg/kg) or vehicle was administered intravenously 20 minutes before initiation of MCAO (or sham MCAO). Bumetanide reduced the MCAO-induced decrease in ADC values, indicating that it reduced edema formation. This effect of bumetanide was present from the start of imaging (30 minutes, not shown) and was sustained throughout the 180-minute experiment. Similar results were found for cortical regions 2 and 3 (data not shown). Depending on the time after start of MCAO, reductions in the decrease of ADC values (i.e., reduction of edema) varied from 59% to 67% for region 1, 40% to 53% for region 2, 51% to 66% for region 3, and 41% to 56% for region 4. In each case, ADC ratios for rats subjected to MCAO plus bumetanide are significantly higher than ADC ratios for rats subjected to MCAO plus vehicle. ADC values for animals subjected to sham surgery were not affected by bumetanide (data not shown). In gravimetric experiments evaluating the effect of 180 minutes of permanent MCAO on brain water in bumetanide-treated rats, we found that the ipsilateral hemisphere brain water was increased by only 1.0% over that of the nonoccluded hemisphere (control) and was significantly reduced compared with the ipsilateral hemisphere of rats subjected to MCAO without bumetanide (Table 1).
In these experiments, we also monitored MABP after intravenous administration of bumetanide or vehicle. We found no significant effect of either bumetanide or vehicle on MABP in the MCAO animals, as shown in Table 2. We also evaluated a number of other physiologic parameters in these experiments. Table 3 shows that no significant differences were found between bumetanide- and vehicle-treated rats with respect to plasma concentrations of Na+, K+, Cl−, HCO3−, glucose, blood urea nitrogen, or hemoglobin. Plasma pH, P
Mean arterial blood pressures of rats subjected to MCAO with or without bumetanide
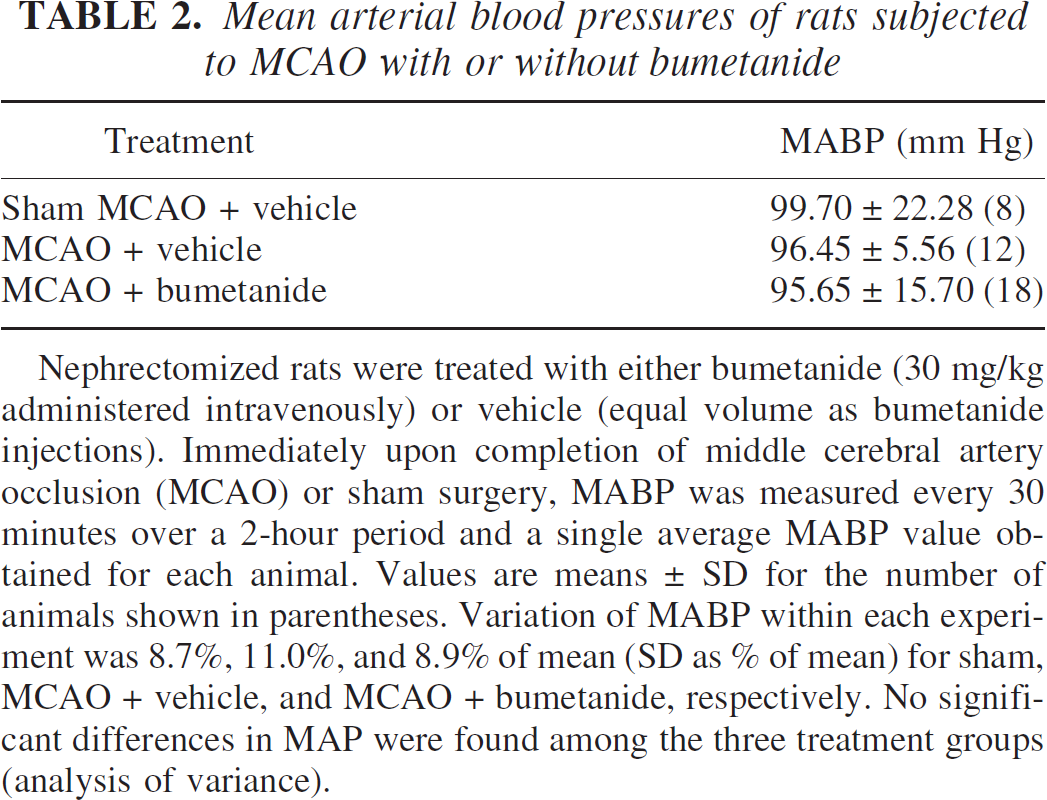
Nephrectomized rats were treated with either bumetanide (30 mg/kg administered intravenously) or vehicle (equal volume as bumetanide injections). Immediately upon completion of middle cerebral artery occlusion (MCAO) or sham surgery, MABP was measured every 30 minutes over a 2-hour period and a single average MABP value obtained for each animal. Values are means ± SD for the number of animals shown in parentheses. Variation of MABP within each experiment was 8.7%, 11.0%, and 8.9% of mean (SD as % of mean) for sham, MCAO + vehicle, and MCAO + bumetanide, respectively. No significant differences in MAP were found among the three treatment groups (analysis of variance).
Physiologic parameters of rats subjected to MCAO with or without bumetanide
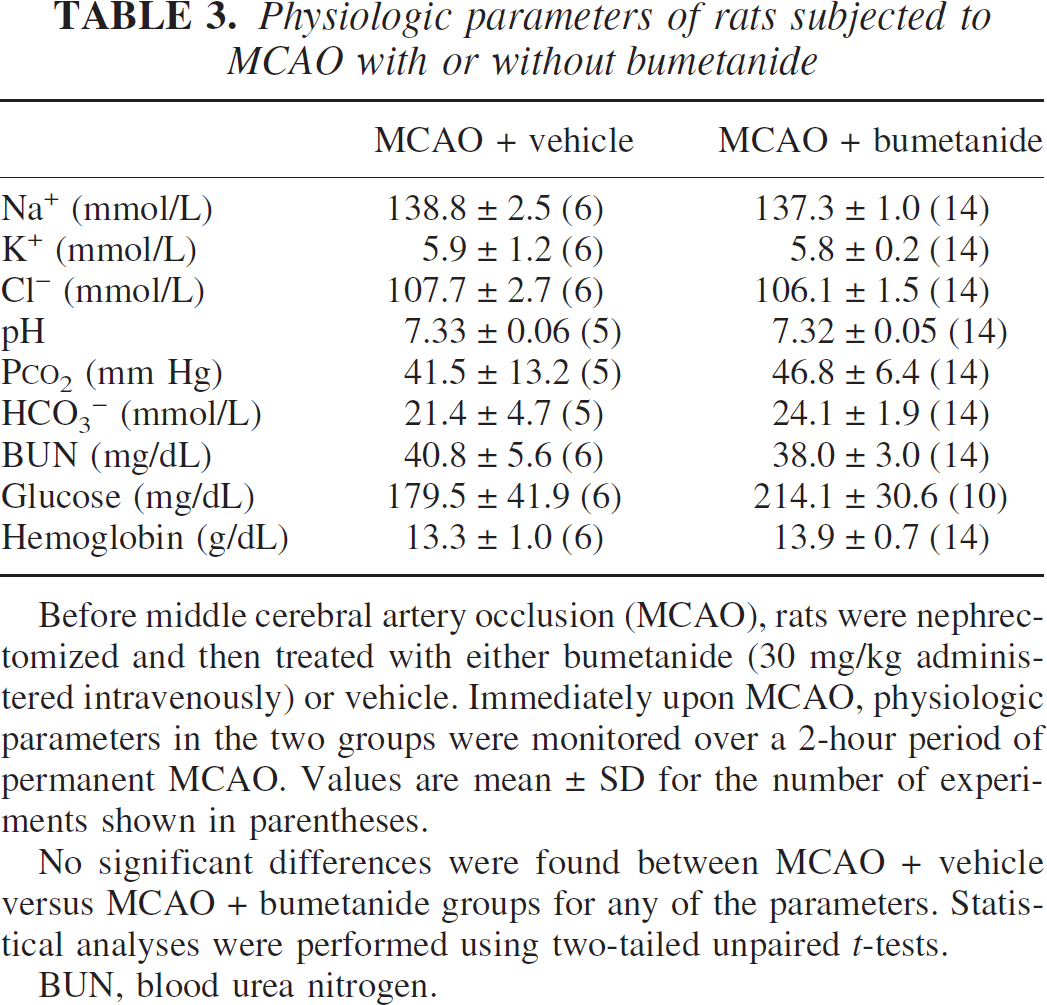
Before middle cerebral artery occlusion (MCAO), rats were nephrectomized and then treated with either bumetanide (30 mg/kg administered intravenously) or vehicle. Immediately upon MCAO, physiologic parameters in the two groups were monitored over a 2-hour period of permanent MCAO. Values are mean ± SD for the number of experiments shown in parentheses.
No significant differences were found between MCAO + vehicle versus MCAO + bumetanide groups for any of the parameters. Statistical analyses were performed using two-tailed unpaired t-tests.
BUN, blood urea nitrogen.
Studies were also conducted to determine whether bumetanide can reduce cerebral edema formation during permanent MCAO in nonnephrectomized animals. Figure 5 shows that, as with the nephrectomized rats, MCAO caused a substantial decrease in ADC values and that bumetanide significantly attenuated the decrease in ADC ratios, indicating a reduction of edema formation. This effect was sustained throughout the 180-minute experiment.
Effect of intravenous bumetanide on infarct volume resulting from permanent middle cerebral artery occlusion
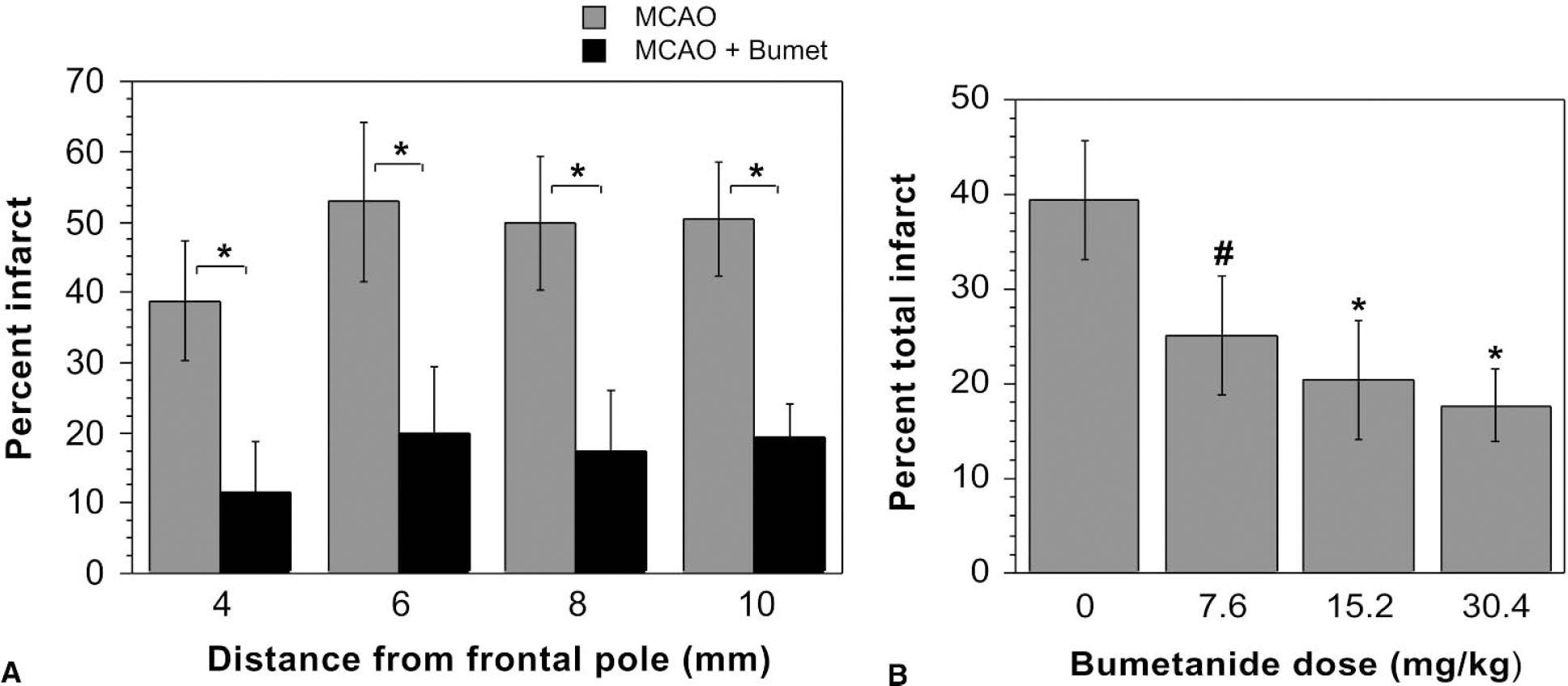
To determine the effect of bumetanide on infarct volume after 180 minutes of permanent MCAO, at the end of the imaging period rats were killed and the brains sliced and stained for infarct detection as described in Materials and Methods. Figure 6 shows representative results of TTC staining in rats subjected to MCAO plus vehicle compared with rats subjected to MCAO plus bumetanide (30.4 mg/kg). An obvious infarct can be seen in the left hemisphere of the rat treated with vehicle, whereas the infarct is noticeably reduced in the rat treated with bumetanide. The data shown in Fig. 7A represent mean infarct volumes resulting from 180 minutes of permanent MCAO in rats treated intravenously with vehicle or bumetanide. Bumetanide markedly reduced the volume of the infarct for all affected brain slices, only four of which are shown here. Figure 7B shows that all three doses of bumetanide used in the experiments (7.6, 15.2, and 30.4 mg/kg) produced a significant reduction of infarct volume. For this figure, all brain slices (eight 2-mm slices starting from the frontal pole) were included in calculation of percent total infarct. In rats treated with vehicle alone, the mean infarct volume was 39% of the ipsilateral hemisphere. In rats treated with 7.6, 15.2, or 30.4 mg/kg bumetanide, the total mean infarct volumes were 25%, 20%, and 18% of the ipsilateral hemisphere, respectively, with no significant differences among these three doses.
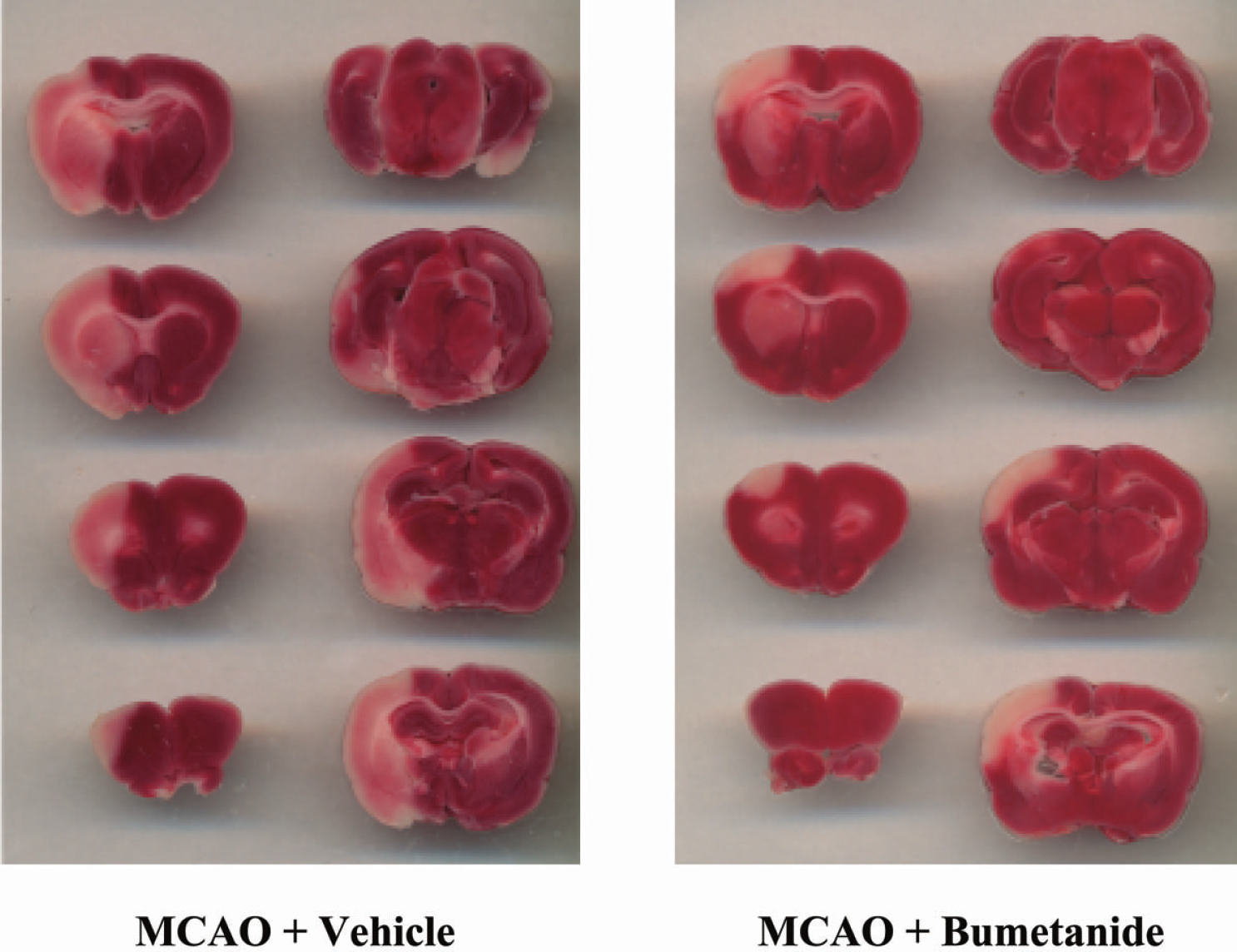
TTC assessment of infarct size: effects of bumetanide. At the conclusion of diffusion-weighted imaging experiments (180–200 minutes), TTC staining of brain slices was performed as described in Materials and Methods. Images shown are from two representative experiments.
(
DISCUSSION
There are two main findings in the present study. The first is that the in situ plasma membrane distribution of the BBB endothelial cell Na-K-Cl cotransporter is asymmetrical, with 80% residing in the luminal membrane. The second finding is that inhibition of the BBB Na-K-Cl cotransporter by intravenous administration of bumetanide reduces cerebral edema formation in ischemia induced by permanent MCAO. These findings support the hypothesis that the BBB Na-K-Cl cotransporter contributes to cerebral edema formation during ischemia.
Immunoelectron micrographs generated using two different antibodies, each over a range of dilutions, all produced the same 80% luminal/20% abluminal result in the present studies. This is consistent with a role for the cotransporter in Na+ and Cl− secretion across the BBB, with the ions entering the cells via the luminal Na-K-Cl cotransporter and then exiting at the abluminal membrane via the Na/K ATPase and a Cl− efflux pathway such as a Cl− channel. It has been suggested previously that an abluminal Na-K-Cl cotransporter might remove K+ from the brain during ischemia (Kawai et al., 1996a; Vigne et al., 1994), a possibility not precluded by our studies because approximately 20% of the plasma membrane cotransporter resides at the abluminal surface. These two functions are not, in fact, mutually exclusive. For K+ absorption, the ion would enter the cells at the abluminal membrane via Na/K ATPase activity and also any Na-K-Cl cotransporter activity present at that membrane. K+ would then exit the cells by the amiloride-sensitive Na+- and K+-selective channel known to be present at the luminal membrane (Vigne et al., 1989). In this manner, NaCl secretion and K+ absorption (transport from brain to blood) are both possible.
Our magnetic resonance diffusion-weighted imaging studies show for the first time that intravenous bumetanide reduces edema formation occurring within the first 3 hours of MCAO-induced cerebral ischemia, i.e., the period before BBB breakdown. This suggests that early edema formation occurs via a mechanism involving the BBB Na-K-Cl cotransporter. A majority of studies on stroke-associated cerebral edema have focused on ischemia/reperfusion, a situation that promotes relatively rapid BBB breakdown. In contrast, our present study examined events occurring during permanent MCAO without the complicating factors of reperfusion. We assessed brain edema via determination of ADC because a decrease in ADC values has been shown to be an excellent indicator of brain edema (Hasegawa et al., 1994; Knight et al., 1994; Liu et al., 2001a; Moseley et al., 1990). This method also allows comparison of specific ipsilateral and contralateral regions, providing an internal control for each experiment. Our studies show that bumetanide reduces edema occurring during ischemia regardless of whether rats are nephrectomized, and thus this effect is independent of any diuretic actions. Bumetanide was effective at doses as low as 7.6 mg/kg, which, by our estimation, corresponds to approximately 100 μM bumetanide in plasma, a concentration that effectively inhibits Na-K-Cl cotransporter activity of brain microvascular endothelial cells (O'Donnell, 1989, 1995a; Palfrey and O'Donnell, 1992). Our studies also show that intravenous bumetanide reduces brain infarct volume. The degree of infarct reduction and attenuation of ADC decrease are in reasonable agreement (43–65% and 40–65% respectively). However, the possibility that the infarct-reducing actions of bumetanide may not be solely attributable to edema reduction needs to be considered. One could propose, for example, that bumetanide improves CBF. However, in LD measurements of CBF made before and after MCAO, we found no evidence that bumetanide significantly alters blood flow.
A previous study showed that intraperitoneal injection of a torasemide derivative (S20390) reduced edema after MCAO in rats (Le Bars et al., 1996). Similarly, torasemide reduced brain swelling and attenuated the increase in brain water after cold injury-induced focal ischemia in rat (Staub et al., 1994). These findings are consistent with our present results. However, torasemide inhibits Cl− channels as well as the cotransporter, and it is therefore unclear whether the effects of torasemide were due to cotransporter inhibition. In spontaneously hypertensive rats subjected to a combined 2-hour ischemia plus 24-hour reperfusion injury, increased brain water and infarct were reduced by intracerebral microdialysis pretreatment with bumetanide (100 μmol/L) (Yan et al., 2001, 2003). The intracerebral bumetanide delivery allowed inhibition of Na-K-Cl cotransport in neurons and astrocytes, and thus supports a role for the cotransporter in cytotoxic edema occurring in astrocytes and/or neurons during ischemia/reperfusion. However, the investigators did not address the question of the role played by the BBB Na-K-Cl cotransporter in edema formation induced by either ischemia/reperfusion or by ischemia alone. In contrast, the present study examined the effects of bumetanide administered intravenously in rats during the first 3 hours of permanent MCAO before the time barrier breakdown begins. Thus, the edema reducing actions of bumetanide observed here should be the result of BBB Na-K-Cl cotransporter inhibition. Previous pharmacokinetic studies have shown that bumetanide and the chemically similar loop diuretics furosemide and torasemide distribute only in the extracellular fluid, indicating that they do not readily cross plasma membranes (Chen, 1996; Friedel and Buckley, 1991). Also, in a study of chemical properties determining the ability of various drugs to cross the BBB, furosemide did not cross the barrier (Fischer et al., 1998). Thus, although bumetanide should readily distribute in extracellular fluid outside the brain, it should not penetrate into the brain in the presence of an intact BBB.
The mechanism whereby ischemia stimulates the BBB Na-K-Cl cotransporter is yet to be determined. There is good evidence, however, that hypoxia and the peptides vasopressin and endothelin, all factors present during ischemia, stimulate activity of the BBB cotransporter. We have found that vasopressin, which is centrally released during ischemia (Dóczi 1993; Landgraf 1992; Ostrowski et al., 1992; Sorensen et al., 1985) and promotes edema formation (Dickinson and Betz, 1992; Dóczi, 1993; Dóczi et al., 1982, 1984; Hertz et al., 2000; Rosenberg et al., 1990), is also a potent stimulator of the brain microvascular Na-K-Cl cotransporter (O'Donnell et al., 1995a), as is endothelin, which is also released during ischemia (Barone et al., 1994; Kawai et al., 1996b, 1997; Spatz et al., 1997). We have shown that vasopressin stimulates the brain endothelial cotransporter by a V1 vasopressin receptor in a manner involving elevation of intracellular [Ca2+] (O'Donnell et al., 1999). Endothelin also elevates intracellular [Ca2+] in these cells, and we know elevated [Ca2+] stimulates the brain microvascular Na-K-Cl cotransporter (O'Donnell et al., 1995a). It is of interest to note that the peptide ANP, which attenuates ischemia-induced brain edema (Naruse et al., 1990), also inhibits activity of the BBB cotransporter (O'Donnell et al., 1995a). With regard to hypoxia, treatment of brain microvascular endothelial cells with oligomycin to induce “chemical hypoxia” stimulates cotransport activity, as does true hypoxia (Kawai et al., 1996a, b ; O'Donnell et al., 1999). Elucidation of the specific mechanisms responsible for hypoxia stimulation of the BBB cotransporter awaits further study. However, this phenomenon has also been observed in both turkey and ferret erythrocytes, where hypoxia is a major stimulus for Na-K-Cl cotransporter activity (Flatman, 2001; Muzyamba et al., 1999). The mechanism appears to involve changes in activity of an unidentified kinase in the cells, resulting in changes in phosphorylation and thus activity of the cotransporter protein (Flatman, 2002).
To our knowledge, the present findings provide the first demonstration that intravenous bumetanide reduces ischemia-induced brain edema occurring in response to permanent MCAO. Our findings suggest that inhibition of the BBB Na-K-Cl cotransporter may be a valuable avenue to pursue for stroke therapy. Our experimental protocol involved pretreatment with bumetanide before MCAO. Future studies will need to evaluate the effectiveness of administering bumetanide after the onset of cerebral ischemia, as would occur in a clinical setting. A previous study showed that intravenous administration of the Na/H exchange inhibitor dimethylamiloride caused a 20% reduction in edema formation induced by permanent MCAO (Betz et al., 1995), and our own recent studies have shown that intravenous administration of the specific Na/H exchange inhibitor HOE-642 can also reduce edema formation in MCAO (Tran et al., 2004). Thus, a luminal Na/H exchanger may also contribute to edema formation during ischemia. Additional studies will need to be performed to address the relative importance of these BBB luminal Na+ transporters in cerebral edema formation.
Footnotes
Acknowledgment
The authors thank Dr. E. B. Jones for providing access to his laboratory cryoembedding facility.