
Abstract
Activation of phospholipase C (PLC) increases intracellular Ca++ and may play a role in delayed neuronal death after ischemia. Because changes in intracellular Ca++ are believed to participate in ischemic neuronal injury, we tested the hypothesis that PLCβ protein levels are temporally altered in brain regions that undergo neurodegeneration after global incomplete ischemia. Dogs (n = 12) were subjected to 20 minutes of global incomplete ischemia followed by recovery of either 1 (n = 5) or 7 days (n = 7). Six sham-operated animals were used as nonischemic controls. In hematoxylin and eosin-stained brain sections, neuronal density at 1 day after ischemia was unchanged relative to nonischemic controls in hippocampus CA1, caudate, and cerebellar cortex (anterior lobule). However, at 7 days after ischemia, neuronal densities were decreased to 56 ± 15% (mean ± SD) and 75 ± 17% of control in CA1 and caudate, respectively. At 1 and 7 days after ischemia, the percentage of neurons showing ischemic injury increased from 13 ± 10 to 40 ± 35% in CA1, 24 ± 25 to 59 ± 16% in cerebellum, and 4 ± 2 to 18 ± 12% in caudate. Densitometric analysis of immunocytochemically stained brain sections from controls (n = 3), 1 day after ischemia (n = 3), and 7 days after ischemia (n = 5) revealed that PLCβ immunoreactivity was increased in cerebellum at 1 day (0.274 ± 0.013 v 0.295 ± 0.005 optical density units [OD] in control and 1 day, respectively) and 7 days (0.108 ± 0.009 v 0.116 ± 0.005 O.D. in control and 7 days, respectively). PLCβ immunoreactivity was unchanged after ischemia in caudate and hippocampus. Western blot analysis of PLCβ immunoreactivity in the cerebellar cortex and hippocampus in the control (n = 3), 1 day (n = 2), and 7 days after ischemia (n = 2) groups showed that PLCβ levels were increased after ischemia in cerebellum 266% and 227% above control at 1 and 7 days, respectively, However, in hippocampus, PLC expression after ischemia was unchanged at 97% and 84% of control at 1 and 7 days, respectively. These results show that delayed neuronal degeneration after global incomplete ischemia is accompanied by regional abnormalities in PLC levels. Elevated PLC levels early may represent an aberrant signal transduction mechanism resulting in delayed cell damage, whereas decreased PLC levels later after ischemia may reflect ongoing neurodegeneration.
Phospholipase C (PLC) is a ubiquitous family of isoenzymes within the brain. Expression of brain PLC varies on a regional and cellular basis (Gerfen et al., 1988; Mizuguchi et al., 1991). Phospholipase C activation is part of an intricate, neuronal intracellular signal transduction cascade and occurs via receptor activation coupled to a G protein. Ischemia modifies the processes involved in receptor activation (Wieloch et al., 1993). For example, changes in PLC activity and expression alter membrane integrity and intracellular Ca++ regulation, and changes in PLC activity can negatively influence the interactions among N-methyl-
Therefore, we sought to determine if PLC expression is altered after brain ischemia. We tested the hypothesis that expression of PLCβ is altered on a temporal and regional basis after global incomplete ischemia in dogs.
METHODS
Temporary global incomplete ischemia
Dogs (n = 12) were anesthetized with halothane (1 to 2% inspired). The trachea was intubated and the lungs were mechanically ventilated. A peripheral intravenous catheter was placed and pancuronium bromide (0.1 mg/kg, intravenously) was injected for muscle paralysis. A femoral artery was cannulated for blood pressure measurement and blood sampling. The left temporalis muscle over the lambdoidal suture was retracted from the skull. After placing a small burr hole, a thermistor was inserted between bone and dura to monitor epidural temperature. A second small burr hole was placed and a Silastic ventricular drain catheter with multiple side ports was inserted into the lateral ventricle for measuring intracranial pressure (ICP) and infusing artificial cerebrospinal fluid (Hurn et al., 1991; Sieber et al., 1994; Palmon et al., 1995). Arterial line placement, intraventricular catheter placement, and production of ischemia were all performed using strict sterile precautions. The body of the dog was wrapped in a plastic bag and placed on a blanket perfused with recirculating warm water. Heat lamps were used, if necessary, to maintain epidural temperature at 37 to 38°C. End-tidal CO2 was monitored and ventilation adjusted to maintain Pa
Incomplete ischemia was produced in dogs (n = 12) by infusing sterile artificial cerebrospinal fluid into the lateral ventricle to produce an ICP 10 mm Hg below the animal's mean arterial blood pressure. The artificial cerebrospinal fluid was prewarmed and maintained at a temperature of 38°C. Six dogs served as sham-operated nonischemic controls. A moderate pressor response resulted, but ICP was easily manipulated to help keep cerebral perfusion pressure constant at 10 mm Hg for 20 minutes. Cerebral blood flow during ischemia ranged from 2 to 11 mL/min/100 g (Sieber et al., 1994). This was sufficient to reduce cerebral O2 uptake, flatten the somatosensory evoked potentials, and reduce phosphocreatine and ATP levels (Sieber et al., 1994; Palmon et al., 1995). To start reperfusion, the cerebrospinal fluid pressure reservoir was disconnected and ICP allowed to normalize.
Arterial blood pressure and ICP were measured continuously with Statham pressure transducers (Quest Medical, Allen, TX, U.S.A.). Arterial P
Conventional neuropathology
For conventional neuropathologic and immunocytochemical analysis, nonischemic controls and ischemic animals were anesthetized with pentobarbital, anticoagulated with heparin, and their brains perfused with phosphate-buffered saline followed by 4% paraformaldehyde. After removal, each brain was divided midsagittally and the hemispheres were cut systematically into 1 cm-thick coronal slabs from anterior to posterior. From the left hemisphere, the midhippocampus and surrounding temporal neocortex, frontal cortex, caudate (head), and cerebellum were sampled consistently for paraffin histology and hematoxylin and eosin (H&E) staining. In this model of ischemia, neuronal damage is similar bilaterally (Sieber et al., 1995). The right cerebrum and cerebellum was systematically cut into 1 cm- thick coronal slabs, cryoprotected in 20% buffered-glycerol, frozen in isopentane, and stored at −80°C for immunocytochemical analyses. Cerebellar and cerebral samples were cut serially into 40-micron sections and transferred into antifreeze buffer for storage until used for immunocytochemistry. Sections of three brain regions were stained with hematoxylin and eosin and evaluated quantitatively for ischemic injury: the hippocampus (CA1), the caudate nucleus, and the anterior cerebellar lobule at the midline. We selected these areas because in other models of ischemia these regions are damaged. In addition, these regions can be sampled and examined consistently. Purkinje cells in the cerebellum were counted in 4 fields at 200× magnification in the rostral cerebellar folia (anterior/rostral vermis) against the pons and above the fourth ventricle. The left hippocampal CA1 region was counted at the midhippocampal level in 6 fields at 600× magnification. The caudate was examined in 5 fields at 1000× magnification. In sections stained with hematoxylin and eosin, the number of neurons were counted and the percent of neurons with ischemic damage was determined using the criterion of microvacuolar change within the cell body, peripheralization of chromatin, perinuclear eosinophilia, or perikaryal shrinkage with eosinophilic cytoplasm. Ischemic neuronal damage was determined by a blinded investigator (LM) who evaluated the three regions of interest separately. The percent of neuronal damage in each region was averaged for each animal and a group mean calculated. The number of neurons/mm2 was calculated for each region in both the ischemic and nonischemic animals and the percent of neurons remaining calculated for the ischemic animals.
Immunocytochemistry
We examined changes in the PLCβ1 isoform. Although PLC has several isoforms in brain and especially neurons, the predominant isoform is PLCβ (Rhee et al., 1991). Immunocytochemistry was performed in hippocampus, striatum, and cerebellum sections using standard immunoperoxidase procedures (Martin et al., 1993; Martin et al., 1993; Martin et al., 1992) to localize PLC β1 (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). Sections of brain from each experimental group were processed concurrently using the same batches of reagents to obviate tissue section variability in antigen localization. These preparations were analyzed using a computer-based image analysis system to determine regional optical density of immunoreactivity (Martin et al., 1991).
Selected sections were incubated with PLCβ1 preabsorbed against a synthetic PLCβ1 antigen (Santa Cruz Biotechnology).
Quantitative immunoblot analysis
Control and ischemic animals were exsanguinated by intraaortic perfusion of phosphate-buffered saline; and the brains were quickly removed. The right cerebrum and cerebellum were frozen unfixed for homogenization; the left hemisphere was cut into 1-cm slabs and fixed by immersion in 5% acrolein prepared in phosphate buffer for paraffin histology. The brains of these animals were prepared in this manner because of the necessity to use fresh tissue for western blotting. However, we also wanted to assess the severity of ischemic damage in these animals. Fresh-frozen brain samples of cerebellar cortex were homogenized and protein assays were conducted as described previously (Martin et al., 1993; Martin et al., 1993; Martin et al., 1992). For western blotting, P2 fractions (10 μg protein) were subjected to SDS-PAGE, electroblotted onto nitrocellulose membrane, and probed with affinity-purified polyclonal antibodies to PLCβ (Martin et al., 1993; Martin et al., 1993; Martin et al., 1992; Fotuhi et al., 1993). We focused on the membrane fraction because this is the fraction of primary PLCβ enrichment. The same blots that were probed for PLCβ were probed with a monoclonal antibody to synaptophysin (Boehringer Mannheim, Indianapolis, IN) to establish that comparable amounts of protein were loaded in each lane. Synaptophysin (p38) was selected because presynaptic proteins are generally preserved after cerebral ischemia (Kitagawa et al., 1992). Immunoblots were quantified densitometrically to evaluate regional brain changes in the levels of PLCβ in postischemic animals relative to controls. Films were scanned using Adobe Photoshop software (Adobe Systems, Buffalo, NY, U.S.A.) for Macintosh (Apple Computer, Inc., Cupertino, CA) and an Agfa Arcus Plus scanner (Agfa Division, Bayer Corp, Ridgefield, NJ, U.S.A.). Densitometric analysis was performed using Signal Analytics IP Lab Gel software (Signal Analytics, Vienna, VA, U.S.A.). PLCβ1 and synaptophysin protein levels were expressed as relative optical density (OD) measurements, determined by comparing the density and area of the immunoreactive band in each lane scanned to control lanes in the same blot. PLCβ1 levels were normalized to synaptophysin measurements to control for errors in loading. The relative values for each animal were replicated in duplicate immunoblotting experiments.
Experimental design
In postischemic and sham animals, we quantitatively and qualitatively evaluated, in cerebellar cortex, hippocampus, and caudate, protein expression of PLCβ by immunocytochemistry and western blotting. We used our model of 20 minutes of global incomplete ischemia to determine protein expression at 3 time points: control nonischemic sham-operated dogs, 1 day after ischemia, and 7 days after ischemia.
The nonischemic, sham-operated animals were anesthetized, underwent similar surgical procedures as their ischemic counterparts, and then were killed (n = 6 total; n = 3 for immunocytochemistry and n = 3 for western blotting). To obtain the 1 day (n = 5 total; n = 3 for immunocytochemistry and n = 2 for western blotting) and 7 day (n = 7 total; n = 5 for immunocytochemistry and n = 2 for western blotting) recovery measurements, animals were anesthetized, followed by sterile placement of mean arterial pressure and ICP catheters. Twenty minutes of global incomplete ischemia was produced, the animal was allowed to recover 1 or 7 days, then was killed. Neuropathologic scores between groups and regions at each time point were analyzed using the Mann-Whitney test. Quantitation of protein expression by immunocytochemistry was analyzed on a regional and temporal basis using T-Test.
RESULTS
Twelve dogs underwent 20 minutes of global incomplete ischemia. Arterial blood gas values immediately before ischemia were pH = 7.39 ± 0.05, P
Delayed neuronal damage after incomplete global ischemia
In the cerebellum, hippocampus, and caudate, the percent of neurons damaged at 24 hours was less than the percent of neurons damaged at 7 days after ischemia. In addition, in all three regions there was minimal neuronal dropout at 24 hours after ischemia. Damage in cerebellum and CA1 was greater than in caudate (Fig. 1).
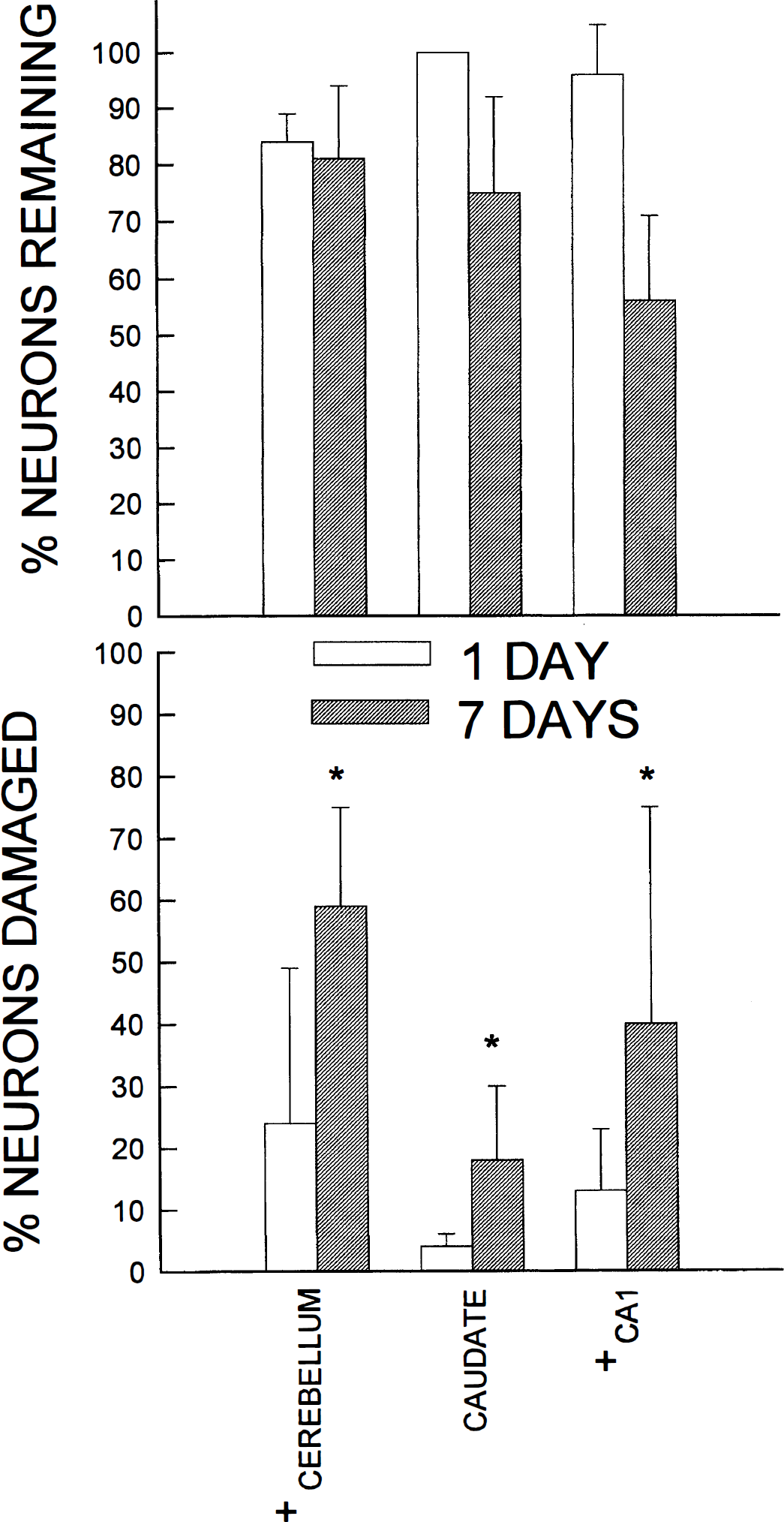
(A) The regional percent of neurons remaining at 1 and 7 days after ischemia. Values are mean ± SD. (B) The percent of neurons with ischemic damage in the regions examined. Values are mean ± SD. + p < 0.05 from caudate, Neuman-Keuls test; * p < 0.05 from respective time within region, Mann-Whitney test.
In the cerebellum, the percentage of damaged Purkinje cells increased from 24% to 59% at 1 and 7 days, respectively. Purkinje cell damage occurred evenly in both the crown and sulci. There was minimal Purkinje cell dropout throughout the postischemic time period.
CA1 pyramidal cell damage increased from 13% to 40% of remaining neurons damaged at 1 and 7 days after ischemia. At 7 days after ischemia, approximately 56% of pyramidal cells remained. The pattern of CA1 damage increased on moving from the subiculum to CA2. Primarily, the neurons in the superficial layers of CA1 showed evidence of ischemic cell change at 1 day after ischemia. This pattern progressed by 7 days to include primarily the pyramidal cell layer.
The caudate was less damaged than the cerebellum and hippocampus. At 24 hours, ischemic cell change was observed in 4% of neurons which increased to 18% damage of the remaining neurons by 7 days. Damage was uniform throughout the region. By 7 days after ischemia, 75% of neurons remained in the caudate.
Time-dependent changes in PLCβ1 protein expression after ischemia
In nonischemic controls, PLC immunoreactivity was enriched in Purkinje cell bodies and their proximal dendritic processes (Fig. 2), with the molecular layer staining more intensely for PLCβ than the granule cell layer (0.547 ± 0.054 v 0.285 ± 0.015 optical units, respectively). Densitometric analysis of immunocytochemically stained brain sections from controls (n = 3), 1 day (n = 3), and 7 days after ischemia (n = 5) revealed that PLCβ immunoreactivity was increased in cerebellum at 1 day (0.274 ± 0.013 v 0.295 ± 0.005 OD in control and 1 day, respectively) and 7 days (0.108 ± 0.009 v 0.116 ± 0.005 OD in control and 7 days, respectively). After ischemia, PLC expression in the molecular layer increased in intensity at 1 day, and remained increased at 7 days after ischemia (0.547 ± 0.094 v 0.570 ± 0.064 OD in control and 7 day after ischemia, respectively). In particular, the arborization of the Purkinje cell dendrites was clearly visualized at 1 and 7 days after ischemia (Fig. 2). Staining of the granule cell layer decreased in intensity by 7 days after ischemia (0.285 ± 0.015 v 0.278 ± 0.015 OD in control and 7 day after ischemia, respectively).
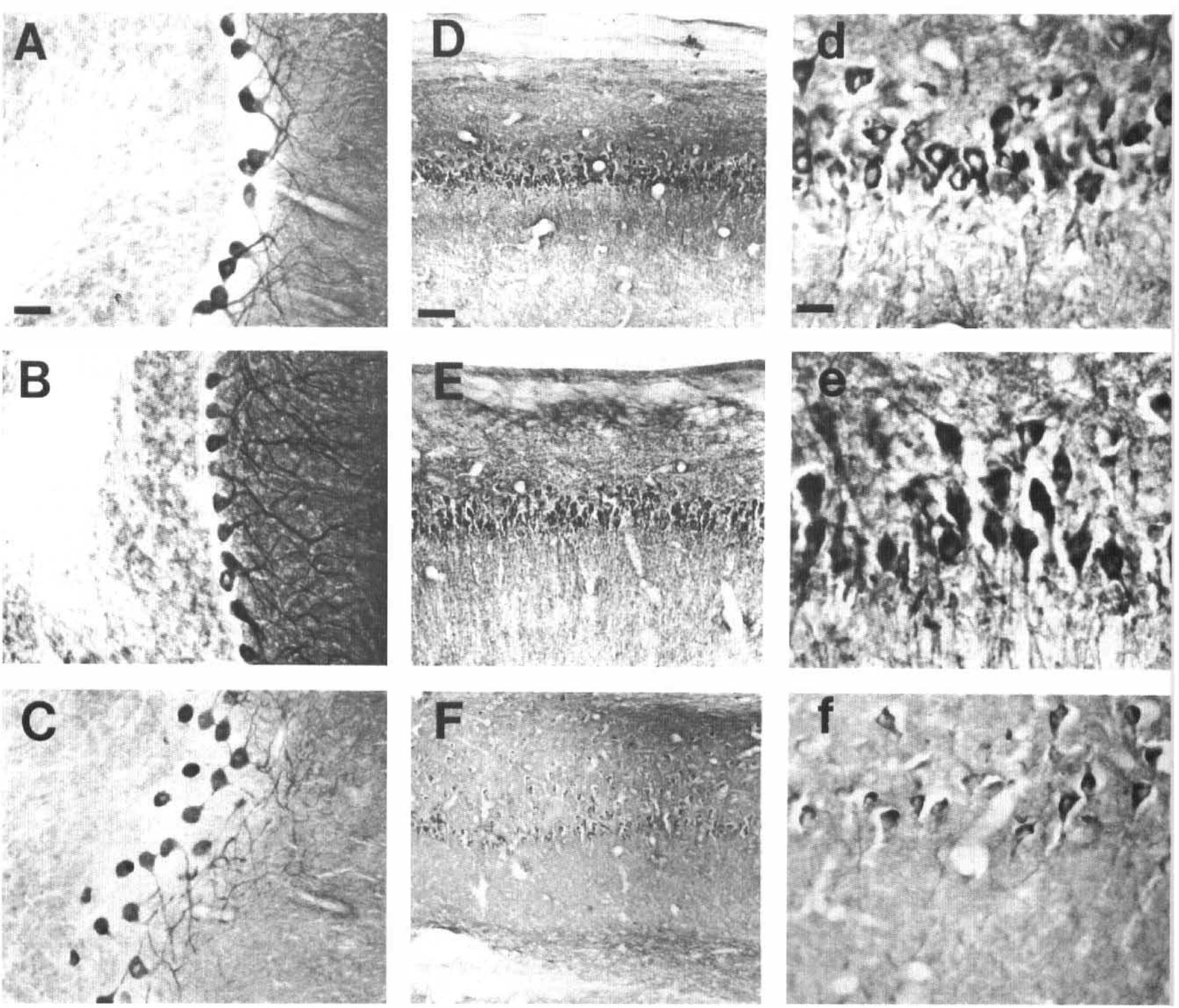
Immunocytochemical localization of PLCβ1 in cerebellum and CA1 hippocampus. (A—C) Cerebellum at 200× magnification. Bar in panel A denotes 50 microns. (D—f) denote CA1 pyramidal cells of hippocampus. (A, B, and C) PLCβ localization in a nonischemic control, 1 day after ischemia, and 7 days after ischemia, respectively. (D—F) show CA1 hippocampus at 100× magnification. Bar in panel D denotes 100 microns. (d—f) CA1 hippocampal pyramidal cells at 400× magnification. Bar in panel D denotes 10 microns. (D—d) Control nonischemic hippocampus. (E—e) CA1 hippocampus at 1 day after ischemia. (F—f) CA1 hippocampus at 7 days after ischemia. There is significant dropout of CA1 pyramidal cells by 7 days after ischemia.
In the nonischemic controls, the pyramidal cell bodies and their proximal dendrites in the CA1 hippocampal region were heavily immunoreactive. PLCβ immunoreactivity was unchanged in hippocampus at 1 day (0.358 ± 0.015 v 0.370 ± 0.019 OD in control and 1 day, respectively) and 7 days after ischemia (0.326 ± 0.019 v 0.324 ± 0.024 OD in control and 7 days, respectively). At 1 day after ischemia, the stratum radiatum became heavily immunoreactive and a modest increase in CA1 immunoreactivity occurred. Yet at 7 days, the neuropil was only modestly immunoreactive (Fig. 2). This loss of immunostaining was related to the degree of neuronal dropout and resulted in no change in CA1 immunoreactivity by 7 days after ischemia (0.136 ± 0.009 v 0.134 ± 0.015 OD in control and 7 days, respectively).
As reported previously (Fotuhi et al., 1993), in nonischemic controls, immunostaining for PLCβ in the striatum occurred predominately in the medium-sized neuronal perikarya and neuropil. At 1 day after ischemia, the principal neurons and neuropil showed enhanced PLCβ immunoreactivity relative to controls. By 7 days, there was still a comparable amount of neuronal cell body immunoreactivity, but decreases had occurred in the neuropil, especially in the putamen. There was no difference in immunoreactivity in the caudate postischemically by densitometry (0.144 ± 0.017 v 0.136 ± 0.026 OD in control and 7 days, respectively).
To determine whether initial increases in PLCβ1 expression observed in CA1 and cerebellum represent a generalized response to ischemia or are specific for selectively vulnerable regions, we performed densitometric analysis on brainstem and thalamic immunocytochemical PLC preparations at 1 day and 7 days after ischemia. These regions were chosen because our initial histologic and physiologic studies of the current model of incomplete ischemia show that significant decreases in regional blood flow occur in these regions during ischemia (Sieber et al., 1994); however, neuronal damage is minimal at 7 days (Sieber et al., 1995). Immunocytochemical optical densities showed no change in PLCβ1 expression from control at 1 day and 7 days after ischemia in both the thalamus and the brainstem.
All immunoreactivity in dog brain sections was completed by addition of the antigen with which the PLCβ1 antibody was made.
By western blotting, PLCβ1 expression significantly increased in cerebellum to 266% and 227% of control at 1 and 7 days after ischemia, respectively. In the whole hippocampus, PLCβ1 expression was 97% control and 84% control at 1 and 7 days after ischemia (Fig. 3).
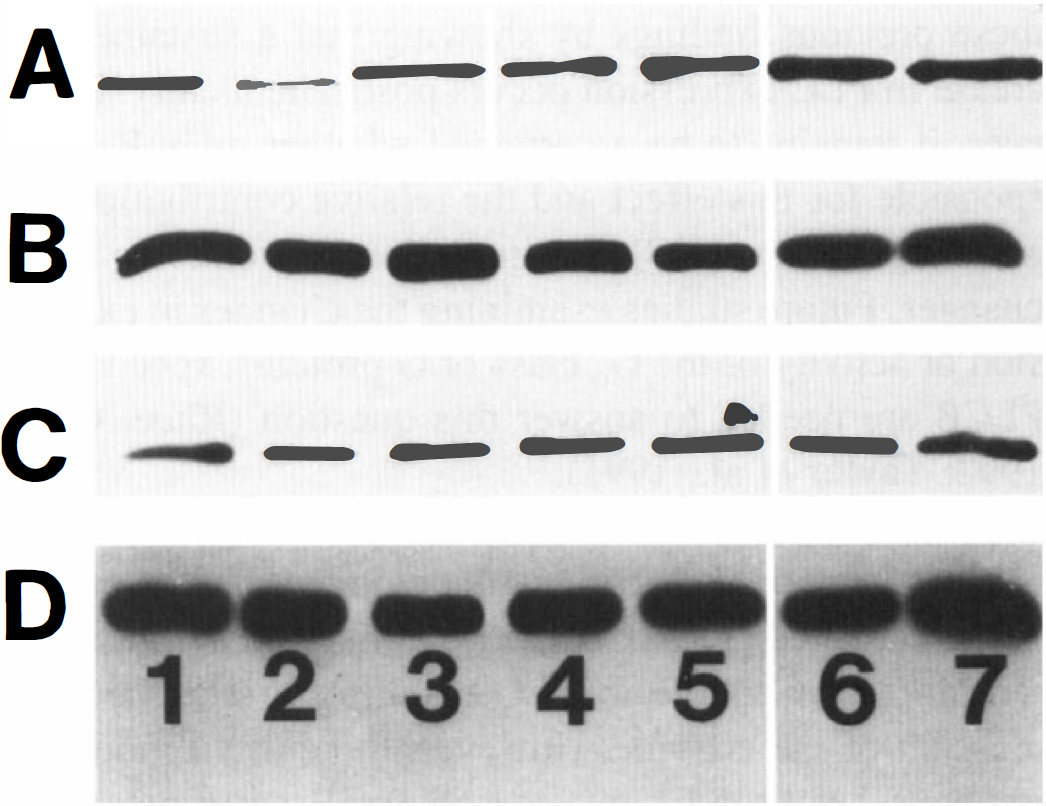
Western blot analysis of PLCβ1 and synaptophysin (p38) levels in cerebellar cortex and hippocampus. PLCβ1 and synaptophysin levels in cerebellar cortex are demonstrated in rows A and B, respectively. PLCβ1 and synaptophysin levels in hippocampus are demonstrated in rows C and D, respectively. Lanes 1, 2, and 3 are nonischemic controls. Lanes 4 and 5 are animals killed 1 day after ischemia. Lanes 6 and 7 are animals killed 7 days after ischemia.
DISCUSSION
These results show that sustained postischemic increases in PLCβ1 protein expression occur on a regional and cellular basis. Specifically, in cerebellum, Purkinje cell and molecular layers show enhanced PLC levels postischemically. Although in the hippocampus, CA1 pyramidal cells express PLC, postischemic increases were not evident at 7 days recovery. Immunocytochemistry revealed that PLC increases occur in the somatodendritic compartment concurrently with neuronal dropout in the hippocampus. Delayed postischemic decreases in regional PLCβ expression probably occur as a result of ultimate degeneration of dendrites. These results are consistent with the loss of cytoskeletal markers for dendrites and spines (e.g., microtubule-associated protein-2 immunoreactivity) postischemically (Matesic, Rick, 1994), and further suggest that postsynaptic perturbations in dendrites may be an early sign of postischemic delayed neuronal death.
PLCβ levels are high in the caudate and CA1 (Gerfen et al., 1988), accounting for greater than 50% of total PLC activity in rat brain (Takenawa et al., 1991). Immunoblotting studies show that the relative level of PLCβ expression in cell culture is greatest in neurons, then oligodendrocytes, and least in astrocytes (Mizuguchi et al., 1991). The current study has not documented the responses of PLC isoforms other than PLCβ1. Thus, we cannot determine from our data whether the early postischemic increases in PLCβ protein levels are a compensatory response to decreases in other PLC isoforms. In addition, our. experiments have not shown whether the early postischemic increase in PLC protein expression is accompanied by a gain in PLC function, or whether increased PLC protein levels reflect a loss of biochemical function of PLC. However, previous investigations suggest that PLC activity is not altered by brain ischemia (Nishida et al., 1994).
It is possible that increases in PLCβ may represent compensatory responses to postischemic decreases in cellular inositol triphosphate (IP3) binding (Kato et al., 1992; Kato et al., 1991). Marked alterations in intracellular signal transduction take place early, within 3 hours, after ischemic insult, and precede delayed neuronal death (Onodera and Kogure, 1989). The IP3 cascade has at least been implicated in the induction of neurodegeneration. Deceases in IP3 cellular binding may reflect primary damage to IP3 receptors in subcellular compartments or downregulation of this receptor (Walton et al., 1991). In addition, stimulation of inositol phospholipid hydrolysis by glutamate and norepinephrine increases by 24 hours after ischemia; an effect that lasts for at least 1 week (Seren et al., 1989). An increase in metabotropic glutamate receptors (mGluR) that function via G proteins coupled to PLC (i.e., mGluR1 and mGluR5) may be a mechanism for the PLC changes. Our study extends these previous findings by showing that a sustained increase in PLC expression occurs postischemically. However, it remains to be ascertained whether mGluR is responsible for this effect and the relative contributions of adrenergic versus mGluR stimulation in producing these changes. Future studies examining the changes in expression or activity of the Gq class of G proteins, specific for PLCβ are needed to answer this question (Rhee, Choi, 1992; Taylor et al., 1991).
A common sequelae of ischemia-producing infarction is gliomesodermal scarring with angiogenesis that occurs in the damaged areas. Capillaries contain many different receptors that couple to PLC resulting in enriched expression of the enzyme. However, the current model of ischemia has little, if any, mesodermal scarring component, and produces no infarction at 7 days recovery (Sieber et al., 1995). Thus, as confirmed by the cellular expression of the immunocytochemical preparations, it is unlikely that the documented PLC changes represent augmented vascular expression of the enzyme.
Phospholipase C may provide a mechanism for delayed neuronal damage. Elevated intracellular Ca++ is a consistent feature of excitotoxic cell damage. Phospholipase C is coupled to mGluR as well as other receptors which act through second messengers to produce postischemic changes in intracellular Ca++. The lack of change in PLC expression in the thalamus and brainstem suggests that regional PLC increases are not a generalized response to ischemia, but rather are associated with ischemic neuronal damage in selectively vulnerable regions. In support of this conclusion, several investigators have shown that PLC inhibition decreases the amount of neuronal dropout in CA1 after ischemia in the rat (Umemura et al., 1992). In addition, pretraumatic PLC inhibition significantly improves neurologic outcome after traumatic brain injury in rats (Golding and Vink, 1994). In summary, this study shows that sustained increases in PLC expression occur up to 7 days after ischemia. These increases occur on a regional and cellular basis. It is unclear what cellular mechanisms are involved in promoting these regional changes. However, PLCβ increases are occurring in the somatodendritic compartment of neurons. In addition, regional PLC increases are not merely a generalized response to ischemia, but are associated with regional ischemic neuronal damage. These data show that increases in PLC expression are associated with delayed neuronal death after incomplete global ischemia.
Footnotes
Acknowledgment:
The authors thank Dawn Spicer, Ann Price, and Freddy Jackson for their technical assistance and Lee Palmer for her expert preparation of this manuscript.