
Abstract
Ischemic preconditioning (IPC) promotes brain tolerance against subsequent ischemic insults. Using the organotypic hippocampal slice culture, we conducted the present study to investigate (1) the role of adenosine A1 receptor (A1AR) activation in IPC induction, (2) whether epsilon protein kinase C (ɛPKC) activation after IPC is mediated by the phosphoinositol pathway, and (3) whether ɛPKC protection is mediated by the extracellular signal-regulated kinase (ERK) pathway. Our results demonstrate that activation of A1AR emulated IPC, whereas blockade of the A1AR during IPC diminished neuroprotection. The neuroprotection promoted by the A1AR was also reduced by the ɛPKC antagonist. To determine whether ɛPKC activation in IPC and A1AR preconditioning is mediated by activation of the phosphoinositol pathway, we incubated slices undergoing IPC or adenosine treatment with a phosphoinositol phospholipase C inhibitor. In both cases, preconditioning neuroprotection was significantly attenuated. To further characterize the subsequent signal transduction pathway that ensues after ɛPKC activation, mitogen-activated protein kinase kinase was blocked during IPC and pharmacologic preconditioning (PPC) (with ɛPKC, NMDA, or A1AR agonists). This treatment significantly attenuated IPC- and PPC-induced neuroprotection. In conclusion, we demonstrate that ɛPKC activation after IPC/PPC is essential for neuroprotection against oxygen/glucose deprivation in organotypic slice cultures and that the ERK pathway is downstream to ɛPKC.
It is now well-established that brief ischemic/anoxic episodes may render brain, heart, and other tissues tolerant against more severe subsequent ischemic insults, which is a phenomenon that is referred to as ischemic preconditioning (IPC). Robust IPC neuroprotection has been studied extensively for more than a decade in a variety of in vivo and in vitro models (Kato et al., 1992; Kitagawa et al., 1990; Schurr et al., 1986).
The specific triggering mechanisms leading to this state of tolerance still remain unknown. However, studies from different laboratories suggest that IPC-mediated neuroprotection is achieved by activation of NMDA (Best et al., 1996; Gonzalez-Zulueta et al., 2000; Kato et al., 1992; Pringle et al., 1996, 1997; Raval et al., 2003) and the adenosine A1 receptor (A1AR) (Blondeau et al., 2000; Cohen et al., 2000; Di-Capua et al., 2003; Heurteaux et al., 1995; Hiraide et al., 2001; Perez-Pinzon et al., 1996; Reshef et al., 2000).
Although the NMDA pathway is better understood, the adenosine pathway has been less characterized. Because both NMDA and A1 receptors have different effects in the CA1 region of the hippocampus, namely post-synaptic excitation (NMDA) and pre-synaptic inhibition (A1AR), the signal transduction pathway that ensues after activation of these two receptors after IPC remains undefined. Recent evidence demonstrates that a common signal transduction pathway for these two disparate receptors involves the translocation of ɛPKC in organotypic hippocampal slices (Raval et al., 2003) and primary rat neuronal cultures (Di-Capua et al., 2003).
It is also necessary to define the signal transduction pathway that ensues after ɛPKC activation. Previous studies in the heart demonstrated that activation of ɛPKC after IPC both in vivo and in vitro protects against ischemia via activation of extracellular regulated kinase (ERK 1/2) and c-Jun N-terminal kinase (JNK) members of the mitogen-activated protein kinase (MAPK) family (Li et al., 2000; Ping et al., 1999a,1999b; Punn et al., 2000). In cortical neurons, the signal transduction pathway after IPC includes the activation of the p21 (RAS)/ERK pathway (Gonzalez-Zulueta et al., 2000). However, it remains to be determined whether ɛPKC and the RAS/ERK pathway are linked together or are two different pathways that promote IPC tolerance in the brain.
The goals of the present study are (1) to elucidate whether A1AR activation is key in the organotypic hippocampal slice model of IPC, (2) to determine whether ɛPKC translocation because of IPC and adenosine receptor activation is mediated by the phosphoinositol pathway, and (3) to determine whether there is a link between ɛPKC translocation and ERK in preconditioning mediated neuroprotection or whether these two pathways are independent of each other.
MATERIALS AND METHODS
Preparation of organotypic slice cultures
All protocols were approved by the Animal Care and Use Committee of the University of Miami. Neonatal (9–11 days old) Sprague-Dawley rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg/pup). Animals were decapitated, and the brains were quickly removed. Organotypic hippocampal slice cultures were prepared as described previously (Raval et al., 2003; Xu et al., 2002). In summary, transverse slices (400 μm) were dissected from the hippocampi and placed in Gey's Balanced Salt Solution (Sigma, St. Louis, MO, U.S.A.) supplemented with 6.5 mg/mL glucose at 4°C. After 1 hour, two slices were placed onto one 30-mm diameter membrane insert (Millicell-CM, Millipore, Bedford, MA, U.S.A.), and inserts were transferred to six-well culture plates with 1 mL of culture medium per well. Culture medium consisted of 50% Minimum Essential Medium, 25% Hank's Balanced Salt Solution, 25% Heat Inactivated Horse Serum (Sigma) supplemented with 6.5 mg/mL glucose and 1 mM glutamine. Slice cultures were incubated (equilibrated at 36°C, 95% O2, 5% CO2, humidity 100%) for 14 to 15 days before experiments were performed.
Induction of ischemia
Our ischemia and preconditioning protocols have been defined in previous studies (Raval et al., 2003; Xu et al., 2002). For oxygen/glucose deprivation (OGD), slices were washed three times with aglycemic Hank's Balanced Salt Solution (AHBSS) (pH 7.4) of the following constitution: CaCl2 · 2H2O 1.26 mM, KCl 5.37 mM, KH2PO4 0.44 mM, MgCl2 0.49 mM, MgSO4 · 7H2O 0.41 mM, NaCl 136.9 mM, NaHCO3 4.17 mM, Na2HPO4 · 7H2O 0.34 mM, and sucrose 15 mM (Sigma). The slices were then transferred into an airtight chamber, which was equilibrated with 95% N2/5% CO2 gas (preheated to 37°C and water saturated) that was blown through the chamber for 5 minutes (4 L/min) to achieve anoxic conditions. Then the chamber was sealed and remained incubated for 10 minutes (for a total of 15 minutes for preconditioning) or 35 minutes (for a total of 40 minutes for ischemic insult). After OGD, slices were placed back in the incubator in plates containing the normal culture medium.
Assessment of neuronal cell death
To determine the extent of neuronal damage, we used the propidium iodide (PI) method (Raval et al., 2003; Xu et al., 2002). Slices were incubated in culture medium supplemented with 2 μg/mL PI (Sigma) for 1 hour. Images were taken using an inverted fluorescence microscope (Olympus IX 50), equipped with a light intensifying SPOT CCD camera (Diagnostic Instruments Inc., Sterling Heights, MI, U.S.A.) and SPOT Advanced software. Images of the cultured slices were taken as follows: (1) as baseline before the preconditioning procedure, (2) 24 hours after the preconditioning, (3) 24 hours after the test ischemic insult to assess ischemic damage, and (4) 24 hours after NMDA treatment to assess maximum damage to neuronal cells. The hippocampal CA1 subfield was chosen as region of interest, and quantification was performed using Scion Image software. The percentage of relative optical intensity (ROI) served as an index of neuronal cell death. Relative cell death was calculated from each ROI as follows: Relative % cell death = (Fexp − Fmin)/(Fmax − Fmin) × 100, where Fexp is the fluorescence of the test condition, Fmax is maximum fluorescence (100 μM NMDA treatment for 1 hour), and Fmin is background fluorescence (before preconditioning or OGD). In all groups, experiments (except Western blot analysis) were terminated by superfusing slices with an overdose of NMDA (100 μM) 24 hours after the end of the experiments to determine total number of neurons using the PI technique (Fmax above).
Cell fractionation and Western blot analysis
To determine whether ɛPKC isozyme was translocated after IPC in organotypic slices, we used Western blot analysis. This method is adapted from one that was described previously (Mackay and Mochly-Rosen, 2001) with some modifications (Raval et al., 2003). Hippocampal organotypic slices were frozen in liquid nitrogen at different time intervals and stored at −80°C until the analysis. At the time of Western blot analysis the hippocampal organotypic slice cultures were separated from the supporting membrane. For one sample, 32 slices were pooled together. Thus, for Western blot analysis, n = 1 represents 32 slices. Also, each slice was obtained from different pups. The pooled slices were washed twice with cold phosphate buffered saline (PBS). Slices were pelleted at 1,000×g and PBS was removed. The pellet was resuspended in 400 μL cell lysis buffer (4 mM ATP, 100 mM KCl, 10 mM imidazole, 2 mM EGTA, 1 mM MgCl2, 20% glycerol, 0.05% Triton X-100, 17 μg/mL PMSF, 20 μg/mL soybean trypsin inhibitor, 25 μg/mL leupeptin, and 25 μg/mL aprotinin). The suspended slices were homogenized using all glass homogenizer. The homogenate was then centrifuged at 4°C at 1000×g for 10 minutes. The supernatant (soluble fraction) was carefully taken off and re-centrifuged at 16,000×g for 15 minutes to get rid of any contaminating pellet material. The initial pellet was resuspended in 250 μL cell lysis buffer containing 1% Triton X-100 and was extracted on ice for 60 minutes. Samples were centrifuged at 16,000×g for 15 minutes. The supernatant is the particulate fraction. Soluble and particulate fractions were analyzed for protein content by the Bradford's assay and 40 μg protein from each fraction separated by 12% SDS-PAGE. Each group consisted of four samples, and each sample was prepared from 32 slices. Protein was transferred to Immobilon-P (Millipore, MA, U.S.A.) membrane and incubated with the primary antibody anti-ɛPKC (Calbiochem, La Jolla, CA, U.S.A.) (1: 500). Immunoreactivity was detected using enhanced chemiluminescence (ECL Western blotting detection kit, Amershampharmacia biotech, UK). Western blot images were digitized at 8-bit precision by means of a charge-coupled-device-based (CCD) camera (8–12 bit, Xillix Technologies Corp., Vancouver, Canada) equipped with a 55-mm Micro-Nikkor lens (Nikon, Japan). The camera was interfaced to an advanced image-analysis system (MCID Model M2, Imaging Research, Inc., St. Catherines, Ont., Canada). The digitized immunoblots were subjected to densitometric analysis using MCID software.
Peptide preparation and delivery
The ɛV1–2 (ɛPKC inhibitor, amino acids 14–21 [EAVSLKPT]) (Gray et al., 1997) and ψɛRACK (ɛPKC activator, amino acids 85–92 [HDAPIGYD]) (Dorn et al., 1999) peptides were synthesized at Stanford's Protein and Nucleic Acid facility and conjugated to Tat (carrier peptide, amino acids 47–57 [YGRKKRRQRRR]) (Schwarze et al., 1999) via a cysteinecysteine bond at the N termini, as previously described (Chen et al., 2001). These peptides are conjugated via disulfide bond to short cell permeable peptides (Souroujon and Mochly-Rosen, 1998). Our previous study clearly showed efficacy of these peptides in the organotypic slice cultures (Raval et al., 2003).
Experimental design
The organotypic slices were divided into five major groups:
Group I: Sham. Slices were incubated for 15 minutes in HBSS solution supplied with an equimolar concentration of glucose instead of sucrose (Sham-IPC) and were subsequently transferred back to regular media. After 48 hours, the same procedure was performed for an extended duration of 40 minutes (sham OGD-ischemia).
Group II: Test ischemia. Sham IPC was induced as in Group I followed after 48 hours by test ischemia (40 minutes of OGD).
Group III: IPC. Slices underwent IPC (15 minutes of OGD) and, 48 hours later, test ischemia (40 minutes of OGD).
Group IV: Pharmacologic preconditioning (PPC) by selective pharmacologic agents hypothesized to emulate IPC. Slices underwent PPC and, 48 hours later, test ischemia (40 minutes of OGD)
Group V: Pharmacologic blockade of IPC.
IPC and PPC experiments were performed in the presence of inhibitors of (A) ɛPKC, (B) phospholipase C (PLC), and (C) mitogen-activated protein kinase kinase (MAPKK) during and after the preconditioning procedure. The slices were subjected to test ischemia 48 hours later.
For Western blot analysis of ɛPKC translocation, slices were frozen 30 minutes after IPC, 30 minutes after R-PIA, and 60 minutes after ψɛRACK (an ɛPKC agonist) treatments.
To provide better controls in these experiments, all six-well plates used contained at least one well for the sham, IPC, and ischemia groups. In addition, for statistical purposes, each insert had two slices obtained from two different pups. Thus every n = 1 (one slice) represents a different animal.
Chemicals
R-PIA (N6-(R)-Phenylisopropyladenosine) (A1AR agonist), DPCPX (1,3 Dipropyl-8-cyclopentylxanthine) (A1AR antagonist), DMPX (3,7-Dimethyl-1-propargylxanthine) (A2AR antagonist), U-73122 (1-(6-[(17β)]-3-Methoxyestra-1, 3,5 [10]-trien-17-yl) amino] hexyl)-1H-pyrrole-2, 5-dione) (PI-PLC inhibitor), and PD-98059 (2–2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) (MAPKK inhibitor) were all purchased from Sigma, St. Louis, MO, U.S.A.
Statistical analysis
The results are expressed as mean ± SD. Statistical significance was determined with an ANOVA test, followed by a Bonferroni's post hoc test.
RESULTS
Confirming our previous findings, IPC was neuroprotective against test ischemia in hippocampal organotypic slices.
To test whether IPC in hippocampal organotypic slice was mediated by the A1AR, slices were superfused with 10 μM of the selective A1AR antagonist DPCPX for the duration of IPC. This treatment diminished the neuroprotection afforded by IPC (P < 0.05). PI fluorescence values after the test ischemic insult were 55.39 ± 16.71% (n = 17) for untreated slices, 22.64 ± 19.32% (n = 12) after IPC, and 50.98 ± 17.23% (n = 12) after combined IPC and DPCPX treatment (Fig. 1A).
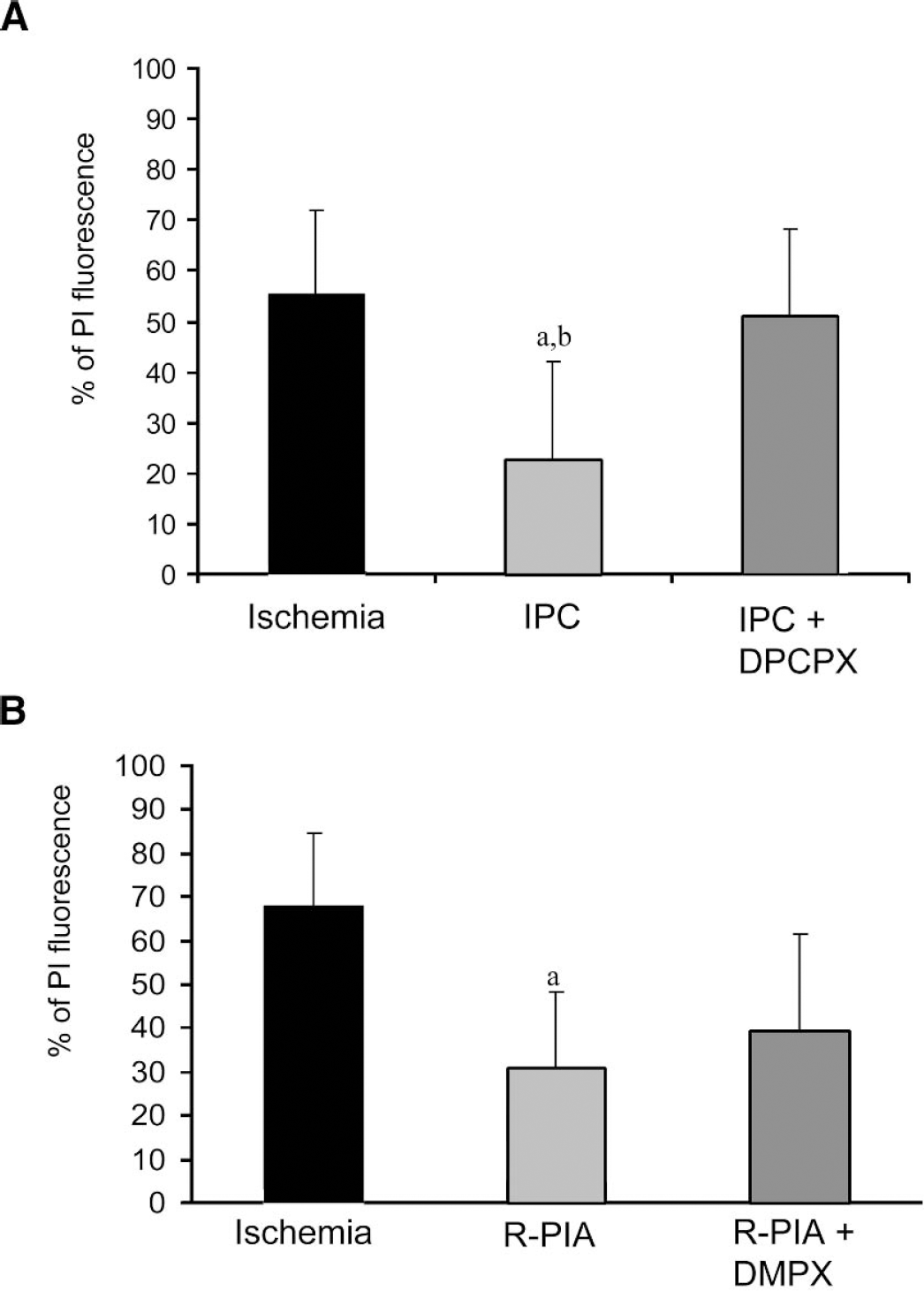
(A) PI fluorescence values measured in the CA1 pyramidal cells in rat organotypic hippocampal slices 1 day after OGD. The selective A1AR antagonist DPCPX reduced IPC-induced neuroprotection. Selective A1AR agonist R-PIA emulated IPC. (B) A2AR did not significantly contribute to R-PIA mediated ischemic tolerance; the selective A2AR antagonist DMPX was superfused during the R-PIA-induced preconditioning 48 hours before a test ischemic insult. Significance as compared with ischemia, aP < 0.05, and IPC + DPCPX, bP < 0.05. PI, propidium iodide; OGD, oxygen/glucose deprivation; A1 AR, adenosine A1 receptor; IPC, ischemic preconditioning; A2 AR, adenosine A2 receptor.
As control experiments, we compared PI fluorescence values between sham IPC, IPC (followed by sham ischemia), and IPC + DPCPX (followed by sham ischemia). PI fluorescence values for sham IPC, IPC, and IPC + DPCPX were determined at 24 and 48 hours. Furthermore, in these experiments, PI fluorescence values were determined in the same groups 24 hours after sham ischemia, which represents 72 hours after the initial insult. PI fluorescence values 24, 48, and 72 hours after sham IPC were 3.15 ± 0.78, 3.33 ± 1.47, and 4.11 ± 0.75 (n = 4), respectively. PI fluorescence values 24, 48, and 72 hours after IPC were 2.97 ± 1.16, 3.06 ± 1.37, and 4.29 ± 1.24, respectively (n = 4). PI fluorescence values 24, 48, and 72 hours after IPC + DPCPX were 3.38 ± 2.18, 3.97 ± 2.53, and 5.37 ± 3.32, respectively (n = 5). No significant differences in PI fluorescence values were found between sham IPC, IPC (followed by sham ischemia), and IPC + DPCPX groups at any of these times.
To further confirm the role of A1AR during IPC, we examined whether IPC could be emulated by the selective A1AR agonist R-PIA. Slices were superfused with R-PIA (at two different concentrations, 50 nM and 100 μM) for 15 minutes, and 48 hours later, the test ischemic insult was induced. Only pre-treatment with 100 μM R-PIA significantly reduced PI fluorescence in hippocampal slices after test ischemia (R-PIA, 25.17 ± 17.52%, n = 18, vs. test ischemia, 55.39 ± 16.71%, n = 17) (P < 0.001) (Fig. 1A). 50 nM R-PIA concentration was not effective in reducing PI fluorescence after test ischemia (62.89 ± 31.81, n = 10). PI fluorescence values 24, 48, and 72 hours after R-PIA treatment alone were 3.06 ± 0.60, 2.56 ± 1.15, and 4.96 ± 2.31, respectively (n = 6). These values were not significantly different than shams or IPC (followed by sham ischemia) groups.
Because 100 μM of R-PIA might also activate the A2AR, we performed an additional set of experiments to determine whether this receptor was involved in the R-PIA induced preconditioning. Slices were subjected to pharmacologic preconditioning with 100 μM R-PIA in presence of the selective A2AR antagonist DMPX (10 μM) for 15 minutes, and 48 hours later, test ischemia was performed. Compared with ischemia (68.06 ± 16.47, n = 5), both R-PIA treatment alone (30.89 ± 17.47%, n = 5) and R-PIA + DMPX (39.46 ± 21.92%, n = 4) exhibited reduction of PI-fluorescence (Fig. 1B) (P < 0.05). There was no significant difference in PI-fluorescence between the R-PIA and the R-PIA + DMPX groups. These results suggest that IPC in rat organotypic hippocampal slices requires A1AR activation. However, we cannot rule out the putative role of the A2AR in the ischemic or R-PIA-induced preconditioning neuroprotection in the organotypic hippocampal slices. As control experiments, we determined PI fluorescence after R-PIA and DMPX treatment alone. PI fluorescence values 24, 48, and 72 hours after R-PIA + DMPX were 2.23 ± 1.33, 3.27 ± 1.46, and 4.49 ± 2.16, respectively (n = 4). These values were not significantly different from shams or IPC (followed by sham ischemia) groups.
To test the hypothesis of whether A1AR induced neuroprotection is mediated by activation/translocation of ɛPKC in the organotypic hippocampal slice, as we previously reported for IPC- and NMDA-induced preconditioning (Raval et al., 2003), we inhibited ɛPKC with ɛV1–2 (20 nM, dosage determined in Raval et al., 2003) during R-PIA (100 μM) induced preconditioning, and 48 hours later, test ischemia was induced. Neuroprotection promoted by R-PIA (20.23 ± 13.17%, n = 8) was significantly reduced in presence of the ɛPKC antagonist (52.46 ± 20.04%, n = 7) (P < 0.01), indicating an essential role of ɛPKC in A1AR induced ischemic tolerance (Fig. 2).
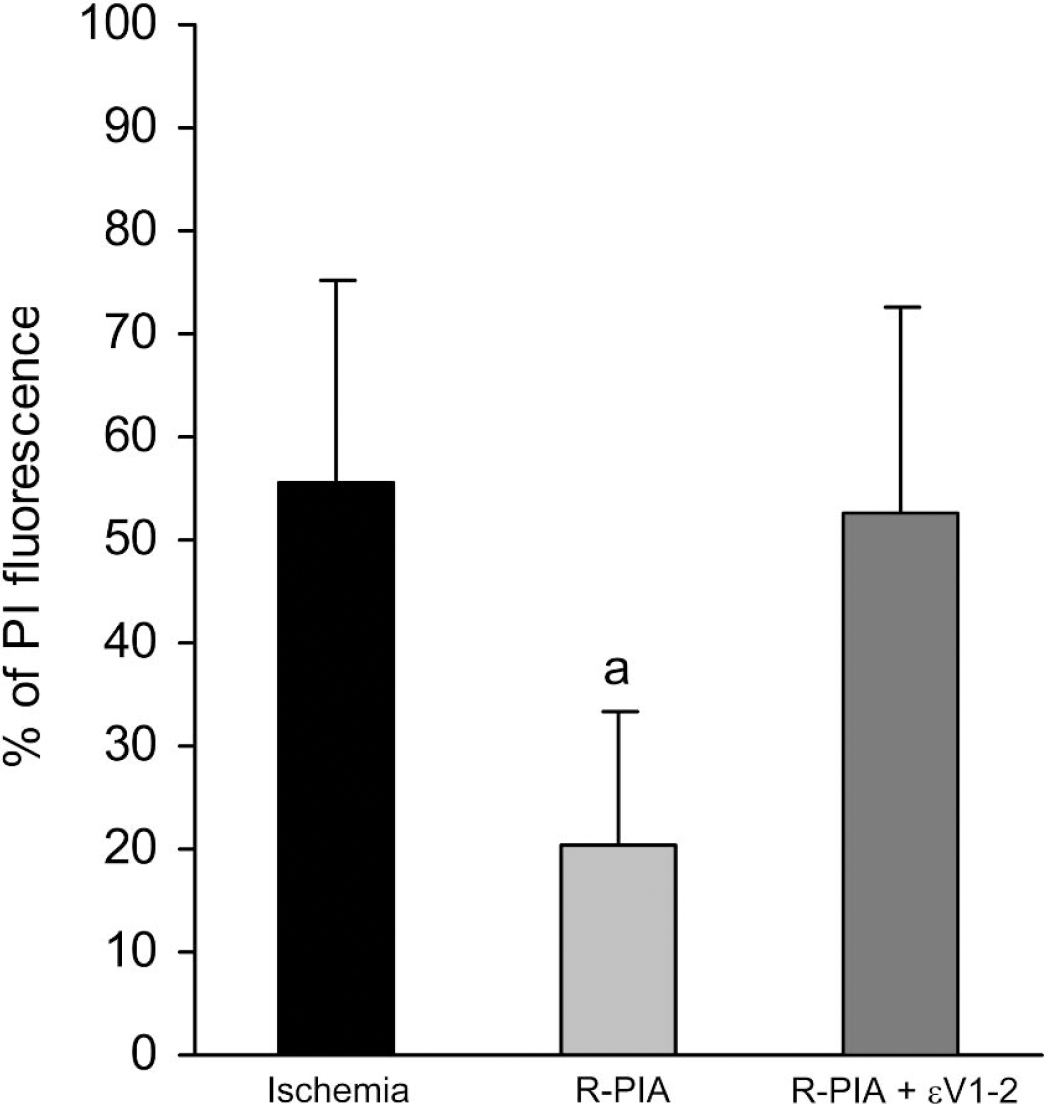
The A1AR-induced ischemic tolerance was mediated by activation of ɛPKC in rat organotypic hippocampal slices. Neuroprotection promoted by the selective A1AR agonist R-PIA was reduced when the selective ɛPKC antagonist peptide ɛV1–2 was administered simultaneously. Significance as compared with ischemia, aP < 0.01. A1 AR, adenosine A1 receptor; ɛPKC, epsilon protein kinase C.
Activation of ɛPKC is typically determined by translocation of ɛPKC from the soluble fraction to particulate fraction (Dorn et al., 1999). To determine that all preconditioning treatments promoted the translocation of ɛPKC, we performed Western blot analysis in slices that underwent IPC or PPC (Fig. 3). When compared with sham, the ratio of ɛPKC isozyme in soluble and particulate fraction decreased by 36% (n = 4, P < 0.05), 41% (n = 4, P < 0.05), and 38% (n = 4, P < 0.01) after IPC, R-PIA, and ɛPKC agonist peptide ψɛRACK treated groups, respectively. ɛPKC levels in the soluble fraction of dimethyl sulfoxide (DMSO) control were similar to the sham group (Fig. 3).
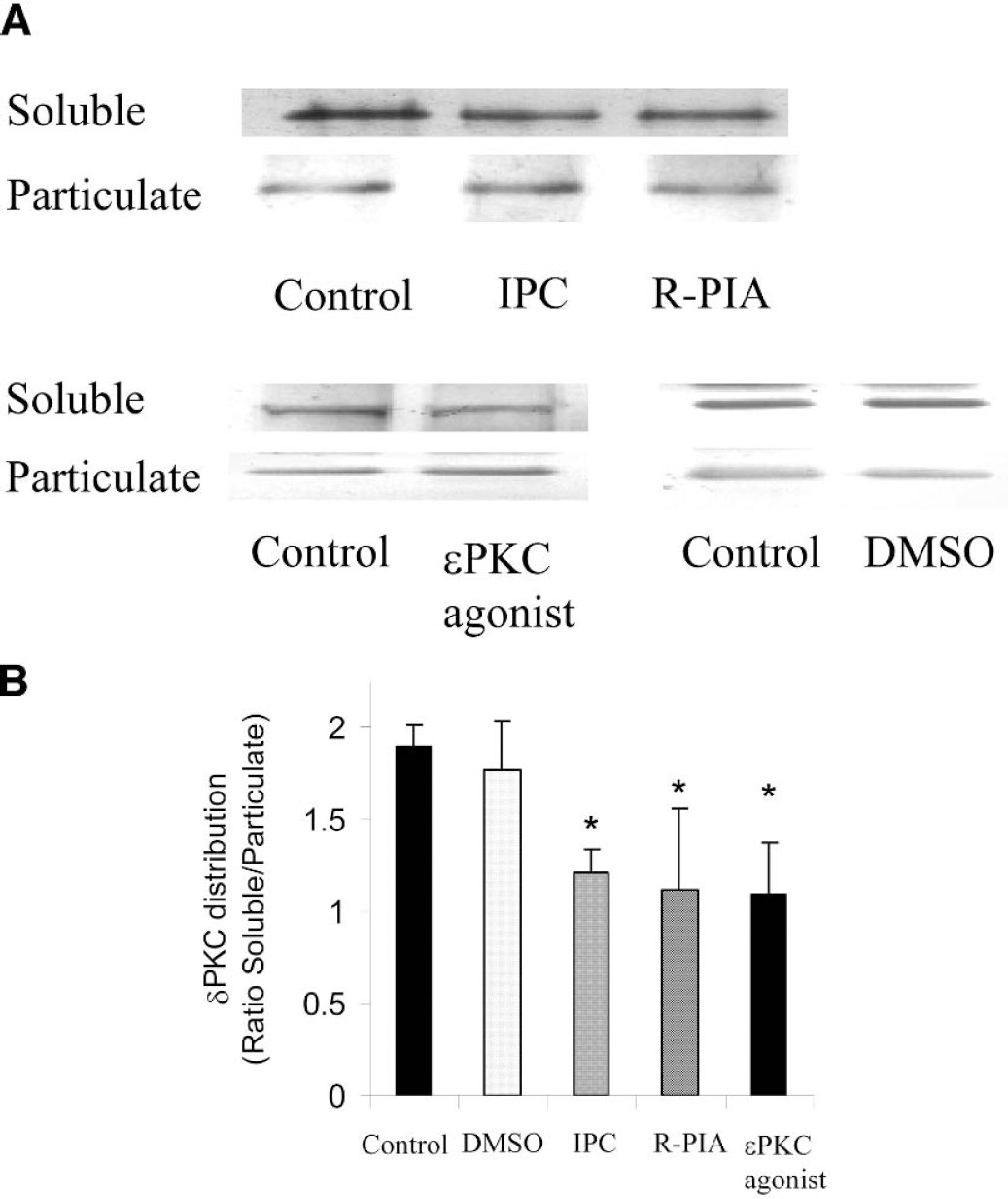
(A) Immunoblots of ɛPKC in soluble and particulate fractions of organotypic slices. (B) The ratio of ɛPKC in soluble and particulate fraction in sham (control), DMSO-treated, IPC, R-PIA, and ɛPKC agonist peptide ψɛRACK-treated slices. *P < 0.05 as compared with sham group. ɛPKC, epsilon protein kinase C; ψɛRACK, ψɛ receptor-activated C kinase.
ɛPKC belongs to the subgroup of novel PKC isozymes, and to our knowledge there are no reports that this isozyme is activated by the A1AR in hippocampus. The novel PKC isozymes are activated by diaceylglycerol (DAG), which is produced by activation of phosphoinositol phospholipase C (PI-PLC) pathway. Therefore, we tested the hypothesis that ɛPKC translocation in both IPC and pharmacologic preconditioning with R-PIA is mediated by activation of the phosphoinositol pathway via activation of the PI-PLC pathway. We incubated slices with the PI-PLC inhibitor U-73122 during IPC and, for 1 hour after preconditioning treatment and 48 hours later, test ischemia was induced. These treatments significantly reduced preconditioning neuroprotection at higher concentrations. The PI fluorescence values for U-73122 treatment at 1, 10, and 100 μM concentrations and IPC were 29.04 ± 19.74 (n = 5), 48.61 ± 20.32 (n = 11) (P < 0.05), 71.39 ± 19.17 (n = 6) (P < 0.05), and 26.02 ± 9.15% (n = 11), respectively. Because the amount of 10 μM of U-73122 was effective at blocking IPC, we incubated slices with the PI-PLC inhibitor U-73122 (10 μM) during R-PIA induced preconditioning, and, for 1 hour after preconditioning treatment and 48 hours later, test ischemia was induced. R-PIA induced preconditioning neuroprotection was significantly diminished by U-73122 treatment. PI fluorescence values for ischemia, R-PIA, and R-PIA + U-73122 were 52 ± 23.32% (n = 9), 22.89 ± 19.27% (n = 9), and 53.06 ± 20.47% (n = 8) (P < 0.01), respectively (Fig. 4).
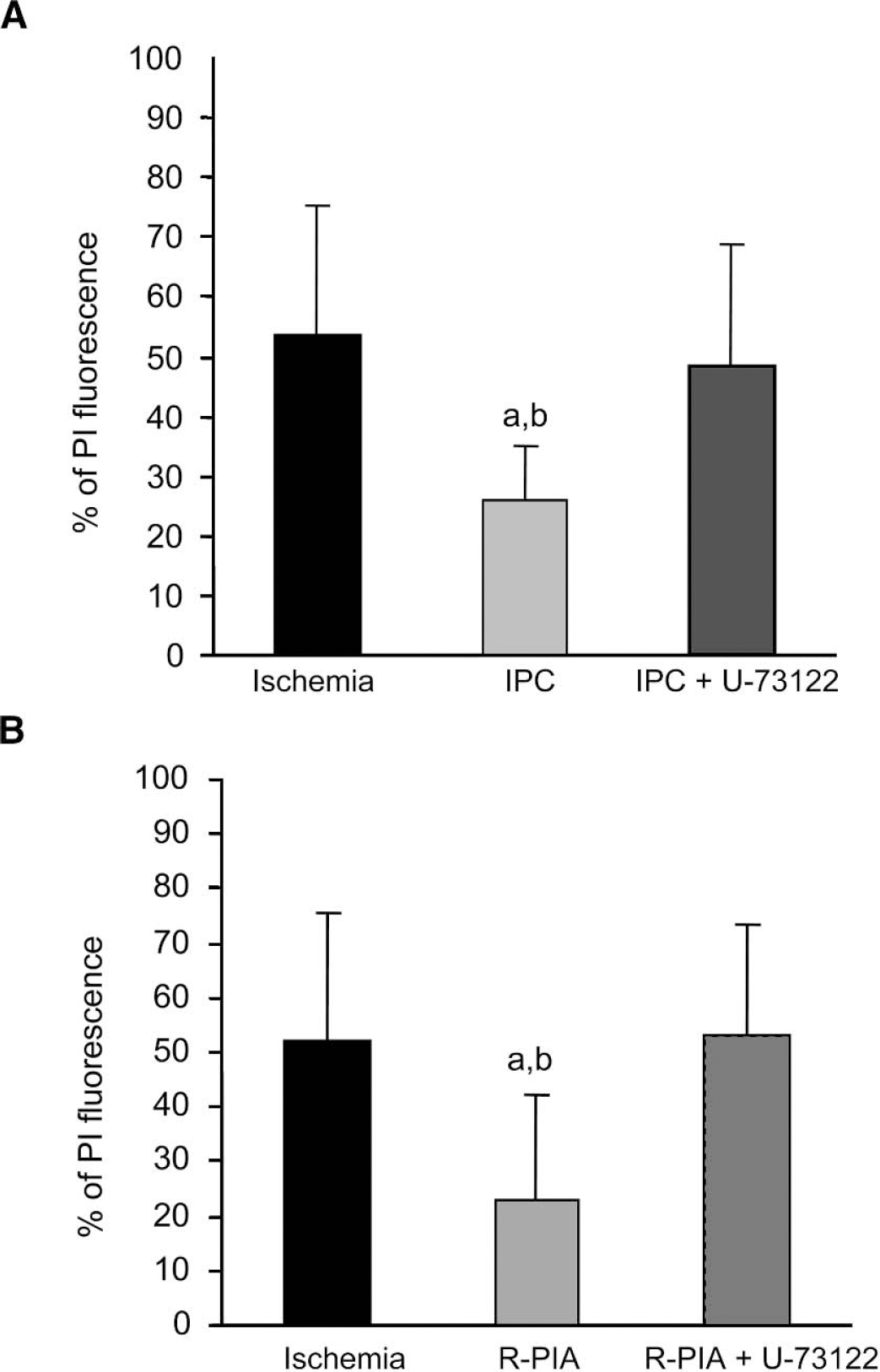
(A) Neuroprotection was achieved by IPC, and (B) A1 AR agonist R-PIA treatment was reduced by the phospholipase C-inhibitor, U-73122 in rat organotypic hippocampal slices. Significance as compared with ischemia, aP < 0.05, R-PIA + U–73122, bP < 0.05, and IPC + U–73122, cP < 0.05. IPC, ischemic preconditioning; A1 AR, adenosine A1 receptor.
To rule out the possibility that PI-PLC inhibitor (U-73122, 10 μM) itself affects neurons, we incubated slices with U-73122 alone (for 75 minutes) 48 hours before test ischemia. This treatment did not affect neuronal survival because PI fluorescence for U-73122 treated (53.32 ± 15.91, n = 6) and ischemic (53.67 ± 21.62%, n = 9) groups were not significantly different. We also performed additional controls to determine that IPC + U-73122 and R-PIA + U-73122 treatments alone did not promote any significant cell death. PI fluorescence values 24, 48, and 72 hours after IPC and U-73122 were 3.16 ± 1.82, 3.53 ± 2.02, and 4.68 ± 1.56, respectively (n = 6). PI fluorescence values 24, 48, and 72 hours after R-PIA and U-73122 were 3.00 ± 0.72, 3.48 ± 1.05, and 5.23 ± 2.45, respectively (n = 6). These values were not significantly different from sham or IPC (followed by sham ischemia) groups.
Because ERK pathway has been implicated in IPC neuroprotection in cortical culture cells (Gonzalez-Zulutea et al., 2000;Shamloo et al., 1999) and PKC appears to be an upstream signaling pathway to ERK, we tested the hypothesis that there is a link between translocation of ɛPKC and activation of ERK after IPC. For that purpose, ERK activation was blocked with the mitogen-activated protein kinase (MAPKK) inhibitor (PD-98059). Inhibition of MAPKK by PD-98059 is a good approach to inhibit the ERK pathway because PD-98059 prevented activation of MAPKK and subsequent phosphorylation of ERK both in vitro and in intact cells (Dudley et al., 1995). The organotypic slices were incubated with PD-98059 (20, 50, and 100 μM) during IPC, and 48 hours later, test ischemia was induced. The PI fluorescence values for different concentrations of the MAPKK inhibitor were 66.14 ± 15.90% (n = 10), 68.54 ± 17.26 (n = 7), and 69.94 ± 27.24% (n = 8) for 20, 50, and 100 μM of PD-98059 treatment, respectively. All concentrations reduced IPC-induced neuroprotection (P < 0.001). The treatment at 20 μM concentration was used for further studies because this low dosage is advantageous in reducing the effects of PD-98059 on NMDA receptors (Pereira et al., 2002). The PI fluorescence values were 30.11 ± 9.68% (n = 5), 66.14 ± 15.90% (n = 10), and 61.50 ± 11.83% (n = 13) for IPC, IPC + PD-98059 treated, and test ischemic groups, respectively (Fig. 5A). As control experiments, we determined that IPC + PD-98059 treatment alone did not promote any significant cell death. PI fluorescence values 24, 48, and 72 hours after IPC + PD-98059 were 2.07 ± 1.28, 3.58 ± 2.20, and 3.93 ± 2.32, respectively (n = 5). These values were not significantly different from sham or IPC (followed by sham ischemia) groups.
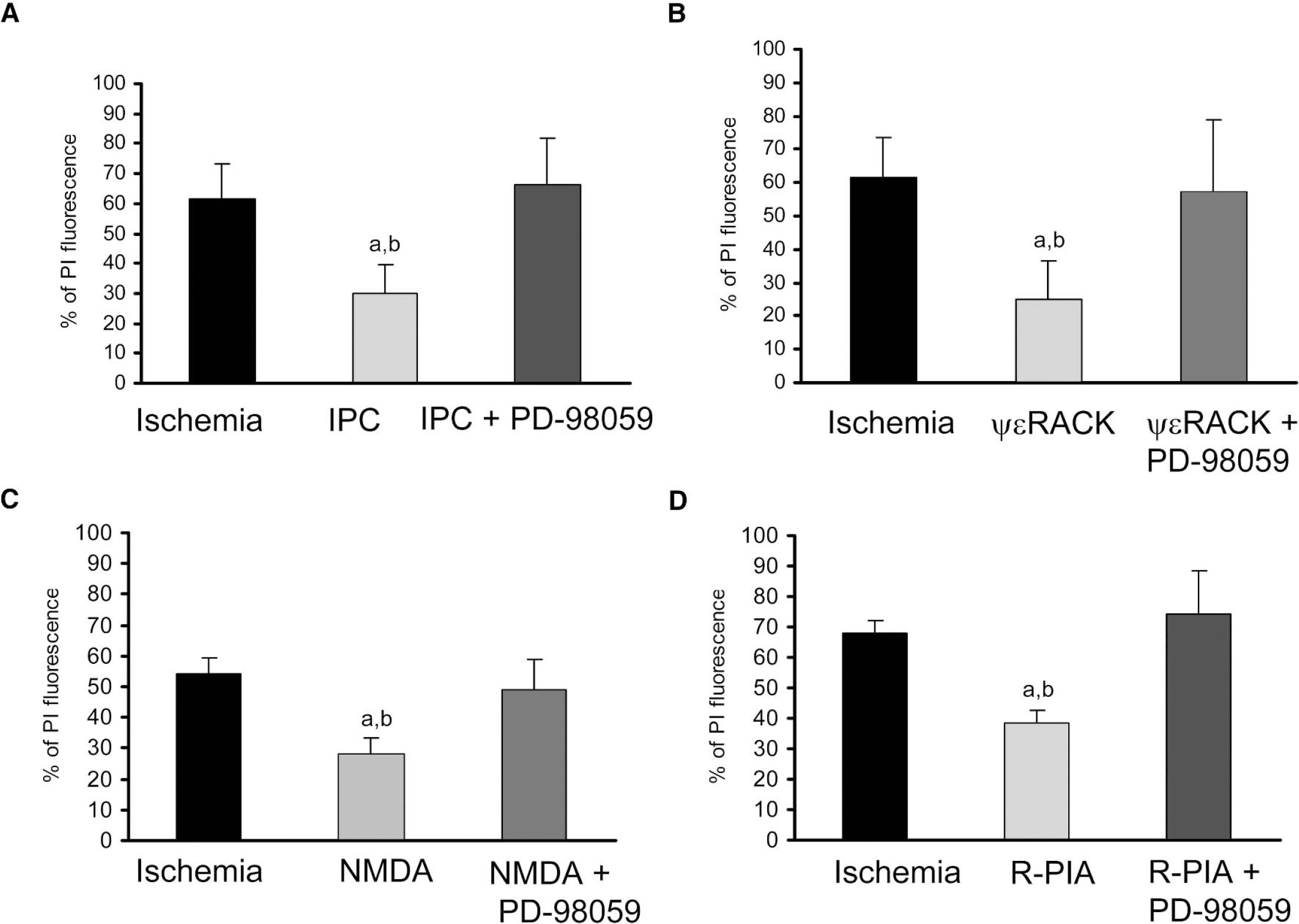
(A) The selective MAPKK antagonist PD-98059 reduced neuroprotection when superfused during IPC in rat organotypic hippocampal slices; significance as compared with ischemia, aP < 0.001, and IPC + PD-98059, bP < 0.001. (B) The selective MAPKK antagonist PD-98059 reduced ψɛRACK-mediated neuroprotection; significance as compared with ischemia (aP < 0.001 and ψɛRACK + PD-98059, bP < 0.001. (C) Pharmacologic neuroprotection afforded by NMDA was reduced by selective MAPKK antagonist PD-98059; significance as compared with ischemia, aP < 0.001, and NMDA + PD-98059, bP < 0.001. (D) The selective MAPKK antagonist PD-98059 reduced R-PIA mediated neuroprotection when superfused for 15 minutes 48 hours before test ischemic insult; significance as compared with ischemia, aP < 0.001, and R-PIA + PD-98059, bP < 0.001. MAPKK, mitogen-activated protein kinase kinase; IPC, ischemic preconditioning; ψɛRACK, ψɛ receptor-activated C kinase.
To further confirm the link between ɛPKC translocation and the ERK pathway in IPC-induced neuroprotection, we blocked ERK activation with PD-98059 (20 μM) when IPC was emulated by superfusion of the ɛPKC agonist in organotypic slices. The PI fluorescence values for ɛPKC agonist treatment and ɛPKC agonist treatment along with MAPKK inhibitor were 25.01 ± 11.57% (n = 8) and 57.00 ± 21.68% (n = 10) (P < 0.001), respectively. These results indicate the involvement of ERK pathway downstream of ɛPKC translocation (Fig. 5B).
We have previously demonstrated that the NMDA receptor mediates IPC neuroprotection via ɛPKC translocation (Raval et al., 2003). In the next two experiments, we tested whether the ERK pathway is involved in NMDA and A1AR mediated preconditioning. In the first experiment, ERK activation was blocked with PD-98059 during NMDA-induced preconditioning. This treatment significantly attenuated the NMDA induced neuroprotection. The PI fluorescence values of NMDA preconditioning and NMDA + PD-98059 treated groups were 28.07 ± 5.52% (n = 4) and 49.08 ± 9.68% (n = 8), respectively (P < 0.001) (Fig. 5C). In the second experiment, ERK activation was blocked during treatment with R-PIA. The PI fluorescence values of R-PIA preconditioning and R-PIA + PD-98059 treated groups were 38.24 ± 4.13% (n = 4) and 74.42 ± 13.92% (n = 14), respectively (P < 0.001) (Fig. 5D).
DISCUSSION
The present study demonstrated that the A1AR is involved in the triggering phase of IPC in the organotypic hippocampal slice preparation. We previously demonstrated that IPC requires activation of NMDA receptors in the same model of ischemic preconditioning (Raval et al., 2003). Both findings are supported by previous studies in other models of cerebral ischemia. For example, in vitro studies from cortical cell cultures (Gonzalez-Zulueta et al., 2000; Grabb and Choi, 1999; Tauskela et al., 1999, 2003) and in vivo studies of global cerebral ischemia by blocking of NMDA receptors during ischemic preconditioning demonstrated the importance of NMDA receptor activation in IPC neuroprotection (Bond et al., 1999; Kato et al., 1992). Additional evidence supporting the role of NMDA receptors emerged from studies on cortical spreading depression induced preconditioning (Douen et al., 2000; Kawahara et al., 1997:Kobayashi et al., 1995; Kunkler and Kraig, 1998; Matsushima et al., 1996). The role of A1AR was also demonstrated in vitro in primary neuronal cultures (Di-Capua et al., 2003; Reshef et al., 2000) and in vivo during global cerebral ischemia (Heurteaux et al., 1995).
A key finding in the current study is that both triggering factors (i.e., NMDA and A1AR) promote IPC neuroprotection in the hippocampal CA1 region by a similar signal transduction pathway. Furthermore, to our knowledge for the first time, we also demonstrate that both receptors activate ɛPKC in hippocampus. A puzzle that emerges from these findings is that both receptors exert opposite actions in the CA1 subregion of the hippocampus. For example, the A1AR mediates inhibition of synaptic potentiation in the CA1 region of hippocampus by reducing Ca+2 influx through voltage gated channels and reducing pre-synaptic neurotransmitter release (Abbracchio and Cattabeni, 1999; Kato et al., 1999; Thompson et al., 1993). In contrast, NMDA receptors in the hippocampal CA1 region are associated with influx of Ca+2 and increase excitability post-synaptically (Purves et al., 2001). Thus a quandary emerges from the disparate actions of these two receptors in this region of the hippocampus. How do pre- and post-synaptic terminals crosstalk to promote ischemic tolerance in all hippocampal cells? Further studies in our laboratory are underway to define putative cross-talk messengers between pre-and post-synaptic terminals in the CA1 region of the hippocampus.
In the present study, we also investigated whether these two disparate receptors activate ɛPKC during IPC. ɛPKC belongs to the subgroup of novel, Ca+2-independent PKC-isozymes that are activated by binding of diacylglycerol (DAG), a second messenger generated by the phosphoinositol pathway (Lester et al., 2000). It is well known that NMDA receptor activation promotes Ca+2 influx, which in turn can activate phospholipase C (PLC), which in turn generates DAG (Nishizuka, 1992). Raval and colleagues (2003) demonstrate that the DAG analogue OAG (oleoylacetyl glycerol) emulated IPC neuroprotection. In the present study, we further corroborate this pathway when ischemic tolerance was reduced by PLC inhibition (Fig. 4B). Furthermore, in the heart, it was demonstrated that protection against ischemia by A1AR-induced preconditioning was linked to increased PLC activity and DAG turnover (Parsons et al., 2000). These data suggest that IPC in the brain triggered by A1AR activation is also coupled to stimulation of the phosphoinositol pathway and subsequent ɛPKC signaling. These findings suggest that PLC may be a common pathway in both pre- and post-synaptic terminals in the CA1 region of the hippocampus.
Taking into account the results of our previous study in which ɛPKC also played a key role in NMDA-induced preconditioning (Raval et al, 2003), we conjectured that signals from different receptors may converge at ɛPKC, underscoring once more the importance of this isozyme for preconditioning neuroprotection. It is important to note that other PKC isozymes may play a role in IPC as well. Translocation of other PKC isozymes, namelyα, γ, and δ after IPC has been reported in the brain (Kurkinen et al., 2001; Shamloo and Wieloch, 1999). However, we could not get translocation of δPKC or γPKC after IPC in this particular model of IPC (organotypic hippocampal slice cultures) (Raval et al., 2003). It will be important for future studies to define whether other PKC isozymes play any role in IPC by using pharmacologic blockers as the ones described in the current study or transgenic species where different PKC isozymes have been knocked down.
Because not much is known about the downstream signals that ensue after ɛPKC translocation, we further investigated whether the ERK pathway was involved in ɛPKC -induced neuroprotection. Previous studies in the heart demonstrated that activation of ɛPKC both in vivo and in vitro protected against ischemia via activation of ERK 1/2 and JNK members of the MAPK family (Li et al., 2000; Ping et al., 1999a, 1999b; Punn et al., 2000). According to Shizukuda and Buttrick (2001), ɛPKC mediated ERK 1/2 regulation in adult rat ventricular myocytes. Furthermore, ɛPKC was implicated in the downstream activation of ERK 1/2 during IPC in the heart (Ping et al., 1999a, 1999b). These results support a putative link between ɛPKC translocation and ERK 1/2 activation leading to neuroprotection after IPC.
Although the effector pathways after activation of ɛPKC-ERK pathways remain undefined in the brain, an essential feature of the activation of ɛPKC during IPC is its subcellular redistribution and binding with its receptor-activated C kinase (RACKs) (Dorn et al., 1999; Kraft and Anderson, 1983; Mochly-Rosen, 1995; Ping et al., 1997, 1999). Subsequent to translocation to specific sites, PKC isozymes exert their effect by phosphorylating specific target proteins. This translocation to the sub-cellular compartments was considered to be an important mechanism for ɛPKC to direct downstream signaling cascades and induce protection (Dorn, 1999). One of the subcellular sites thought to play a role in the induction of neuroprotection is the mitochondria. Ischemic preconditioning has been shown to preserve mitochondrial function in the late window of IPC (Dave et al., 2001; Fryer et al., 2000). There is lack of direct evidence suggesting translocation of ɛPKC to brain mitochondria; however, formation of mitochondrial ɛPKC -ERK 1/2 modules coupled to the inactivation of BAD, a pro-apoptotic molecule was demonstrated in the heart (Baines et al., 2002). ERK 1/2 was also reported to phosphorylate the pro-apoptotic protein BAD and dissociate it from BCL-XL. This balance was postulated to prevent apoptosis and enhance cell survival (Scheid et al., 1999). In the brain, similar pro-survival influence after the phosphorylation of ERK 1/2 has been reported (Gonzalez-Zulueta et al., 2000; Shamloo et al., 1999). Further studies will be required to determine whether ɛPKC + ERK 1/2 interacts in the same manner in the hippocampus after IPC.
Other pathways involved in ɛPKC + ERK 1/2-induced neuroprotection after IPC may involve expression of neuroprotective genes. For example, activation of ERK 1/2 results in phosphorylation of downstream targets including p90rsk (Wood et al., 1992), as well as several transcription factors (Treisman, 1996).
CONCLUSION
We demonstrate here that the ischemic tolerance induced by IPC and two of its key triggering factors, the adenosine A1 and NMDA receptors, was mediated by ɛPKC -ERK pathway via PLC activation. Future studies are required to define the exact subcellular sites targeted by this signal transduction pathway that leads to this naturally intrinsic state of ischemic tolerance in the brain.