
Abstract
Few studies have examined the signaling pathways that contribute to early brain injury after subarachnoid hemorrhage (SAH). Using a rat SAH model, the authors explored the role of vascular endothelial growth factor (VEGF) and mitogen-activation protein kinase (MAPK) in early brain injury. Male Sprague-Dawley rats (n = 172) weighing 300 to 350 g were used for the experimental SAH model, which was induced by puncturing the bifurcation of the left anterior cerebral and middle cerebral arteries. The blood–brain barrier (BBB), brain edema, intracranial pressure, and mortality were evaluated at 24 hours after SAH. The phosphorylation of VEGF and different MAPK subgroups (ERK1/2, p38, and JNK) were examined in both the cortex and the major cerebral arteries. Experimental SAH increased intracranial pressure, BBB permeability, and brain edema and produced high mortality. SAH induced phosphorylation of VEGF and MAPKs in the cerebral arteries and, to a lesser degree, in the cortex. PP1, an Src-family kinase inhibitor, reduced BBB permeability, brain edema, and mortality and decreased the phosphorylation of VEGF and MAPKs. The authors conclude that VEGF contributes to early brain injury after SAH by enhancing the activation of the MAPK pathways, and that the inhibition of these pathways might offer new treatment strategies for SAH.
Early brain injury after aneurysmal rupture strongly determines the mortality and morbidity of patients suffering from subarachnoid hemorrhage (SAH) (Adams et al., 1981; Kirsch et al., 1989; Ogungbo et al., 2001; Weir 1979). Few studies, either clinical or experimental, have examined the mechanisms, especially the signaling pathways that mediate early brain injury (Kassell and Drake, 1983). One of the early brain injuries is the alteration of blood–brain barrier (BBB) permeability after SAH (Doczi et al., 1986; Germano et al., 2000; Imperatore et al., 2000; Johshita et al., 1990a; Nakagomi et al., 1989; Peterson and Cardoso, 1983; Sasaki et al., 1986; Zuccarello and Anderson, 1989). Dysfunction of BBB contributes to brain edema (Doczi et al., 1995; Johshita et al., 1990b; Laszlo et al., 1995) and prolongs elevated intracranial pressure (ICP) values after SAH (Fornezza et al., 1990; Fukuhara et al., 1998; Hayashi et al., 1977; Voldby and Enevoldsen, 1982). Brain edema, elevated ICP, and microcirculation compromise (Doczi 2001; Hall et al., 1996; Ohkuma et al., 2000; Vollmer et al., 1992) all contribute to early brain injury, which results in death or disability after SAH.
Subarachnoid hemorrhage elicits a wide range of stress responses in human brain tissues and results in the activation of several intracellular signaling pathways, including tyrosine kinase and its substrate mitogen-activated protein kinase (MAPK) pathways (Fujikawa et al., 1999; Tibbs et al., 2000; Vollrath et al., 1998). Tyrosine and MAPK regulate tissue stress response which leads to tissue differentiation, proliferation, death, or survival (Eguchi and Inagami, 2000; Nozaki et al., 2001; Page and Doubell, 1996; Wolf and Seger 2002). Three major MAPK subgroups are the extracellular signal-regulated kinase (ERK), c-jun-N-terminal kinase (JNK), and p38 (Zhu et al., 1999). These MAPKs are activated by various stimulants such as growth factors, including vascular endothelial growth factor (VEGF), oxidative stress, and inflammatory cytokines (Chakraborti and Chakraborti, 1998; Chow et al., 2001; Parker et al., 2002; Sugden and Clerk, 1998).
Vascular endothelial growth factor is an endothelial mitogen which is expressed in neuronal and vascular tissues in the brain (Ogunshola et al., 2002). VEGF binds with its receptor (a receptor tyrosine kinase) and activates MAPK signaling pathways (Chow et al., 2001; Ogunshola et al., 2002). The levels of VEGF in the bloody cerebral spinal fluid and the expression of VEGF in the brain tissues are increased after SAH (Borel et al., 2003; Josko et al., 2001; McGirt et al., 2002), which might enhance BBB permeability (Mayhan, 1999; Zhang et al., 2000). Src tyrosine kinase is downstream of VEGF activation (Irving and Bamford, 2002), and Src is activated after experimental SAH (Marton et al., 1996; Patlolla et al., 1999; Vollrath et al., 1998). Src regulates VEGF-mediated BBB permeability alteration, and inhibition of Src attenuates vascular permeability, decreases brain edema, and reduces brain infarct after ischemic stroke (Paul et al., 2001). In addition, VEGF fails to enhance vascular permeability in c-Src knockout mice (Paul et al., 2001). In the present study, we examined the hypotheses that Src tyrosine kinase represents a key molecular factor in early brain injury after SAH, and the inhibition of Src interrupts the VEGF–MAPK pathways in the ischemic cortex and cerebral arteries.
MATERIALS AND METHODS
This protocol was evaluated and approved by the Animal Care and Use Committee at Louisiana State University Health Sciences Center in Shreveport, Louisiana.
Subarachnoid hemorrhage rat model
Male Sprague-Dawley rats (N = 172) weighing 300 to 350 g were used in this study. The animals were anesthetized with an intraperitoneal injection of α-chloralose (40 mg/kg) and urethane (400 mg/kg). Polyethylene catheters were placed in the left femoral artery for continuous arterial blood pressure and blood gas monitoring. The animals were intubated, and respiration was maintained with a small animal respirator (Harvard Apparatus, Holliston, MA, U.S.A.). Blood gas was measured and maintained within the physiological range. Rectal temperature was maintained at 36°C with a heating pad.
The left common carotid artery, including its bifurcation, was exposed. In rats subjected to SAH, the external carotid artery was isolated and severed, leaving a stump of approximately 3 to 4 mm. The pterygopalatine artery was ligated using a silk suture. The internal carotid artery was clamped with a small vascular clip, and the common carotid artery was clamped with a small 5-mm aneurysm clip. The stump of the external carotid artery was reopened, and a 4.0 monofilament nylon suture was inserted up through the internal carotid artery for about 1.8 mm. A small resistance was usually felt, and the suture was then advanced 2 mm further to perforate the artery. It was immediately withdrawn through the internal carotid artery into the external carotid artery, allowing reperfusion to produce SAH. Sham-operated rats underwent identical procedures except the suture was withdrawn just after the resistance was felt. After surgery, the rats were returned to their cages to recover. The endotracheal tube was left in place for self-extubation as the animals awakened from anesthesia. We have conducted this animal model of SAH previously (Gules et al., 2002). It was modified from a previous report (Bederson et al., 1995).
Treatment groups
To evaluate the effect of PP1, an Src-family kinase inhibitor, on early brain injury after SAH, intraperitoneal injections of PP1 (Biomol, Plymouth Meeting, PA, U.S.A.) were delivered (0.5 mg/kg or 1.5 mg/kg) 2 hours before and 6 hours after the induction of SAH. PP1 was dissolved in 1% dimethyl sulfoxide (DMSO) and was further diluted in phosphate-buffered saline (PBS) (final < 0.01% DMSO). Other rats were treated with the same volume of vehicle (DMSO diluted in PBS) at 2 hours before and 6 hours after SAH.
Mortality
Mortality was measured at 24 hours after SAH. The number of animals in each group designated for mortality study was SAH+DMSO (n = 16), SAH + PP1 0.5 mg/kg (n = 14), SAH+PP1 1.5 mg/kg (n = 14), and sham (n = 6).
Brain water content (brain edema)
The brains were removed at 24 hours after SAH. The entire brain was weighed immediately after removal (wet weight) and weighed again after drying in an oven at 105°C for over 24 hours as described by others (Xi et al., 2001). The percent of water content was calculated as [(wet weight − dry weight)/wet weight] × 100%. The number of animals used in each group for brain edema study was SAH+DMSO (n = 5), SAH+PP1 0.5 mg/kg (n = 4), SAH+PP1 1.5 mg/kg (n = 4), and sham (n = 4).
Intracranial pressure
The cannula (polyethylene tube no. 10) was placed into the subarachnoid space of L5 through the dura mater after laminectomy and was secured with glue. The distal end of the catheter was fitted to a pressure transducer. The cerebrospinal fluid pressure was measured before SAH, immediately after SAH (the maximum pressure was recorded for 5 minutes after puncture), and 3, 6, and 24 hours after SAH. The number of animals used in each group for ICP study was SAH+DMSO (n = 8), SAH+PP1 0.5 mg/kg (n = 9), SAH+PP1 1.5 mg/kg (n = 12), and sham (n = 6). In all of the rats, ICP was simultaneously recorded from a catheter in the subarachnoid space at the cisterna magna as described by others (Barth et al., 1992). The ICP values from both the lumbar catheter and cisterna magna catheter were almost identical. Since blood clots occluded ICP catheters in the cisterna magna in several rats, only the ICP values from the lumbar catheter were reported in this study.
BBB permeability
At 24 hours after SAH, under general anesthesia (as described previously) and under an operating microscope, the left skull bone was widely exposed by a U-shaped skin incision, followed by a wide hemicraniectomy. The dura mater was carefully removed without brain contusion. Immediately after craniotomy, the rat was placed on the stage of a Nikon fluorescence microscope, and a cover glass (7 mm diameter) was put on the rat's brain. Artificial CSF consisting of Na+ 147.8 mEq/L, K+ 3.0 mEq/L, Mg2+ 2.3 mEq/L, Ca2+ 2.3 mEq/L, Cl− 135.2 mEq/L, HCO3− 19.61 mEq/L, lactate 1.67 mEq/L, phosphate 1.1 mmol/L, and glucose 3.9 mmol/L was used to keep the brain surface wet. The number of animals used in each group in the BBB study was SAH+DMSO (n = 5), SAH+PP1 0.5 mg/kg (n = 5), SAH+PP1 1.5 mg/kg (n = 5), and sham (n = 5).
Intravital fluorescence microscopy
An upright Nikon microscope equipped with a silicon-intensified target camera (C2400 to 08, Hamamatsu Photonics, Hamamatsu, Japan) and a mercury lamp were used to observe the cerebral vessels. With x10 objectives, the magnifications on the video screen (Sony, Tokyo, Japan) were x280. The microscopic images were received by a CCD video camera that was attached to an image intensifier. The images were recorded on a video recorder (Sony, Tokyo, Japan) equipped with a video timer (Time-Date Generator, WJ-810, Panasonic, Tokyo, Japan). Other equipment included a xenon lamp, an objective with a magnification of x20, two filter blocks with an excitation filter for 450 to 490 nm (Nikon, B-2A, dichroic mirror for 510 nm, and barrier filter of 520 nm), and an excitation filter for 510 to 560 nm (Nikon, G-2A, dichroic mirror for 580 nm and barrier filter of 590 nm). To quantify albumin leakage across 30- to 50-μm-diameter vessels that were randomly selected on the surface of a parietal cortex, 50 mg/kg of fluorescein isothiocyanate (FITC)-labeled bovine serum albumin (Sigma, St. Louis, MO, U.S.A.) desorbed with saline (10 mg/mL) was administered intravenously (1 mL/min). Extravasation of FITC-albumin (FITC-A) fluorescence intensity was measured and compared to the intensity of vessels immediately after the injection of FITC-A.
Western blotting
For Western blot analysis, the rats were anesthetized, killed with an overdose of pentobarbital (150 mg/kg), and perfused with ice-cold PBS from the left ventricle. The brains were removed and the basal cortex (adjacent to the basal cistern) and major cerebral arteries (basilar, middle cerebral, anterior cerebral arteries) were removed under a stereoscopic microscope. These samples were frozen and stored at −80°C until protein extraction.
The method for Western blot has been described in our previous publications (Satoh et al., 2002; Zubkov et al., 2000, 2001). The frozen samples were homogenized for 20 minutes at 4°C with an ultrasonic wave (10 seconds, three times) in 100 μL of extraction buffer (containing 50-mmol/L Tris-HCl, pH 7.6; 1% nonylphenol ethoxylate [Igepal]; 0.25% sodium deoxycholate; 150-mmol/L NaCl; 1-mmol/L EGTA; 1-mmol/L phenylmethylsulfonyl fluoride; 1-μg/mL aprotinin, leupeptin, pepstatin; 1-mmol/L Na3VO4; and 1-mmol/L NaF). The insoluble material was removed by centrifugation at 16,000g at 4°C for 30 minutes. The samples (for VEGF, ERK1/2 and JNK: 20 μg proteins from cerebral cortex, 10 μg proteins from cerebral arteries; for p38: 60 μg proteins from cerebral cortex, 15 μg proteins from cerebral arteries), in nonreducing condition, were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis with 12% polyacrylamide gel. After electrophoretic transfer of the separated polypeptides to the nitrocellulose membranes, the membranes were blocked with 5% nonfat milk in Tween-TBS (TBST). The membranes were then washed and incubated with the primary antibodies at 4°C using 1% nonfat milk in TBST. The following primary antibodies were obtained from commercial sources: rabbit polyclonal anti-JNK phosphospecific, rabbit polyclonal anti-P38 phosphospecific, rabbit polyclonal anti-VEGF, and goat polyclonal antiactin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). The rabbit polyclonal anti-ERK1/2 phosphospecific antibody was purchased from Biosource International. After incubation with the primary antibodies, the nitrocellulose membranes were washed with TBST and incubated with appropriate horseradish peroxidase-labeled secondary antibodies (Biosource International, Santa Cruz Biotechnology) using 1% nonfat milk in TBST for 1 h at room temperature. An enhanced chemiluminescence system (Amersham, Piscataway, NJ, U.S.A.) was used to visualize the protein bands. Quantity One software (BioRad, Hercules, CA, U.S.A.) quantified the results. As an internal control, actin was blotted by goat polyclonal antiactin antibody in the same membrane. The number of animals in each group used for Western blot analysis was SAH+DMSO (n = 16), SAH+PP1 0.5 mg/kg (n = 14), SAH+PP1 1.5mg/kg (n = 14), and sham (n = 6).
Data analysis
Data are expressed as mean ± SEM. Statistical differences between the control and other groups were compared by one-way analysis of variance (ANOVA) and then, if significant differences were found, the Tukey-Kramer multiple comparison procedure. The neurologic evaluations were compared by the Kruskal-Wallis one-way ANOVA on ranks, and then, if significant differences were found, by the Dunn multiple-comparison procedure. A probability value of P < 0.05 was considered statistically significant.
RESULTS
Mortality
None of the rats in the sham-operated group died within 24 hours. In the SAH treated with vehicle (DMSO) group, 43.8% of the rats died within 24 hours after SAH. Autopsies revealed massive cerebral bleeding occurred in these animals. PP1 treatment at 0.5 mg/kg and 1.5 mg/kg reduced the mortality in a concentration-dependent manner and decreased the mortality rate to 28.6% and 21.4%, respectively.
Brain water content (brain edema)
The mean values of brain water content in the sham-operated, SAH treated with vehicle (DMSO), SAH treated with PP1 at 0.5 mg/kg, and SAH treated with PP1 at 1.5 mg/kg groups were 77.635%, 78.150%, 77.740%, and 77.567%, respectively. The brain water content in the DMSO-treated group was significantly higher than the sham-operated or PP1-treated groups (P < 0.05, ANOVA) (Fig. 1). No concentration-dependent effect of PP1 on brain edema was observed (P > 0.05).
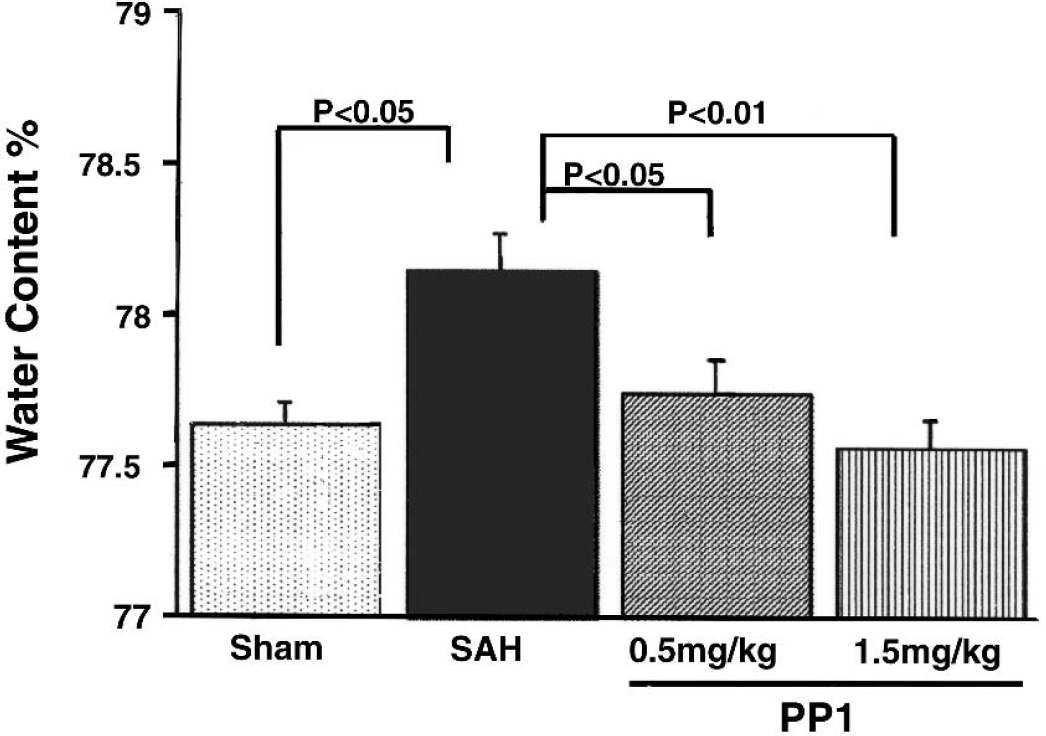
Brain edema. The brain water content was increased significantly (P < 0.05 vs. sham-operated, ANOVA) at 24 hours after subarachnoid hemorrhage (SAH). PP1 treatment markedly reduced (P < 0.05) brain water content.
ICP
The mean baseline ICP values of sham-operated, SAH treated with vehicle (DMSO), SAH treated with PP1 at 0.5 mg/kg, and SAH treated with PP1 at 1.5 mg/kg were 6.7, 7.1, 7.1, and 7.3 mm Hg, respectively. These numbers were not statistically different (P > 0.05, ANOVA). This observation indicates that PP1 did not affect normal ICP values. Immediately after SAH, the mean peak ICP values were 6.7, 58.3, 61.6, and 66.4, respectively (Fig. 2). Even though all of the SAH groups (SAH+DMSO, SAH+PP1 to 0.5mg/kg, and PP1 to 1.5 mg/kg) were significantly higher than the sham-operated group (P < 0.05, ANOVA), there were no statistical significances among these three SAH groups (P > 0.05).
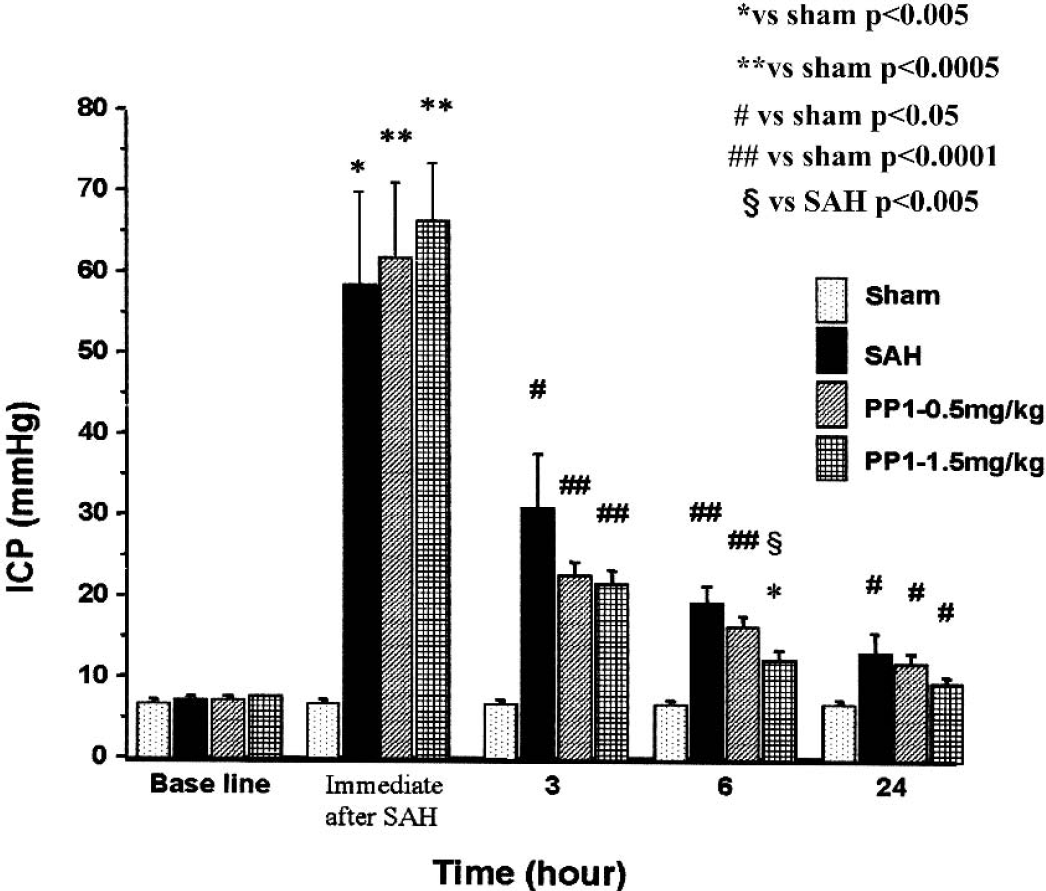
Intracranial pressure (ICP) values. Similar baseline ICP was observed in all groups. ICP increased more than 10 times after SAH in all of the groups, regardless of treatment. No significant differences were observed among the subarachnoid hemorrhage (SAH) groups. The plateau phases of ICP at 3, 6, and 24 hours were lower in all PP1-treated groups, even though statistical significance (P < 0.05 vs. SAH treated with DMSO, ANOVA) was obtained only in PP1 (1.5 mg/kg) at 6 hours after SAH.
However, the mean ICP values at 3, 6, and 24 hours had a tendency to be lower in the PP1-treated groups than the SAH treated with vehicle (DMSO) group, even though only the ICP value of PP1 at 1.5 mg/kg at 6 hours was significantly lower (P < 0.05, ANOVA) than the SAH with vehicle treatment group. Even at 24 hours after SAH, in all of the SAH groups, regardless of treatment, the ICP values were markedly higher (P < 0.05) than in the sham-operated group.
Blood–brain barrier permeability
The method for the calculation of BBB permeability is shown in Fig. 3. Frames of vessels recorded for comparison of the background (BG), immediately after SAH (IM), and 30 minutes after SAH (30 min) were grabbed onto a computer. Using image software (NIH Image), rectangles measuring either 10 × 50 μm (as shown), the center of the vessel (Ii: the mean fluorescent intensity in the interstitium), or 10 μm away from the vessel wall (Iv: the mean fluorescent intensity in the vessel) were drawn and calculated. The albumin leak index (%) was calculated by using the following formula: 100 × [(Ii at 30m − Ii at BG) − (Ii at IM − Ii at BG)]/(Iv at IM − Iv at BG) = 100 × (Ii at 30m − Ii at IM)/(Iv at IM − Iv at BG).
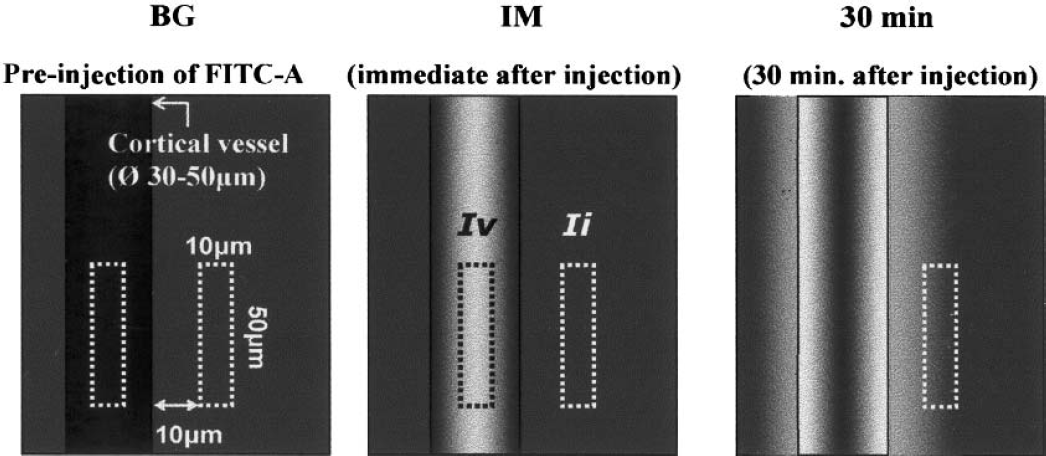
Schematic blood–brain barrier measurement. Frames of vessels recorded for comparison of the background (BG), immediately after SAH (IM), and 30 minutes after subarachnoid hemorrhage (30 minutes) were grabbed onto a computer. Using image software (NIH Image), rectangles measuring either 10 × 50 μm (as shown), the center of the vessel (Ii: the mean fluorescent intensity in the interstitium), or 10 μm away from the vessel wall (Iv: the mean fluorescent intensity in the vessel) were drawn and calculated. The albumin leak index (%) was calculated by using the following formula: 100 × [(Ii at 30m − Ii at BG) − (Ii at IM − Ii at BG)]/(Iv at IM − Iv at BG) = 100 × (Ii at 30m − Ii at IM)/(Iv at IM − Iv at BG).
The pictures from fluorescent microscopy showed the degree of extravasation of the FITC albumin. In the sham-operated rats, no extravasation was observed. However, in the SAH treated with vehicle (DMSO) group, remarkable extravasation was observed. PP1 treatment reduced extravasation of the FITC albumin (Fig. 4).
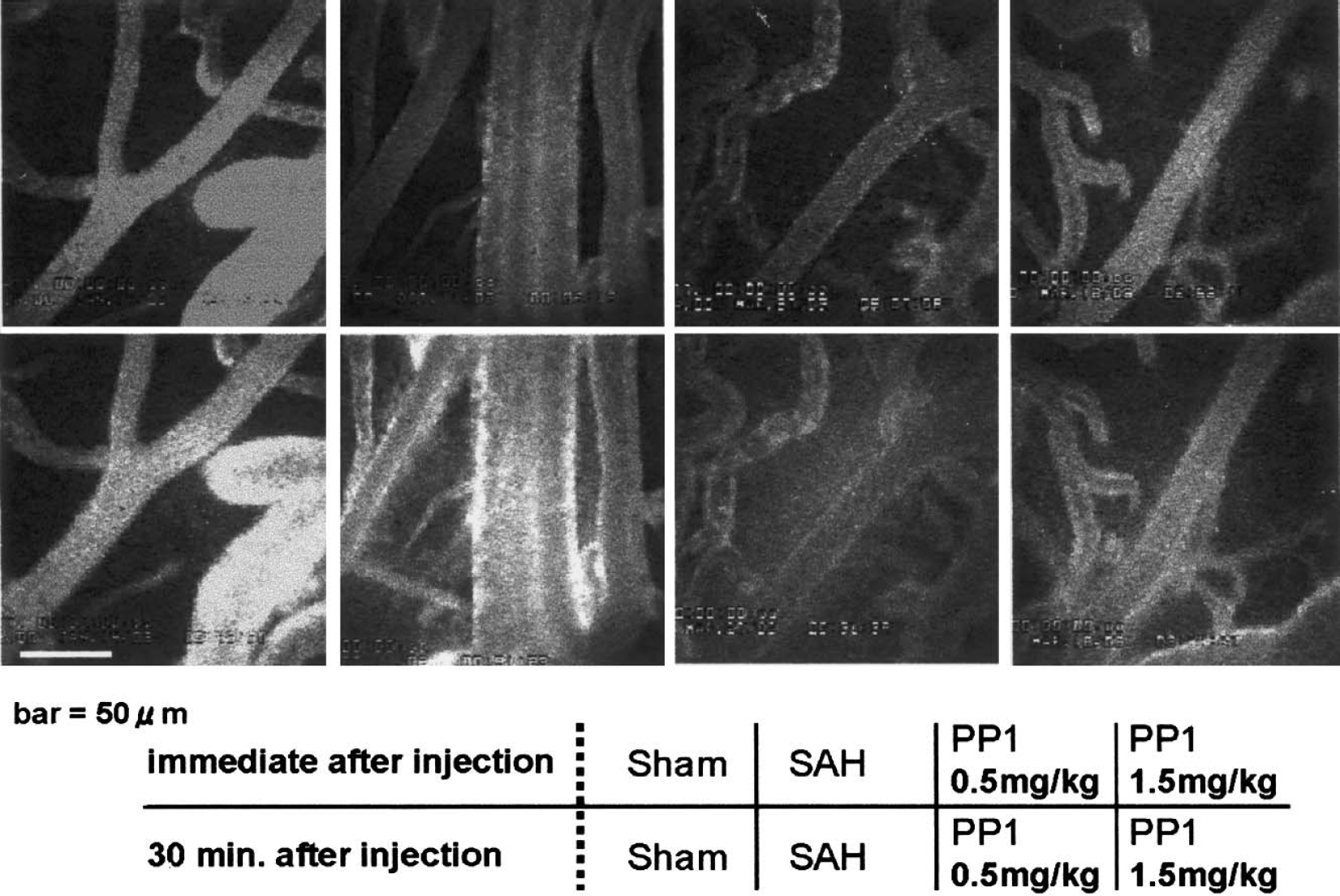
Blood–brain barrier imaging. The top four panels represent imaging pictures showing the dye in the blood vessels immediately after the dye injection in the sham-operated, subarachnoid hemorrhage (SAH), and SAH treated with PP1 animals. The lower panel shows the imaging 30 minutes after the dye injection. Significant enhanced fluorescent intensity occurred at the edge of the vessels in the SAH animals but not in the sham-operated or PP1-treated animals.
The albumin leak index is shown in Fig. 5. In the SAH treated with vehicle group, the index significantly increased compared to the sham group (from 3.5% to 56.7%, P < 0.001). PP1 reduced the albumin leak index in a concentration-dependent manner (P < 0.05).
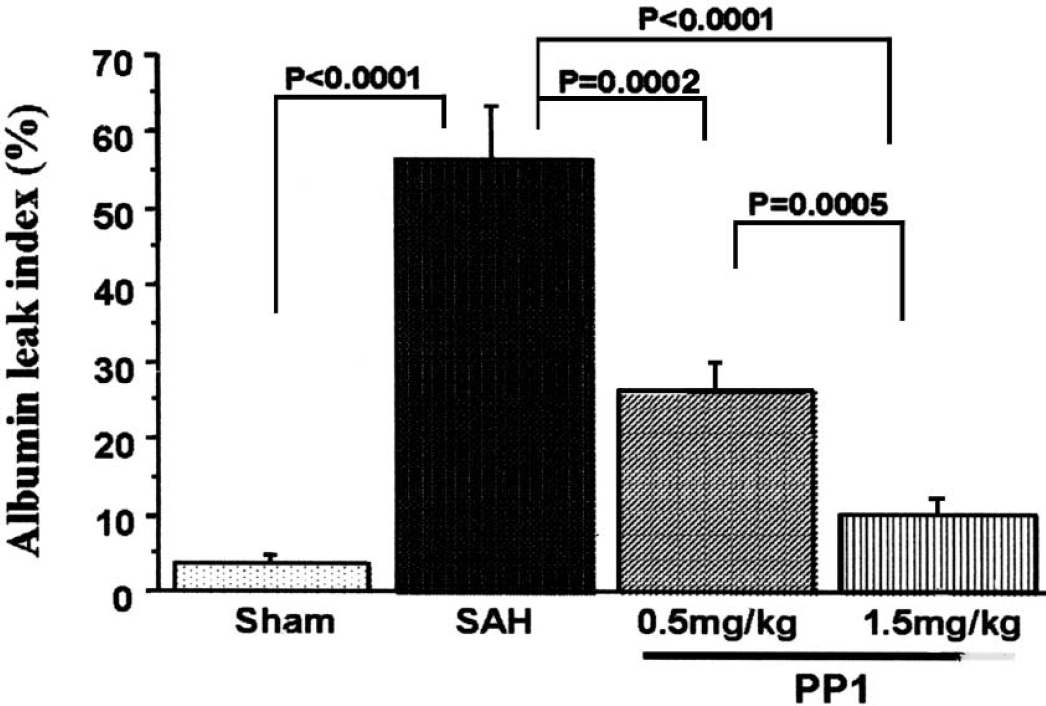
Blood–brain barrier permeability (measured by albumin leak index) was enhanced up to 50% of the baseline level (P < 0.0001 vs. sham-operated, ANOVA). PP1 reduced blood–brain barrier leakage in a concentration-dependent manner (P < 0.05 vs. subarachnoid hemorrhage [SAH], ANOVA).
Western blot analysis
Vascular endothelial growth factor expression was significantly increased (P < 0.05, ANOVA) at 24 hours after SAH in both the cerebral cortex adjacent to the basal cistern and in the cerebral arteries when compared to the sham-operated group. PP1 reduced VEGF expression enhanced by SAH (P < 0.05, Fig. 6). No marked concentration-dependent effect of PP1 was observed (P > 0.05).
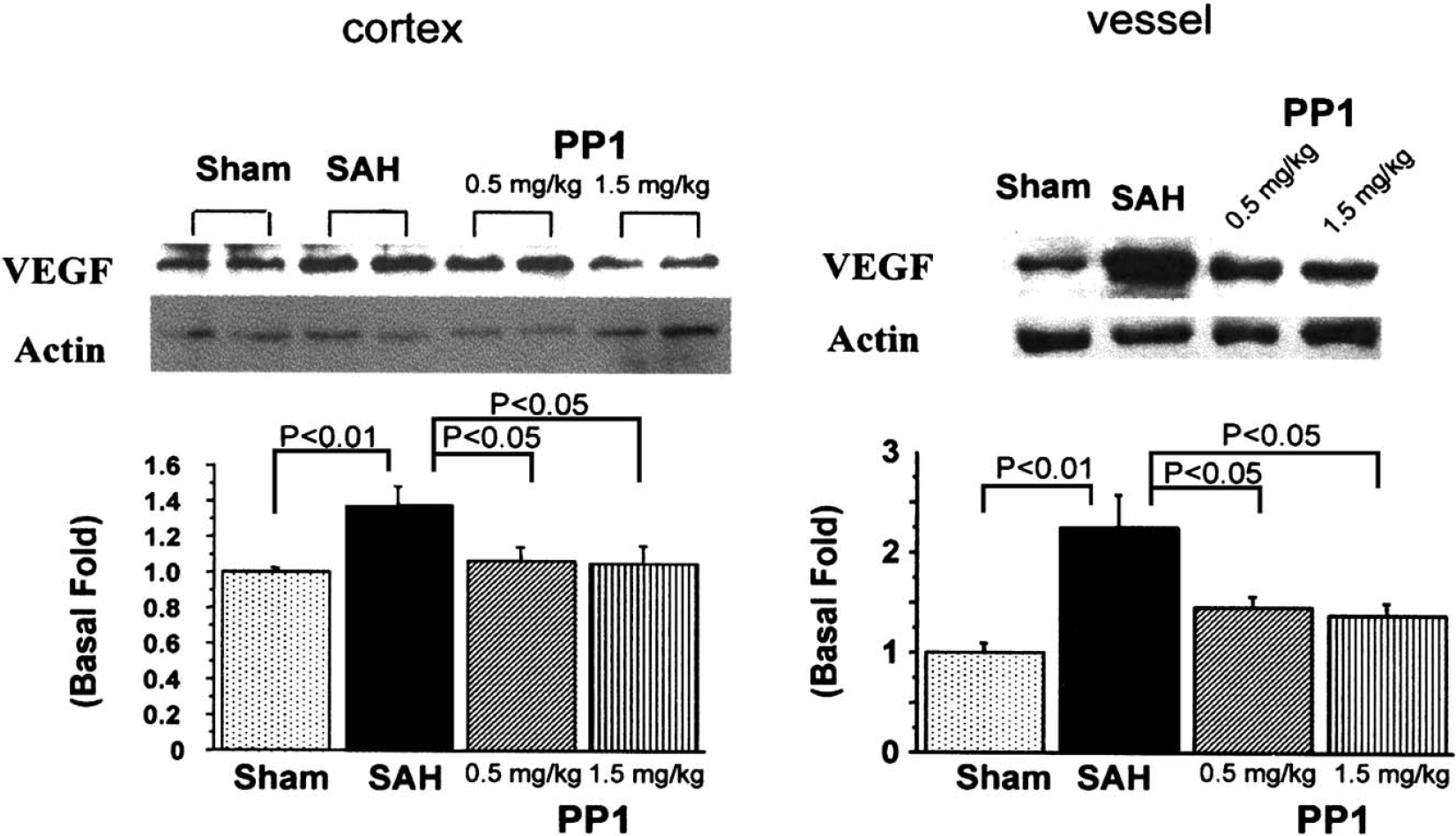
Vascular endothelial growth factor (VEGF) expression. Sample bands from the cortex (left side) and cerebral arteries (right side) from the sham, subarachnoid hemorrhage (SAH), SAH+PP1 (0.5 mg/kg), and SAH+PP1 (1.5 mg/kg) groups are shown in the top part of the figure. The band density values were calculated as a ratio of VEGF/actin, and the values from the sham group were used as 100%. Western blot demonstrated that VEGF expression was enhanced in the cortex, especially in the cerebral arteries (P < 0.05 vs. sham-operated, ANOVA). PP1 treatment at both concentrations reduced VEGF expression (P < 0.05 vs. SAH, ANOVA).
Activation of ERK1/2 was observed in the cerebral arteries, and PP1 reduced ERK1/2 phosphorylation in a concentration-dependent manner (P < 0.05) (Fig. 7). No significant activation of ERK1/2 in the cortex was obtained (P > 0.05), even though the phosphorylation of ERK1/2 in the SAH vehicle-treated group was significantly higher than in the PP1-treated groups (P < 0.05).
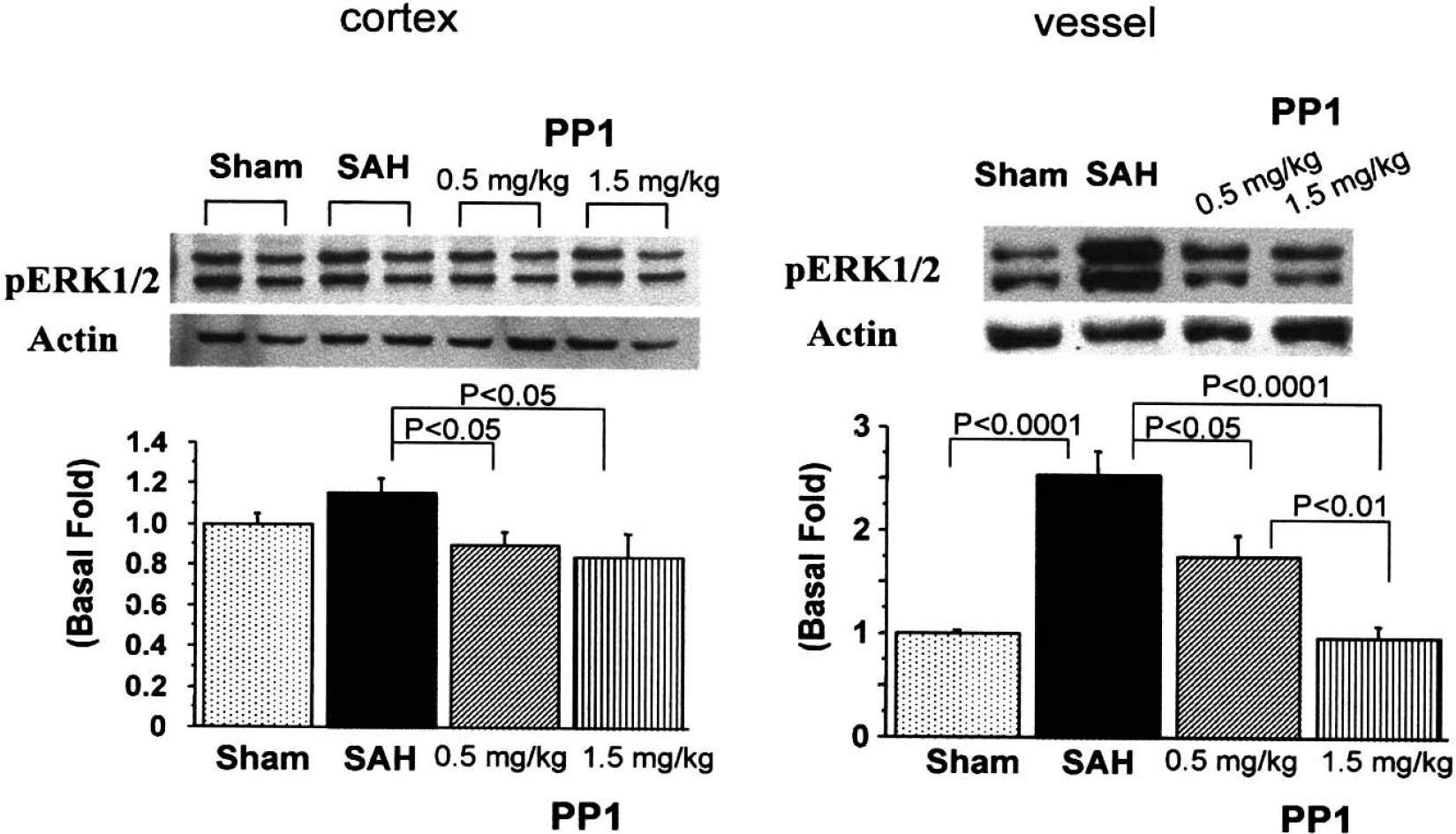
ERK1/2 activation. Sample bands from the cortex (left side) and cerebral arteries (right side) from the sham, subarachnoid hemorrhage (SAH), SAH+PP1 (0.5 mg/kg), and SAH+PP1 (1.5 mg/kg) groups are shown in the top part of the figure. The band density values were calculated as a ratio of pERK1/2/actin, and the values from the sham group were used as 100%. Phosphorylated ERK1/2 levels were enhanced in the cerebral arteries (P < 0.0001) and lightly in the cortex (P < 0.05). PP1 treatment at both concentrations reduced ERK1/2 activity in the cerebral cortex and arteries (P < 0.05, ANOVA).
JNK activation after SAH was observed in the cerebral arteries (P < 0.01), but not in the cerebral cortex (P > 0.05, Fig. 8). PP1 abolished the phosphorylation of JNK in the cerebral arteries (P < 0.05).
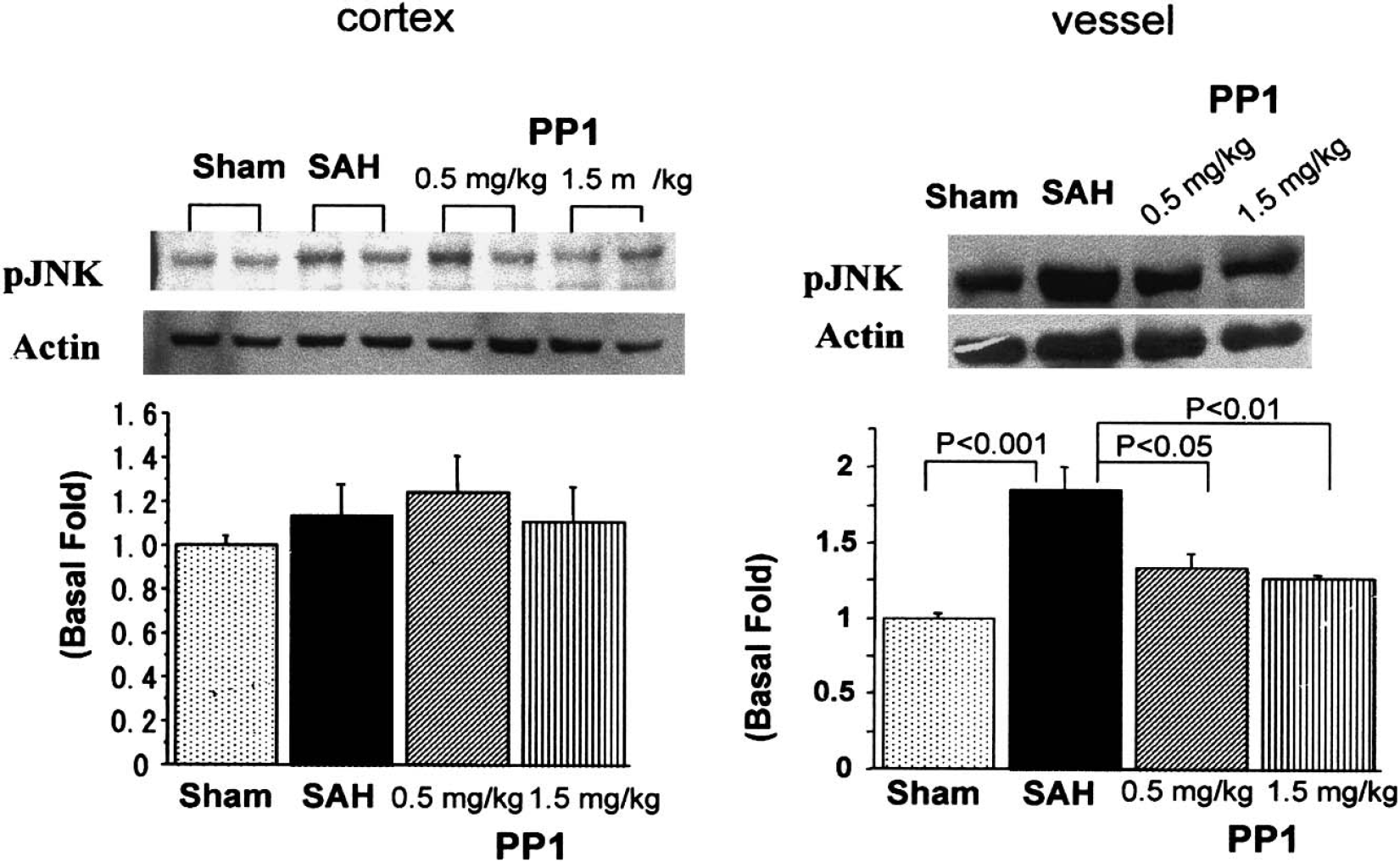
JNK activation. Sample bands from the cortex (left side) and cerebral arteries (right side) from the sham, subarachnoid hemorrhage (SAH), SAH+PP1 (0.5 mg/kg), and SAH+PP1 (1.5 mg/kg) groups are shown in the top part of the figure. The band density values were calculated as a ratio of pJNK/actin, and the values from the sham group were used as 100%. Phosphorylated JNK levels were enhanced in the cerebral arteries (P < 0.001 vs. sham-operated, ANOVA) but not in the cortex (P < 0.05). PP1 treatment at both concentrations reduced JNK activity in the cerebral arteries (P < 0.05, ANOVA).
A slight but significant activation of p38 was obtained in the cerebral cortex, and a marked activation of p38 was observed in the cerebral arteries (P < 0.05) (Fig. 9). PP1 abolished the phosphorylation of p38 in the cerebral arteries (P < 0.05).
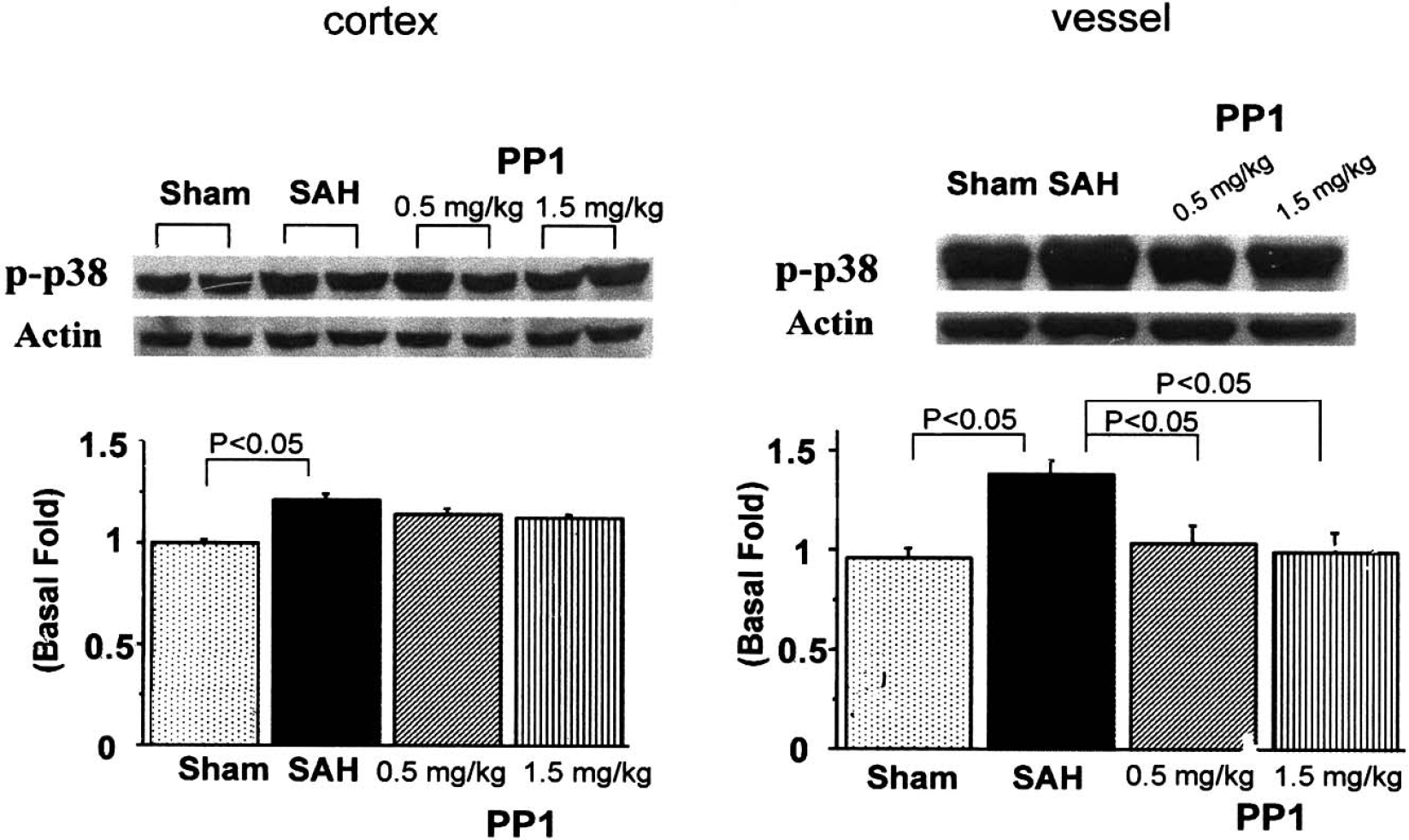
p38 activation. Sample bands from the cortex (left side) and cerebral arteries (right side) from the sham, subarachnoid hemorrhage (SAH), SAH+PP1 (0.5 mg/kg), and SAH+PP1 (1.5 mg/kg) groups are shown in the top part of the figure. The band density values were calculated as a ratio of p-p38/actin, and the values from the sham group were used as 100%. Phosphorylated p38 levels were enhanced in the cerebral arteries (P < 0.05 vs. sham-operated, ANOVA) and in the cortex (P < 0.05). PP1 treatment at both concentrations reduced JNK activity in the cerebral arteries (P < 0.05, ANOVA). No clear effect of PP1 on p38 activity was observed in the cerebral cortex.
DISCUSSION
In the present study, we have found that experimental SAH increased VEGF activity and activated MAPK pathways in the cerebral arteries and, to a lesser degree, in the cerebral cortex adjacent to the basal cistern. These molecular changes contributed to the altered permeability of the BBB, enhanced brain edema, and elevated ICP. These pathologic events led to neurologic deficit (not shown) and eventually death in rats after SAH. Inhibition of the Src-family tyrosine kinase abolished these molecular changes, reduced BBB rupture, brain edema, and ICP values, and decreased mortality. An interesting observation in the present study was that PP1, the Src-family tyrosine kinase inhibitor, also reduced the expression of VEGF in the cerebral cortex and arteries. This new observation indicates that Src is not only downstream of VEGF receptor activation (Irving and Bamford, 2002) and leads to MAPK signaling cascades, but is also upstream of VEGF and may be involved in the signaling pathways that lead to the activation of VEGF. In addition to serving as the downstream effector for VEGF, MAPK activation, which mediates the action of VEGF, promotes other molecular changes, especially related to apoptotic cell death. Besides ERK1/2, JNK and p38 are specifically involved in stress-induced cell death (Irving and Bamford, 2002).
A striking observation in the present study is the reduction of mortality after PP1 treatment. PP1 reduced mortality from 43.8% to 21.4% at 1.5 mg/kg. Since, in this rat model most mortality occurs within 24 hours, the mortality at 48 or 72 hours after SAH was not studied. Two treatments of PP1 were conducted in each rat at 2 hours before and 6 hours after SAH. The reason that we used PP1 prior to SAH is that Src was activated within hours after ischemia (Hattori et al., 2001). Even though pretreatment with PP1 proves the concept of a key role of Src in early brain injury, the therapeutic effect of PP1 at post-SAH application requires further investigation. The concentrations of PP1 used in the present study are consistent with its action on Src kinase inhibition as reported previously (Hattori et al., 2001; Paul et al., 2001). At these concentration ranges, PP1 apparently passed through the BBB because PP1 reduced cerebral infarct by systemic (intraperitoneal) administration (Paul et al., 2001). The reduction of mortality by PP1 resulted, at least in part, from decreased BBB permeability, reduced brain edema, and lowered ICP values. The mortality after SAH in patients varies among reports but ranges from 20% to 60% (Adams et al., 1981; Broderick et al., 1994; Kassell et al., 1981). No effective therapy, especially selective targeting treatment for early brain injury, is available because of the lack of understanding of the molecular mechanisms (Kassell et al., 1981). The inhibition of Src signaling pathways may open a new avenue for the clinical treatment of SAH.
Subarachnoid hemorrhage increased the expression of VEGF in the cerebral arteries and cerebral cortex adjacent to the basal cistern. Activation of VEGF and its receptors (receptor tyrosine kinase) leads to the phosphorylation of Src tyrosine kinase and MAPK (Irving and Bamford, 2002). Src and MAPK mediate the effect of VEGF on the permeability of the BBB as well as brain edema and cell death (Paul et al., 2001). We have found that the phosphorylation of all three groups of MAPK was enhanced, especially in the cerebral arteries after SAH. All these MAPKs are involved in apoptosis even though ERK1/2 is also part of the survival pathways in different cell types (Irving and Bamford, 2002). In particular, p38 and JNK are activated primarily by stress stimulations and inflammatory cytokines, which are highly elevated in the bloody cerebral spinal fluid and in cerebral arteries after SAH (Aihara et al., 2001; Bavbek et al., 1998; Fassbender et al., 2001; Gruber et al., 2000; Hirashima et al., 1997; Kikuchi et al., 1995; Mathiesen et al., 1997). PP1 inhibited the phosphorylation of JNK and p38, both of which are determining factors for apoptosis after cerebral ischemia (Irving and Bamford, 2002). Indeed, apoptotic changes occurred in the neuronal and cerebral vascular tissues after SAH (Matz et al., 2000; Meguro et al., 2001). Therefore, it can be speculated that part of the neuroprotective action of PP1, especially on mortality, may be mediated by its antiapoptosis effect, which needs to be established in future studies. It needs to be noted that not only VEGF, but also other growth factors, cytokines, glutamate, and free radicals all stimulate Src–MAPK pathways (Irving and Bamford, 2002). One of the issues surrounding the specificity of PP1 is whether its action is directly on Src, an important factor in VEGF signaling pathway, or is indirect and needs other organs or metabolites. The injection of VEGF intracranially produces Evans blue extravasation whereas injection of VEGF together with PP1 abolishes it (Paul et al., 2001), indicating that metabolic processing in other organs is not required for the action of PP1. Another issue is the ability of PP1 to cross the BBB. Besides all of the indirect evidence that PP1 reduces BBB permeability and decreases brain edema, the systemic application of PP1 blocks the ischemia-induced increases in phosphorylation of focal adhesion kinase, a substrate of active Src (Src activity) (Eliceiri et al., 1999; Paul et al., 2001). These studies indicate that PP1 crosses the BBB when used via systemic injection. Finally, the selectivity of PP1 as a Src-family kinase inhibitor raises concerns that PP1 not only inhibits Src but also inhibits Lck and Fyn at concentrations significantly lower than those required to inhibit other protein kinases (Hanke et al., 1996). PP1 is a power tool for the study of the Src-family kinase since it has a low selectivity for other protein kinases, but it does not distinguish Src from Fyn or Lck (members of the Src family) (Irving and Bamford, 2002). A recent observation, however, indicates that Fyn was not involved in the BBB damage or brain edema after ischemic stroke (Paul et al., 2001).
An interesting observation in the present study is that PP1 (Src-family kinase inhibitor, Src is downstream of VEGF [Irving and Bamford, 2002]) reduced the phosphorylation of VEGF in the cerebral cortex and arteries. It has been established that VEGF is a substrate of hypoxia-inducible factor-1α (HIF-1α), which accumulates in the cells after hypoxia resulting from cerebral ischemia, including focal cerebral ischemia (Bergeron et al., 1999), global ischemia (Chavez and LaManna, 2002), and cerebral hemorrhage (Jiang et al., 2002). The activation of HIF-1α is regulated by MAPK (Mottet et al., 2002; Sang et al., 2003). MAPK p38 is involved in HIF-1α–dependent (Gao et al., 2002) or HIF-1α–independent activation of VEGF (Duyndam et al., 2003). ERK1/2 MAPK activates the VEGF promoter at the proximal region under normal conditions and stabilizes HIF-1α under hypoxia to enhance VEGF activation (Berra et al., 2000). Even though further evidence is needed, it is possible that SAH activates Src-MAPK (Marton et al., 1996; Vollrath et al., 1998; Zubkov et al., 2001), which enhances HIF-1α-VEGF (Berra et al., 2000; Sang et al., 2003), which activates Src-MAPK (Irving and Bamford, 2002) to contribute to early brain injury. PP1 inhibited Src-MAPK pathways both upstream and downstream of HIF-1α-VEGF and reduced MAPK as well as VEGF expression, as observed in the present study. This observation indicates the importance of Src as a key molecule in the signaling pathways in early brain injury after SAH.
We used an established rat model of SAH in the present study. Numerous animal models have been created and tested for SAH and cerebral vasospasm after SAH studies (Megyesi and Findlay, 2001). The most widely used SAH models in rats are the endovascular puncture model (Bederson et al., 1995) and blood injection through the cisterna magna model (Solomon et al., 1985). We have tested these models previously and found that even though the double-hemorrhage model (blood injection via the cisterna magna) is better for the study of cerebral vasospasm, the endovascular puncture model is superior for the study of early brain injury after SAH (Gules et al., 2002). The endovascular puncture model leads to 43.8% mortality, increased ICP and BBB permeability, and deteriorated neurologic function that closely resembles SAH in patients (Adams et al., 1981; Bederson et al., 1995). These pathologic parameters, however, cannot be produced in blood-injection models in rats. The changes of these pathologic parameters offer a unique opportunity to evaluate the therapeutic effect of drug treatment for SAH, which remains a problem for neurosurgeons (Ogungbo et al., 2001; van Gijn and Rinkel 2001).