
Abstract
The effect of propentofylline, an adenosine uptake inhibitor, on ischemic tolerance was investigated in the gerbil global ischemia model. Propentofylline was administered 24 hours after short preconditioning ischemia, and animals were subjected to 5-minute ischemia 24 hours thereafter. Propentofylline at a dose of 20 mg/kg intraperitoneally, but not at a dose of 10 mg/kg, significantly potentiated the protective effect of preconditioning ischemia in the CA1 hippocampal neurons. This effect was completely abolished by simultaneous administration of theophylline (20 mg/kg), an adenosine receptor blocker. This finding suggests the involvement of adenosine receptor for the development of ischemic tolerance.
Keywords
A brief period of cerebral ischemia produces neuronal damage in the vulnerable regions of the brain, such as the CA1 sector of the hippocampus (Kirino, 1982; Pulsinelli et al., 1982). In contrast, pretreatment of the brain with sublethal ischemia has been reported to induce neuronal resistance to otherwise lethal ischemia, a phenomenon designated ischemic tolerance (Kitagawa et al., 1990; Kirino et al., 1991). This neuronal tolerance to lethal ischemia also can be induced by a variety of sublethal stresses other than ischemia, such as hyperthermia (Kitagawa et al., 1991), chronic hypoperfusion (Ohtsuki et al., 1993), oxidative stress (Ohtsuki et al., 1992), and spreading depression (Kawahara et al., 1995).
The mechanism of ischemic tolerance is unknown. Although induction of 72-kd heat shock protein (hsp72) has been suggested to be a possible mechanism of ischemic tolerance in previous investigations (Kirino et al., 1991; Liu et al., 1993; Ohtsuki et al., 1993), its causal relation has been doubted by other studies (Kato et al., 1992). Recently, Heurteaux and colleagues (Heurteaux et al., 1995) suggested a role of adenosine, an endogenous neuromodulator (for review, see Rudolphi et al., 1992), in the development of ischemic tolerance. They demonstrated that an adenosine receptor antagonist markedly decreased the protective effect of preconditioning ischemia, thereby suggesting a significant involvement of adenosine receptor stimulation during preconditioning ischemia.
Prompted by this finding, we hypothesized that further stimulation of adenosine receptor after preconditioning ischemia may potentiate its protective effect on subsequent lethal ischemia. In this study, therefore, we used propentofylline (PPF), an adenosine uptake blocker (Fredholm and Lindström, 1986; Ohkubo et al., 1991), to increase endogenous adenosine after preconditioning ischemia, and examined its effect on ischemic tolerance in the gerbil hippocampal CA1 neurons.
MATERIALS AND METHODS
Male Mongolian gerbils (Meriones unguiculatus, 60 to 80 g) were used. Propentofylline was a gift from Hoechst Marion Roussel Ltd., and theophylline was purchased from Research Biochemicals (Natick, MA, U.S.A.). The experiment was divided into three different series (Table 1). In the first experiment, gerbils were subjected to either 2- or 5-minute single ischemia as described later (groups B and C). Normal animals were used as controls (group A). In the second experiment, animals first were subjected to sham operation, administered with either vehicle (saline, group D) or PPF (20 mg/kg, group E) intraperitoneally 24 hours later, and then subjected to 5-minute ischemia 48 hours after the sham operation. In the third experiment (groups F to I), animals were subjected first to preconditioning ischemia for 2 minutes, and then to 5 minutes of ischemia 48 hours later. In these groups, either vehicle (group F) or PPF (10 mg/kg in group G, 20 mg/kg in groups H and I) was administered intraperitoneally 24 hours after the first ischemia. In group I, theophylline (20 mg/kg intraperitoneally) was simultaneously administered.
CA1 neuronal density and postischemic temperature
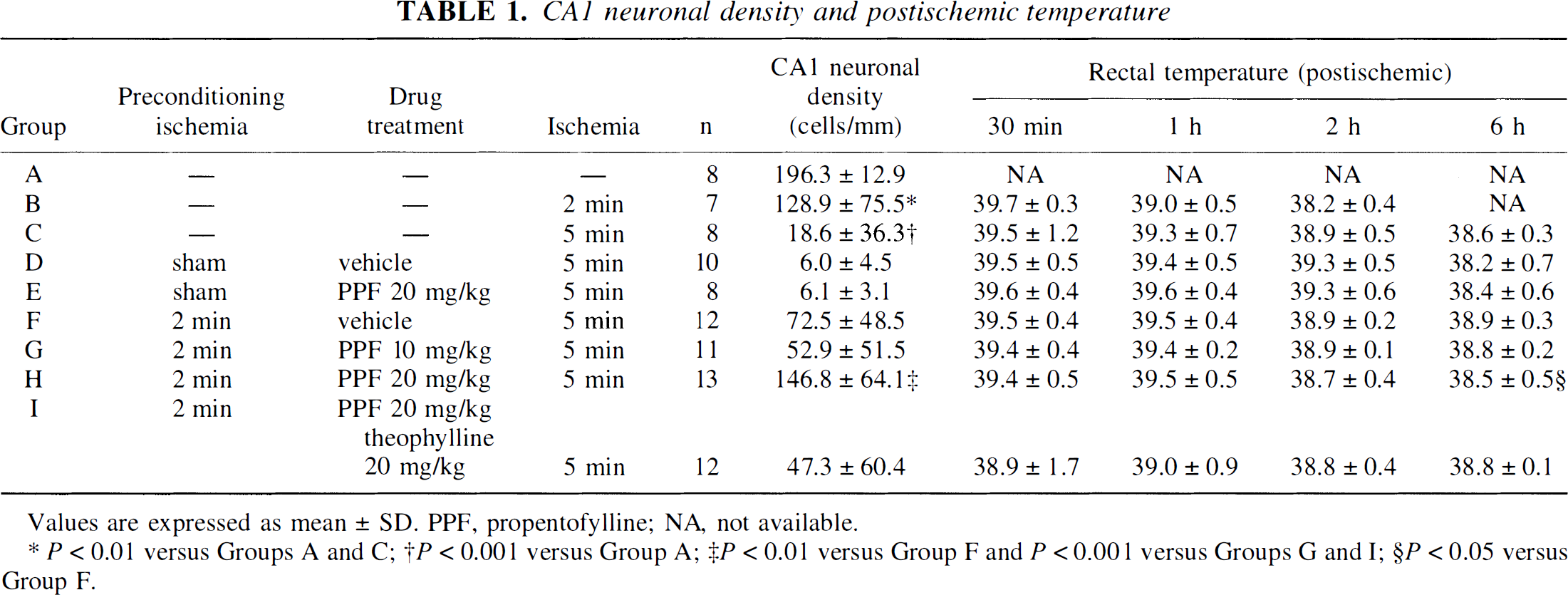
Values are expressed as mean ± SD. PPF, propentofylline; NA, not available.
P<0.01 versus Groups A and C;
P<0.001 versus Group A;
P<0.01 versus Group F and P<0.001 versus Groups G and I;
P<0.05 versus Group F.
For preconditioning ischemia, gerbils were subjected to 2 minutes of ischemia under an awake condition using Tone's method (Tone et al., 1987). Briefly, a nylon thread, which was looped around the carotid arteries the day before, was pulled and bilateral common carotid arteries were occluded for 2 minutes in awake state. For the sham operation, a nylon thread, which was not looped around the carotid arteries, was removed. For 5-minute ischemia, both carotid arteries were transiently occluded for 5 minutes with aneurysm clips under halothane anesthesia (0.75%) in 30% O2/70% N2O mixture. Rectal and temporal muscle temperatures were maintained close to 37.5°C during ischemia using a heating pad and a lamp. Postoperatively, rectal temperature was recorded at 30 minutes, 1 hour, 2 hours, and 6 hours after reperfusion, and those animals who failed to restore the rectal temperature above 38°C were excluded.
Seven days after the last ischemia, the brains were perfusion fixed with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) under halothane anesthesia and embedded in paraffin. Sections (4-mm thickness) containing the dorsal hippocampi were stained with hematoxylin and eosin. The number of intact neurons in the hippocampal CA1 sector was counted in blind fashion. The mean value of the neuronal density (cells per millimeter) on each side was used for statistical analysis.
The differences in the neuronal density of the CA1 sector and temperature within each experimental series were analyzed using one-way analysis of variance (ANOVA) with Bonferroni's post hoc test with significance set at P<0.05.
RESULTS
Results are summarized in Table 1 and depicted in Fig. 1. In the first experiment (groups A to C; Fig. 1, Table 1), effect of single ischemia in the CA1 sector was evaluated in our experimental paradigm. Unexpectedly, single 2-minute ischemia alone resulted in significant neuronal damage by approximately 35% compared with normal controls. This was caused by severe damage observed in two of seven gerbils. The other five animals had almost normal neuronal density. After 5-minute ischemia, more than 90% of the CA1 neurons were lost. The differences among these groups were statistically significant (Fig. 1).
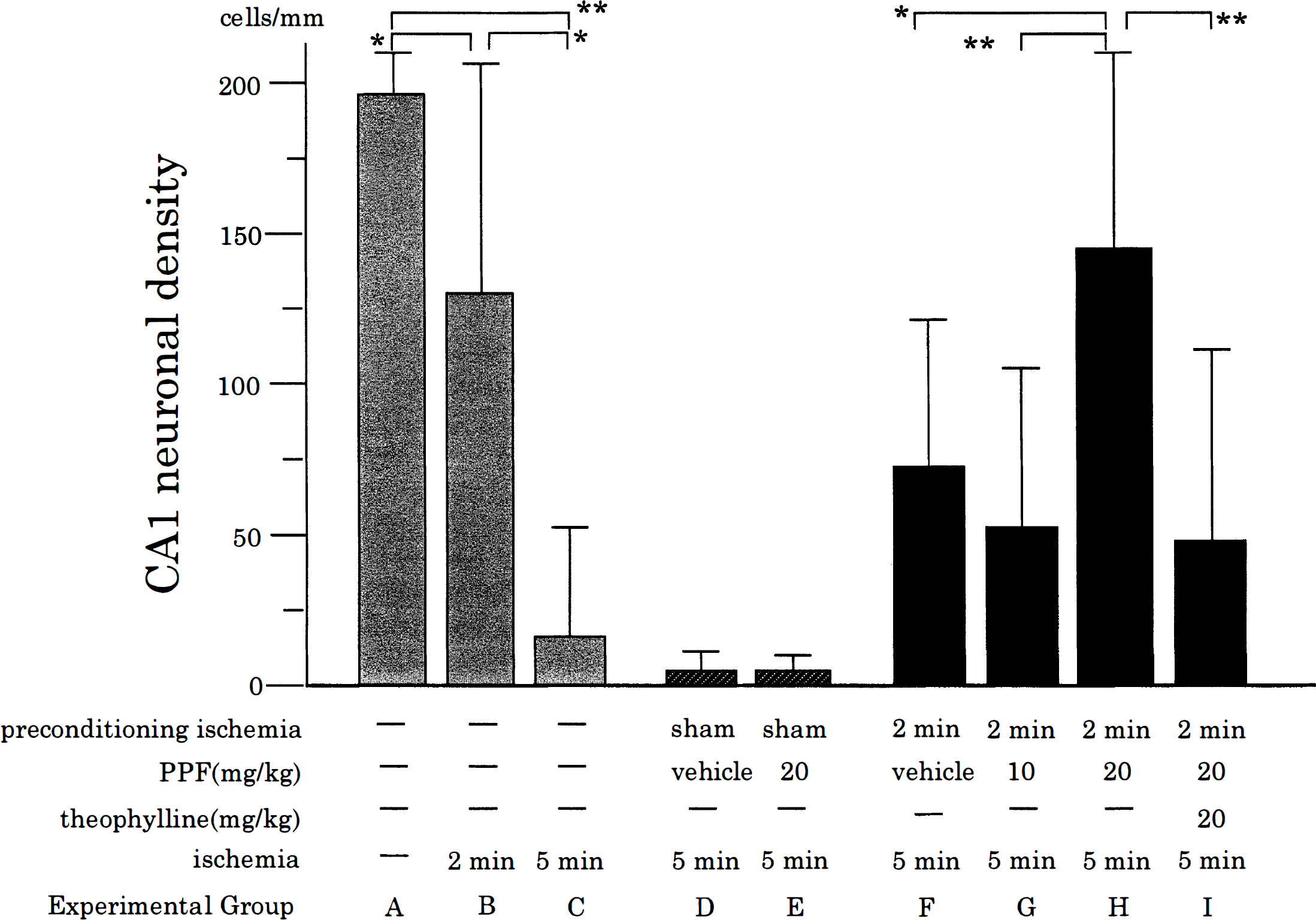
CA1 neuronal density in the experimental groups. Each experimental group corresponds to the same group shown in Table 1. Error bars represent SD. PPF, propentofylline, *P<0.01, **P<0.001.
In the second experiment, the effect of PPF administered 24 hours before ischemia on neuronal survival after 5-minute ischemia was evaluated. As shown in Table 1 and Fig. 1, there was no significant difference in the CA1 neuronal density between the vehicle-treated (group D) and PPF-treated (20 mg/kg, group E) animals. Neither did postischemic rectal temperatures reveal any significant difference (Table 1).
In the third experiment (groups F to I), the effect of PPF on endogenous neuroprotection conferred by preconditioning ischemia was examined. As reported previously (Kirino et al., 1991), 2-minute ischemia, when induced 48 hours before 5-minute ischemia, protected CA1 pyramidal neurons (group F) when compared with sham operation (group D). Since our 2-minute ischemia was slightly severe, resulting in 35% damage, this indicates that preconditioning ischemia protected 57% of neurons that survived 2-minute ischemia. The effects of PPF and theophylline treatments in this experimental condition on the CA1 neuronal damage are shown in Table 1 and Fig. 1 as groups F, G, H, and I. Statistical analysis using ANOVA revealed that the CA1 neuronal densities were significantly different (P<0.0002). Post hoc analysis disclosed that the CA1 neuronal density in animals treated with PPF (20 mg/kg, group H) was significantly higher than that in animals treated with either vehicle (group F) or PPF (10 mg/kg, group G), indicating the protective effect of PPF and its dose dependency. This protective effect was completely abolished by an adenosine receptor antagonist, theophylline, as shown in group I. Concerning post ischemic temperature, there was a minor but statistically significant difference at 6 hours (ANOVA, P<0.033). By post hoc analysis, the differences among these groups were significant only between groups F and H.
DISCUSSION
Propentofylline (20 mg/kg) markedly attenuated the ischemic neuronal injury in the hippocampal CA1 sector in the double ischemia experiment in a dose-dependent manner. This effect was completely abolished by an adenosine receptor blocker, theophylline, strongly indicating involvement of adenosine receptor stimulation in neuroprotection by PPF. The protective effect of PPF on ischemic injury has been reported in various studies. Pretreatment and early posttreatment with PPF attenuates the ischemic injury in global (DeLeo et al., 1988; Dux et al., 1990) and focal ischemia models (Park and Rudolphi, 1994). The mechanism of neuroprotection has been considered to be related to reinforcement of neuroprotective actions of endogenous adenosine (Fredholm and Lindström, 1986) by inhibiting adenosine uptake (Ohkubo et al., 1991) during and immediately after ischemia, thereby decreasing the release of glutamate (for review, see Rudolphi et al., 1992). However, this direct protective effect on 5-minute ischemia is not the likely mechanism of neuroprotection in the current study, since PPF (20 mg/kg) administered 24 hours before 5-minute ischemia had no effect on CA1 neuronal injury. Rather, the protective effect could be attributed to potentiation of endogenous mechanism of ischemic tolerance conferred by preconditioning ischemia.
The importance of adenosine receptor stimulation for endogenous induction of ischemic tolerance by preconditioning ischemia has been suggested by Heurteaux and colleagues (Heurteaux et al., 1995). They proposed a cascade of adenosine, adenosine A1 receptor stimulation, and opening of ATP-sensitive K+ channels for induction of ischemic tolerance. It is, therefore, conceivable that the observed potentiating effect of PPF is mediated through this pathway by further stimulating adenosine receptor. Lack of neuroprotection by PPF without preconditioning ischemia might imply that this pathway becomes “sensitized” to adenosine after preconditioning ischemia.
There is, however, evidence that PPF provides beneficial effect on ischemia by reducing free radical formation (Banati et al., 1994), or by increasing synthesis of nerve growth factor in glial cells (Shinoda et al., 1990), presumably through adenosine receptor (Schubert et al., 1992). These mechanisms might be partly involved for the potentiation of ischemic tolerance by PPF. Indeed, a free radical scavenger, bifemelane, has been reported to enhance ischemic tolerance in the same experimental paradigm (Ohtsuki et al., 1996). Finally, we could not exclude completely the effect of mild hypothermia observed between the PPF-treated (20 mg/kg) and vehicle-treated groups at 6 hours after ischemia. Nevertheless, this possibility is less likely because there were no significant differences with other groups, and PPF per se had no hypothermic effect (groups D and E).
In summary, we demonstrated that PPF could potentiate the effect of endogenous ischemic tolerance through adenosine receptor stimulation. This finding would provide further relevance for its clinical use and might offer a clue to elucidate the mechanism of ischemic tolerance.
Footnotes
Acknowledgment
The authors thank Ms. Reiko Matsuura for her technical assistance in this study.