
Abstract
Current knowledge regarding the pathophysiology of cerebral ischemia and brain trauma indicates that similar mechanisms contribute to loss of cellular integrity and tissue destruction. Mechanisms of cell damage include excitotoxicity, oxidative stress, free radical production, apoptosis and inflammation. Genetic and gender factors have also been shown to be important mediators of pathomechanisms present in both injury settings. However, the fact that these injuries arise from different types of primary insults leads to diverse cellular vulnerability patterns as well as a spectrum of injury processes. Blunt head trauma produces shear forces that result in primary membrane damage to neuronal cell bodies, white matter structures and vascular beds as well as secondary injury mechanisms. Severe cerebral ischemic insults lead to metabolic stress, ionic perturbations, and a complex cascade of biochemical and molecular events ultimately causing neuronal death. Similarities in the pathogenesis of these cerebral injuries may indicate that therapeutic strategies protective following ischemia may also be beneficial after trauma. This review summarizes and contrasts injury mechanisms after ischemia and trauma and discusses neuroprotective strategies that target both types of injuries.
Incidence and Prevalence of TBI and Stroke
Ischemic and traumatic brain injuries are global health problems. Traumatic brain injury (TBI) is a leading cause of death and disability among children and young adults. Each year, an estimated 1.5 million Americans sustain head trauma (National Center for Injury Prevention and Control, 2001). As a consequence, 230,000 are hospitalized and survive, 50,000 die, and 80–90,000 experience the onset of long-term disability. As a cumulative result of past TBI, an estimated 5.3 million men, women, and children are living with a permanent TBI-related disability in the United States today. The annual economic burden in the United States is approximately $56.3 billion for TBI alone. Also, with increased longevity of the general population, head trauma due to falls and other mechanisms has led to increased frequency of TBI in the elderly. Currently, no pharmacological treatments have been proven to protect against the detrimental consequences of TBI (Narayan et al., 2002).
Stroke is the third most common cause of death in most industrialized countries with an estimated global mortality of 4.7 million per year. A stroke occurs every 53 seconds in North America and by 2002 is projected to become the fourth-leading burden of disease worldwide (Michaud et al., 2001). Stroke killed 283,000 people in 2000 and accounted for about one in every 14 deaths in the United States. Each year, about 700,000 people suffer new or recurrent stroke. It is the major cause of serious, long-term disability, with more than 1,100,000 American adults reporting functional limitations resulting from stroke. Also, recent evidence suggests that the presence of small strokes, or of local chronic ischemia may be much more common in aging populations than previously thought. According to the NHLBI Framington Heart Study (Wolf et al., 1977), the incidence of stroke more than doubles in each successive decade for people over the age of 55, with the chance of having a stroke before age 70 being 1:20 for both sexes. Stroke is more common in men than women, although at older ages the incidence is higher in women than in men. Unlike TBI, there is one treatment that is somewhat successful in a subpopulation of stroke victims. The thrombolytic, tissue plasminogen activator (tPA) has been proven effective in treating stroke when given within 3 hrs after onset of neurological symptoms. Although stroke and traumatic brain primarily affects different age groups, both result in a significant number of individuals with long-term deficits (Table 1).
Primary Insults
Stroke and traumatic insults arise from very different initial insults. Nevertheless, similarities exist in the molecular and cellular mechanisms underlying cell death (Dirnagl et al., 1999; McIntosh et al., 1997). There are three major categories of stroke: subarachnoid hemorrhage, intracerebral hemorrhage and ischemic stroke. The most common variety of stroke is atherothrombotic brain infarction (61%), with the next most common being cerebral (24%) (Bamford et al., 1991). Cerebral ischemia results from severe reductions in cerebral blood flow (CBF), after cardiac arrest, the occlusion of cerebral and extracerebral vessels supplying nervous tissues, or periods of prolonged systemic hypotension. Severe and/or prolonged reductions in CBF lead to deprivations in oxygen and glucose delivery as well as the build-up of potentially toxic substances. Because nerve cells do not store alternative energy sources, these hemodynamic reductions can result in the reduction in metabolites such as ATP, leading to metabolic stress, energy failure, ionic perturbations, and ischemic injury (Dirnagl et al., 1999; Siesjo et al., 1989). Cells that undergo severe ischemia may die within minutes of the insult or display a delayed vulnerability. Ischemic insults can be both focal or global as well as permanent or transient, leading to reperfusion of post-ischemic areas. Depending on how early reperfusion is initiated, metabolic and ionic homeostasis can return and cell survival maintained.
Traumatic brain injury (TBI) generally results from mechanical impact or acceleration-deceleration insults that cause skull fractures, compression of cerebral tissues, and tearing of white and gray matter and subsequent hemorrhage (Adams, 1992; Graham, 1996). TBI can lead to a spectrum of histopathological changes, including hemorrhagic contusion, intracerebral hemorrhage, subarachnoid hemorrhage, and widespread white matter damage (Graham, 1996; Graham and Adams, 1971; Graham and Gennarelli, 1997). Thus, histological damage after TBI can be both focal and diffuse. Also, contrecoup contusions, most common after deceleration of the head, can occur away from the point of injury (Gennarelli and Graham, 1998). The primary insult initiates a wide range of secondary injury mechanisms that critically participate in the pathogenesis of TBI (Graham, 1996; Gennarelli and Graham, 1998). Histopathological data from autopsy specimens, for example after fatal head injury, are consistent with ischemic processes contributing to the pathological consequences (Adams et al., 1982a; Graham and Adams, 1971). The severity of the secondary mechanisms of TBI depends upon injury severity (mild, moderate, severe) or location of the primary insult. In conditions of severe TBI, reductions in CBF have been reported to reach ischemic levels (Marion et al., 1991; Zauner et al., 1996). Thus, cerebral ischemia is discussed as one secondary injury mechanism that may participate in brain trauma (Bouma et al., 1991; Marion et al., 1991).
Epidemiology of Stroke and Traumatic Brain Injury (USA statistics)

Cellular Vulnerability
In both cerebral ischemic and traumatic insults, patterns of neuronal vulnerability are well described (Adams, 1992; Graham, 1996; Dirnagl et al., 1999; Pulsinelli et al., 1982). The neuron has classically been shown to be very sensitive to periods of cerebral ischemia. Flow reductions reaching 25ml/100g/minute in rodents are considered severe enough to lead to eventual cell death (Bolander et al., 1989). In addition to the severity of the ischemic insult, the duration of ischemia also determines vulnerability patterns (Heiss and Rosner, 1983). For example, a brief period of severe ischemia may lead to selective neuronal damage, with minor cellular changes observed in glia and blood vessels (Pulsinelli et al., 1982). However, with longer ischemic periods, other cellular responses can be observed, ultimately producing ischemic infarction (Petito et al., 1982). With reperfusion injury, damage to cerebral blood vessels and the activation of inflammatory processes can produce hemorrhagic transformation of infarcted tissue and severe brain swelling (del Zoppo and Mabuchi, 2003; Hallenbeck et al., 1986).
In models of cardiac arrest and transient forebrain ischemia, specific populations of neurons have been shown to be vulnerable (Kirino et al., 1982; Pulsinelli et al., 1982). After cardiac arrest, the CA1 sector and dentate hilus of the hippocampus, dorsolateral striatum, and Purkinje neurons in the cerebellum are most vulnerable. In contrast, focal ischemia produces a core of severe hypoperfusion where flow is reduced to 15–18ml/100g/minute (Mies et al., 1991). This core is surrounded by a rim of viable tissue where blood flow may be only mildly depressed (Hossmann, 1994). The ischemic core, if not reperfused, generally evolves into an ischemic infarct where all cellular elements are necrotic. With severe focal ischemia, neuronal cell death can be demonstrated within a few hours after the insult (Plum, 1983). However, following periods of transient global or focal ischemia, neuronal death can take as long as 2–3 days to mature (Du et al., 1996; Kirino et al., 1984; Pulsinelli et al, 1982). This maturation phenomenon implies a secondary injury process and may suggest a more prolonged therapeutic window for treatment strategies.
The penumbra region surrounding the ischemic core characteristically contains a preserved ionic state and may display scattered ischemic neurons within an intact neuropil (Back, 1998). The evolution of these cellular events may take hours to days to mature, depending upon the nature of the ischemic insult and presence of perineuronal depolarizations (Hossmann, 1996). Following focal ischemia, multiple episodes of cortical spreading depression have been demonstrated in the penumbra as well as more remote regions of the brain (Back, 1998; Dietrich et al., 2000; Hossman, 1996). These propogating waves are caused by intracellular calcium that depolarizes cells and cause metabolic stress and the induction of a variety of genes and proteins in areas away from the infarct (Kariko et al., 1998).
Patterns of both diffuse and focal neuronal injury can also be identified following human and experimental TBI (Adams, 1992; Cortez et al, 1989; Dietrich et al, 1994b; Graham, 1996).Acommon consequence of severe trauma is the generation of a hemorrhagic contusion that results from blunt head injury or a gliding insult. Both insults can directly damage blood vessels and neuronal membranes, including those of cell bodies and axonal processes. Severe injuries can lead to damage of glial cells including astrocytes and oligodendrocytes. Indeed, one of the earliest cellular changes observed after contusion injury is glial swelling.
Contusions are commonly associated with hemodynamic changes including focal reductions in local cerebral blood flow (lCBF) (DeWitt et al., 1986; Dietrich et al, 1998a). In both clinical and experimental studies, the degree of flow reductions has been reported to correlate with injury severity (Dietrich et al., 1998a; Graham and Adams, 1971). Thus, following mild to moderate TBI in experimental animals, reductions in flow are commonly 70–80% of normal (Dietrich et al., 1996a). However, with more severe injury, CBF values approach ischemic levels (Dietrich et al., 1998a). Currently a controversial issue is whether ischemic events represent a primary cause of cell injury or only a secondary consequence of tissue damage (Bouma et al., 1991; Diringer et al., 2002). Clinical studies have reported that ischemic levels of flow are only observed in irreversibly damaged tissue (Diringer et al., 2002; Zazulia et al., 2001). In contrast, other studies in head injured patients have demonstrated histopathological changes consistent with hypoxic/ischemic insults (Graham and Adams, 1971; von Oettingen et al., 2002) and severe flow reductions early after TBI (Bouma et al., 1991; Marion et al., 1991; von Oettingen et al., 2002). Following TBI, energy demand and tissue PO2 in the traumatized tissue are reduced and, therefore, moderate reductions in blood flow may have severe consequences for cellular survival (Giri et al., 2000). Experimental studies have reported reduction in ATP content in traumatized cortical regions and elevated lactate levels after severe TBI (Lee et al., 1999).
To investigate the importance of CBF reductions in the pathogenesis of brain trauma, recent studies have utilized Positron Emission Tomography (PET) in patients after TBI and cerebral hemorrhage (Diringer et al., 2002; Zazulia et al., 2001). In these studies, cerebral blood flow and cerebral metabolic rate of oxygen (CMRO2) and oxygen extraction fraction (OEF) were evaluated to assess the O2 availability in injured tissue. Although both CBF and CMRO2 were significantly reduced in the injured hemisphere, OEF was also reduced in areas showing hemodynamic reductions. Because OEF normally increases in cerebral ischemic insults, the question of whether these hemodynamic changes are due to an ischemic insult or secondary to metabolic dysfunction and necrosis is critical (Zazulia et al., 2001). In another study, hyperventilation after severe TBI produced large reductions in CBF but not energy failure, as measured by CMRO2 (Diringer et al., 2002). This low metabolic rate of the brain after TBI may also indicate that low CBF values do not necessarily indicate a critical limitation of O2 availability. More experimental work is required to clarify the role of ischemia in secondary injury processes after TBI.
Focal areas of reduced CBF after brain trauma can be surrounded by regions exhibiting milder reductions in flow (DeWitt et al., 1986; Dietrich et al., 1996a). This surrounding area may thus correspond to the penumbral region surrounding an ischemic core (Back, 1998; Hossmann, 1994). This border zone area contains scattered damaged neurons within an intact neuropil (Dietrich et al., 1994b). Importantly, this area is sensitive to therapeutic interventions as well as at risk for secondary insults (Bramlett et al, 1999).
Although much is known about structural changes that occur early after cerebral ischemia or trauma, less is known about the progressive nature of these acute insults to the brain. Clinically, progression of damage has been observed in stroke and TBI patients using MRI (Anderson and Bigler, 1995; Baird et al., 1997). Experimentally, progressive atrophy of gray and white matter structures has been reported in models of TBI (Bramlett et al., 1997a; Bramlett and Dietrich 2002; Dixon et al., 1999; Smith et al., 1997). In one study, severe atrophy of multiple white matter tracts was described 1 year after TBI (Bramlett and Dietrich, 2002). A greater understanding of the pathophysiological processes in this underlying progressive injury cascade could lead to better strategies for neuroprotection and reparative processes.
White Matter Vulnerability
In addition to damage to gray matter structures, white matter vulnerability is observed in both ischemic (Dewar et al., 2003; Gillard et al., 2001; Pantoni et al., 1999) and traumatic (Christman et al., 1994; Gentleman et al., 1995; Maxwell et al., 1993; Strich, 1961) conditions. Because the normal function of the brain is dependent on the communication between brain regions, the clarification of mechanisms contributing to acute and/or progressive white matter damage is critical to our understanding of brain injury (Goldberg and Ransom, 2003). In this regard, an explanation for the lack of successful human stroke trials is that neuroprotective strategies have primarily targeted gray but not white matter structures (Dewar et al., 1999). Following focal cerebral ischemia, damage to white matter tracts can be observed using a variety of immunocytochemical markers.
Abnormal beta amyloid precursor protein (βAPP) immunoreactivity, an indicator of abnormal axonal transport has been reported in white matter structures in models of focal ischemia (Irving et al., 2001; Yam et al., 1998) and thromboembolic stroke (Dietrich et al., 1998b) (Fig. 1A). These white matter changes may underlie some of the behavioral consequences of the ischemic insult not explained by gray matter pathology alone (Stagliano et al., 1997). In addition to axonal damage, abnormal tau staining of oligodendrocytes after MCAO has been reported (Dewar and Dawson, 1995). Therefore, not only are there white matter perturbations but the axonal support cells are vulnerable as well to ischemia.
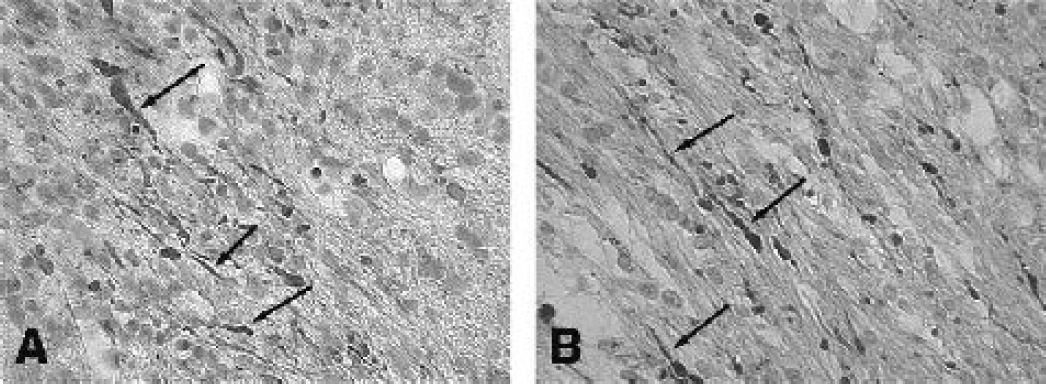
Both cerebral ischemia and traumatic injury lead to white matter vulnerability. Focal areas of axonal pathology can be demonstrated using beta amyloid precursor (β-APP) immunohistochemistry. In these figures, immunoreactive β-APP profiles (arrows) are observed in the external capsule of rats following thromboembolic stroke (A) and moderate fluid-percussion brain injury (B). Although the initial insult is different in the two cerebral insults (metabolic stress, mechanical damage), axonal damage can lead to circuit dysfunction and functional abnormalities. (x1200)
Diffuse axonal injury (DAI) is a common consequence of TBI (Adams et al., 1982b; Christman et al., 1994; Gentleman et al., 1995; Povlishock, 1992). In terms of neurological deficits, DAI has been reported to significantly contribute to poor outcome. The pathophysiology of axonal pathology after TBI is complex and has been reviewed in detail (Graham, 1996; Graham and Gennarelli, 1997). Primary axotomy can occur with severe insults, leading to disconnection of axon processes and to axonal swelling. Secondary axotomy can result in an inhibition of axoplasmic flow as evidenced by β-APP accumulation after human (Gentleman et al., 1993; Sheriff et al., 1994) and experimental TBI (Bramlett et al., 1997b; Pierce et al., 1996) (Fig. 1B). More recent studies have emphasized other mechanisms underlying progressive axon damage that include permeability abnormalities (Pettus et al., 1994), calcium-induced calpain mediated proteolysis (Buki et al., 1999a; Buki et al., 1999b; Wolf et al., 2001) and caspase activation (Keane et al., 2001). Mitochondrial abnormalities have also been implicated in axonal perturbations following TBI (Okonkwo and Povlischock, 1999). Based on current evidence, white matter changes as a result of ischemic or traumatic insults play a major role in the neuropathological and functional consequences of these injuries and are an important target for therapeutic intervention.
Vascular Perturbations
The vascular endothelium has classically been considered to be resistant to cerebral ischemia (Garcia et al., 1971; Petito et al., 1982). Petito and colleagues reported that alterations in the fine structure of endothelial cells was observed in brain regions showing signs of ischemic infarction (Petito et al., 1987). However, using scanning electron microscopy to illustrate endothelial surface alterations, increased frequency of microvilli can be observed during periods of transient forebrain ischemia (Dietrich et al., 1984). Because the vascular endothelium plays an important role in blood-brain barrier (BBB) maintenance as well as to responsiveness to vasoactive substances (Faraci and Heistad, 1998), the effects of trauma and ischemia on endothelial function is currently a critical area of investigation (del Zoppo and Mabuchi, 2003). Increased vascular permeability is observed in many models of brain ischemia (Chan et al., 1984; del Zoppo, 1997; Hatashita and Hoff, 1990). Acute alterations in BBB are observed during early periods of post=ischemic reperfusion (Yang and Betz, 1994) or embolic stroke (Dietrich et al., 1988). In models of focal ischemia, such as MCAO, increased BBB is seen with reperfusion of the MCA territory or following chronic, permanent ischemia (Yang and Betz, 1994). In these cases, increased BBB is frequently associated with vasogenic edema and subsequent brain swelling (del Zoppo, 1997). In addition to ischemic severity, intraischemic brain temperature has also been reported to be an important factor in the early BBB consequences of ischemic insults (Dietrich et al., 1990).
Acute abnormalities in the vascular endothelium and increased BBB permeability are also documented after TBI (Cortez et al., 1989; DeWitt et al., 1986; Dietrich et al., 1994b; Hekmatpanah and Hekmatpanah, 1985; Povlishock and Kontos, 1985). Increased pinocytotic activity within the endothelial cells, endothelial microvilli projections, large vacuoles and craters are reported (Povlishock and Kontos, 1985). Because of the shearing forces generated by head trauma, mechanical damage to endothelial cells can lead to acute BBB permeability and the extravasation of various barrier tracers, including horseradish peroxidase (HRP) (Cortez et al., 1989; Dietrich et al., 1994). Areas of BBB breakdown can be associated with contusion formation in both gray and white matter areas. Also, brainstem nuclei appear to be susceptible to head trauma, in terms of permeability abnormalities (Povlishock et al., 1978). In both cerebral ischemia and trauma, abnormalities in vascular permeability participate in the early pathogenesis of these insults (Povlishock and Dietrich, 1992).
Cortical Spreading Depression
As previously mentioned, reductions in lCBF resulting in metabolic stress can lead to a variety of ionic imbalances and glutamate release, leading to neuronal cell death. TBI has been reported to cause depolarization similar to that observed in cerebral ischemia (Katayama et al., 1990; Rogatsky et al., 1996). Propagated or non-propagated depressions of electrical activity commonly called cortical spreading depression (CSD) or periinfarct depolarization (PID) have been described in a variety of experimental models of focal cerebral ischemia and traumatic brain injury (Back et al., 1994; Dietrich et al., 1994a; Hansen and Lauritzen, 1984; Leao, 1944; Sunami et al., 1989; Zhang et al., 2002). Importantly, these ionic events contribute to neuronal damage in vulnerable brain regions such as the ischemic penumbra (Back et al., 1996; Busch et al., 1996; Mies et al., 1993). Although documented in various injury models, until recently, limited clinical evidence was available to support the presence of these potentially deleterious events in patients with brain injury (Back et al., 2003; Mayevsky et al., 1996; McLachlan and Girvin, 1994).
In this regard, Strong and colleagues (2002) have recently provided the first evidence for spread of depression of cortical activity in the human neocortex. Using electrocortical graphic electrodes placed near foci of damaged cortical tissue, evidence for propagation of depressed electrical activity across the cortex at rates consistent with CSD was presented. Based on previous experimental and recent clinical data, the peri-infarct polarizations in CSD appear to occur in a variety of brain injured patients. In reference to the present discussion of common disease states, these abnormal events may participate in both ischemic and traumatic injury processes and continue to merit consideration as being clinically relevant therapeutic targets (Strong, 2003).
Platelet Aggregation
In clinical stroke, platelet aggregation leading to vascular thrombosis and subsequent embolization are common mechanisms involved in the production of ischemic insult (Millikan, 1965). Abnormal platelet function is seen in patients at risk for stroke and following transient ischemic events. Platelets may accumulate in areas of abnormal flow characteristics, including heart valves and specific cerebral arterial branch points. Platelet events can lead to severe but transient hemodynamic perturbations that may result in mild, moderate, or severe morphologic changes. Transient platelet accumulation can also lead to vascular perturbations, including leakage of BBB and abnormalities in vascular reactivity (Danton et al., 2002; Dietrich et al., 1993). In stroke, the thrombolytic agent, tissue plasminogen activator (tPA), is currently the only therapeutic strategy shown to be beneficial in acute stroke therapy.
In brain trauma, abnormal platelet activation has also been reported and may participate in the pathogenesis (Dietrich et al., 1994; Dietrich et al. 1998a; Maeda et al., 1997). Following moderate F-P injury, platelet accumulation was reported primarily in venules showing normal endothelial ultrastructure (Dietrich et al., 1994). Microthrombosis has also been reported in the peripheral regions of a contusion (Maeda et al., 1997). Studies using Indium-labeled platelets to autoradiographically visualize patterns of platelet accumulation have demonstrated the importance of injury severity on the platelet response. Severe F-P injury leads to the accumulation of labeled platelets in the contusion site and overlying subarachnoid spaces (Dietrich et al., 1996a). Importantly, these areas of prominent platelet accumulation were associated with severe reductions in lCBF. Thus, TBI induced vascular injury resulting in local platelet thrombosis and focal perfusion deficits may lead to cerebral ischemia under some traumatic conditions. In this regard, treatment with the anti-platelet agent, prostacycline, was reported to decrease overall contusion volume and improved cortical perfusion after F-P injury (Bentzer et al., 2003). In TBI patients, prostacyclin treatment improved tissue oxygenation and reduced pericontusion cell membrane degradation (Grande et al., 2000). Strategies targeting abnormal platelet function and cerebral perfusion would therefore benefit both ischemic and traumatic injuries (Naredi et al., 2001).
Cell Death Mechanisms
Both necrotic and apoptotic cell death mechanisms have been implicated in the pathogenesis of ischemic (Graham and Chen, 2001; Kirino, 1982; Liu et al., 1999; Martin et al., 2000; Snider et al., 1999) and traumatic brain injury (Clark et al., 2000; Conti et al., 1998; Eldadah and Faden, 2000; Kotapka et al., 1991; Yakovlev et al., 1997). The brain is vulnerable to oxidative stress due to its high rate of oxidative metabolic activity (Maier and Chan, 2002). Oxidative stress leading to calcium accumulation, mitochondrial dysfunction and the production of reactive oxygen radicals is an important mechanism of cell death following both ischemic (Chan, 2001; Globus et al., 1995b) and traumatic insults (Globus et al., 1995a; Lewen et al., 2001; Mikawa et al., 1996). After cerebral ischemia and trauma, evidence for the generation of reactive oxygen species (ROS) has been demonstrated in a variety of injury models (Kim et al., 2002; Siesjo et al., 1989). In addition, transgenic models where antioxidants including superoxide dismutase (SOD) are overexpressed have been shown to provide neuroprotection from both ischemia and traumatic insults (Chan et al., 1995; Mikawa et al., 1996).
Following global ischemia, morphological and molecular techniques have been used to obtain evidence for apoptotic cell death (Chen et al., 1998; MacManus et al., 1993). Linnik and colleagues (1993) first provided evidence for programmed cell death by showing that inhibition of protein synthesis was neuroprotective in a model of transient global ischemia. Subsequent studies identified DNA laddering and sensitive indicators of DNA damage associated with apoptosis (Heron et al., 1993; Petito et al., 1997). In addition, histopathological verification of injury models supports the hypothesis that at least a subpopulation of neurons die by apoptotic mechanisms. Clearly, the identification of molecular markers of apoptosis has provided additional evidence for this injury mechanism being active in adult models of injury. The exact percentage of cells dying of apoptosis vs. necrosis depends upon several factors including ischemic severity and duration (Du et al., 1996; Gobbel and Chan, 2001). Importantly, while necrotic neuronal damage is commonly observed early after severe ischemic insults, apoptotic cell death may occur with more mild insults and with longer survival periods (Chen et al., 1998; Choi et al., 1996; Du et al., 1996).
In models of cardiac arrest and transient global ischemia, CA1 hippocampal neurons may die by both necrotic and apoptotic mechanisms. Although ultrastructural studies have failed to identify apoptotic bodies in neurons undergoing delayed cell death in the hippocampus, (Colbourne et al., 1999), immunocytochemical studies and molecular approaches have apoptotic neuronal cell death (Katz et al, 2001). After transient or permanent focal ischemia, neurons that are immunoreactive to caspase-3 and other pro-apoptotic proteins have been identified in the penumbral regions (Matsushita et al, 1998).
Apoptotic cell death has also been implicated in the pathogenesis of TBI (Eldadah and Faden, 2000; Kaya et al., 1999; Keane et al., 2001; Raghupathi et al., 2000; Rink et al., 1995). McIntosh and colleagues first obtained evidence for apoptotic cell death in a TBI model by the ultrastructural identification of apoptotic markers (Rink et al., 1995). Subsequent studies have reported caspase-3 activation and TUNEL staining in neurons after cortical impact injury (Clark et al., 2000; Conti et al., 1998) and fluid-percussion (F-P) injury (Keane et al., 2001; Yakovlev et al. 1997). Evidence for apoptotic cell death has also been reported in human tissues obtained by biopsy (Clark et al., 1999). Recent studies have identified caspase-3, 8, and 9-immunoreactive cells after F-P brain injury (Keane et al, 2001). Several members of the IAP (inhibitors of apoptosis) family have been measured following brain injury (Keane et al., 2001). The IAPs may be playing a role in an attempt to provide neuroprotection by regulating cell death and the understanding of IAPs after injury may provide new strategies for treatment (Lotocki and Keane, 2002). Thus, the continued investigation of apoptotic cell injury mechanisms following cerebral ischemia and trauma merits consideration.
Apoptotic cell death after an ischemic or traumatic insult may differ from classical programmed cell death mechanisms active during normal neuronal development (Mattson and Barger, 1995). In fact, studies have emphasized the importance of age when considering apoptotic cell injury. Hu and colleagues (2000) reported a decline in the frequency of TUNEL-positive cells and caspase-3-activated neurons with increasing age following hypoxic/ischemic insults. Evidence for apoptotic cell death requires the use of various immunological and molecular probes to study the complicated mechanisms of apoptotic cell death. Recently, immunological localization of various caspases, including 8 and 9 as well as anti-apoptotic proteins, i.e. XIAP, have been identified following acute asphyxial cardiac arrest (Katz et al., 2001) and traumatic brain injury (Keane et al., 2001).
Several studies in cerebral ischemia and trauma have also implicated calcium-induced calpain-mediated proteolysis in the pathogenesis (Bartus et al., 1995; Buki et al., 1999a; Buki et al., 1999b; Hong et al., 1994; Kampfl et al., 1997; Posmantur et al., 1997; Saatman et al., 1996) of injury. The substrates for calpain effects have been shown to include many cytoskeletal proteins, membrane proteins, and various regulatory and signaling proteins. Uncontrolled proteolysis of these multiple substrates lead to serious consequences, in terms of cell survival and death. Calpain-mediated spectrum proteolysis has been demonstrated in both cell bodies as well as cellular processes in the area of TBI (Newcomb et al., 1997). For example, the importance of post-traumatic cleavage of spectrin by calpain has been implicated in the pathology of DAI (Buki et al., 1999b). Also, this pathway has been implicated in necrotic neuronal death and ischemic and excitotoxic neuronal injury (Yuen and Wang, 1998). Because both caspase and calpain are activated in apoptotic neurons, the importance of these two neuronal death pathways and their interactions are an area of active investigation (Wang, 2000). Thus, in addition to apoptosis, calpain-mediated proteolysis is an important target for therapeutic interventions in both cerebral ischemia and trauma.
Inflammation
Inflammation, a host defense mechanism that is initiated by injury or infection, is a process through which blood-leukocytes (neutrophils, monocytes/macrophages, T-cells) and soluble factors (cytokines, chemokines, complement, lipid by-products) attempt to restore tissue homeostasis (Bethea and Dietrich, 2002). The inflammatory response in the CNS may have various consequences on outcome, depending upon the degree of inflammatory response and when it occurs (Blight, 1985; Benveniste, 1992). Both acute and chronic inflammatory processes have been shown to influence outcome in various experimental models of cerebral ischemia and trauma (Arvin et al., 1996; Clark et al., 1994; del Zoppo et al., 2000; Fan et al., 1995; Fan et al., 1996; Feuerstein et al., 1998; Kochanek and Hallenbeck, 1992). While acute inflammatory events may participate in secondary injury processes, more delayed inflammatory events may be reparative (Bethea and Dietrich, 2002; Kerschensteiner et al., 1999). Thus, the importance of the inflammatory response to functional outcome is an area of active investigation.
Following cerebral ischemia and endothelial damage, activated platelets interact with the endothelium and facilitate its conversion from an anti-inflammatory, anti-thrombotic state to a pro-inflammatory, pro-thrombotic state (del Zoppo et al., 2000). These changes are mediated through the altered expression of released and cell surface signaling molecules (Feuerstein et al., 1998). Altered adhesion molecule expression promotes cellular interactions that are critical for inflammatory and thrombotic processes. Increased endothelial adhesion molecule expression has been observed in various models of ischemic stroke (del Zoppo et al., 2000). Baboon models of transient MCAO have also demonstrated increases in adhesion molecules, including P-selectin and ICAM-1, with sustained levels up to 24 hours after reperfusion (del Zoppo et al., 2000). Leukocytes may contribute to stroke damage by re-occluding vessels after reperfusion or by entering infarcted tissue and exacerbating cell death through cytotoxic interactions (del Zoppo, 1997). Evidence that leukocytes may contribute to infarction has been obtained by many investigators (Barone et al., 1991; Hallenbeck et al., 1986; Zhang et al., 1994).
In models of TBI, similar evidence exists that posttraumatic inflammatory processes contribute to the pathogenesis of this injury (Biagas et al., 1992; Carlos et al., 1997; Schoettle et al., 1990). Neuroprotective strategies, including antibody treatments and therapeutic hypothermia, have been reported to decrease the acute inflammatory response and lead to histopathological protection (Carlos et al., 1997; Chatzipanteli et al., 2000). However, a recent review by Emerich and colleagues (2002) concluded that no study has successfully demonstrated that leukocytes satisfy any criteria for causing cell death or increased infarction due, in part, to a lack of sufficient controls. A better understanding of the timecourse and consequences of neuro-inflammation may aid in therapeutically promoting beneficial and reducing harmful aspects of neuro-inflammation following cerebral ischemia and traumatic brain injury (Leker and Shohami, 2002).
Biochemical Responses
Severe ischemia results in metabolic stress, resulting in the depletion of essential energy metabolites, including ATP and phosphocreatine (Back, 1998; Dirnagl et al., 1999). Anoxic injury can lead to potassium-induced release of excitatory amino acids, the opening of glutamate receptor associated ion channels and the influx of Ca2+ (Choi et al., 1987; Choi and Rothman, 1990; Siesjo and Bengtsson, 1989). With reperfusion, these metabolite levels may normalize, indicating normalization of cellular functions and ion homeostasis. In models of TBI like those of cerebral ischemia, the degree of metabolic stress is dependent upon injury severity (Katayama et al., 1990; Nilsson et al., 1990). Mild or moderate injury is usually not associated with severe metabolic changes such as prolonged decreases in ATP and increase in lactate levels (Lee et al., 1999). However, after more severe TBI, disturbances in ion homeostasis including cellular efflux of potassium, influx of calcium and sodium ions occur (Kawamata et al., 1995). A complex pattern of glucose metabolism has been reported after TBI in animal models (Hovda et al., 1991; Yoshino et al., 1991) as well as in patient studies (Bergsneider et al., 1997). In several investigations, acute increases in glucose metabolism followed by prolonged reductions in glucose utilization have been documented (Passineau et al., 2000; Yoshino et al., 1991). The degree of metabolic stress as well as tissue oxygen delivery appear to be critical factors in determining whether or not cerebral ischemia plays an important role in cell injury mechanisms following TBI.
The neurochemical response to cerebral ischemia (Benveniste et al., 1984; Globus et al., 1988a; Globus et al., 1988b; Globus et al., 1995b) and TBI (Globus et al., 1995a, Katayama et al., 1990; Nilsson et al., 1990; Zauner et al., 1996) also has similar characteristics (Choi, 1988). Following global and focal ischemia, extracellular levels of various neurotransmitters, including glutamate, glycine, and GABA, and dopamine have been reported using microdialysis techniques (Benveniste et al., 1984; Globus et al., 1988a; Globus et al., 1988b). Patterns of neurotransmitter release are dependent upon brain regions being analyzed with or without reperfusion (Globus et al., 1991). Similar neurochemical responses have been documented in some models of TBI (Faden et al., 1989; Globus et al., 1995a; Hayes et al., 1992; Katayama et al., 1990) with extracellular levels of glutamate being elevated after cortical impact injury or F-P trauma. Elevations in glutamate are seen as early as five minutes after experimental trauma, with normalization over a period of several hours (Globus et al., 1995a). More prolonged increased levels of extracellular glutamate have also been reported in human TBI (Bullock et al., 1995; Vespa et al., 1998). Importantly, secondary ischemic brain injury, focal contusions and seizures were most significantly correlated with high levels of glutamate in TBI patients (Bullock et al., 1998).
The role of nitric oxide (NO) in the pathophysiology of cerebral ischemia (Ashwal et al., 1998; Dalkara and Moskowitz, 1994; Iadecola, 1997; Stagliano et al., 1997) and trauma (Wada et al., 1998a; Wada et al., 1998b) has also been documented. In ischemic models, abnormal NO production may be both beneficial and detrimental, depending upon when and where this diffusible neurotransmitter is released (Iadecola, 1997). Studies using knockout mice have demonstrated that NO production by neuronal NOS (nNOS) contributes to damage by producing radicals and lipid peroxidation (Huang et al., 1994). In contrast, NO produced by endothelial NOS (eNOS) can be of benefit by improving vascular dilation and perfusion (Huang et al., 1996). Generally, NO produced by the inducible form of NOS (iNOS) has been reported in models of cerebral ischemia (del Zoppo et al., 2000) and trauma (Wada et al., 1998b) to have detrimental effects on outcome. Increased iNOS activity generally occurs in a delayed fashion after brain ischemia and trauma and is associated with inflammatory processes. Pharmacological techniques targeting iNOS activity, such as treatment with aminoguanidine, have been shown to be neuroprotective in both animal models of cerebral ischemia (Iadecola et al., 1995) and trauma (Wada et al., 1998b). However, studies using iNOS knockout mice have implicated iNOS as also being involved with reparative strategies in the more chronic TBI injury setting (Sinz et al., 1999). Obviously, more research needs to be conducted in this important area to enhance our understanding of what pathophysiological events should be targeted for therapeutic interventions both in the acute and more chronic injury setting (Barone et al., 2000; McIntosh et al., 1998).
Gender
Previous epidemiological reports have noted that premenopausal women have a lower incidence of stroke compared to men. After traumatic brain injury (TBI) women have also shown better recovery than men (Groswasser et al., 1998). These reported differences between males and females could be the result of hormonal influences on outcome. Recent hormone studies targeting cerebral ischemia and CNS trauma have opened new avenues of investigation to promote recovery following injury. Several studies both past and present have demonstrated sex differences in lesion size, neuronal cell loss, and mortality rates after ischemia and trauma (Alkayed et al., 1998a; Bramlett and Dietrich, 2001; Hall et al., 1991; Sadoshima et al., 1988; Zhang et al., 1998). These findings have led to the hypothesis that hormones, in particular progesterone and estrogen, have the ability to provide neuroprotection. This results in either endogenous protection afforded to females or exogenous protection to both males and females after injury with hormone treatment (Roof and Hall, 2000).
Recent investigations in cerebrovascular models of damage have documented the influence of estrogen. In particular, several investigators have examined the efficacy of 17β-estradiol in models of stroke (Alkayed et al., 1998a, 2001; Carswell et al., 2004; Dubal et al., 1998; Fukuda et al., 2000; Hurn et al., 1995; McCullough et al., 2001; Toung et al., 1998; Yang et al., 2000). Hurn and colleagues (1995) first described in a model of global cerebral ischemia the effects of chronic 17β-estradiol treatment. They demonstrated an increase in cerebral blood flow (CBF) and a decrease hyperemia in treated females compared to untreated females and males. In a model of transient middle cerebral artery occlusion (MCAO), Alkayed et al. (1998a) reported a gender difference in intact females versus ovariectomized females and males. Infarct size was reduced in both the cortex (CTX) and caudate putamen (CP) of intact females compared to males and ovariectomized females. CBF was also affected by hormones in this study. Higher measurements were observed in the CP of intact females compared to males and ovariectomized females but not in the CTX. Estrogen treatment in male rats has also been explored after MCAO (Toung et al., 1998). Neuropathological outcome was improved with both acute and chronic 17β-estradiol treatment in male animals including those castrated prior to injury and treatment. Therefore, testosterone may not be involved in the reduction of pathology with estrogen treatment. In contrast to these positive studies with 17β-estradiol treatment, some studies have shown an exacerbation of injury with this treatment paradigm (Carswell et al., 2004; Harukuni et al., 2001). Carswell and colleagues report that low or high dose 17β-estradiol pre-treatment for two weeks before permanent MCAO produces an increase in damaged tissue compared to placebo treatment in ovariectomised rats. This was observed in normotensive Wistar Kyoto rats but not in spontaneously hypertensive stroke-prone rats. These findings emphasize that the mechanisms of this treatment strategy are not fully understood and require further investigation.
While the majority of estrogen studies have demonstrated neuroprotection in various ischemia models, two recent papers studying progesterone in stroke models present contrasting results (Kumon et al., 2000; Murphy et al., 2000). Kumon and colleagues (2000) reported an improvement in neurological deficits and reductions in infarct volume with an administration of progesterone as late as 7 days after transient MCAO. In contrast, Murphy et al. (2000) showed an exacerbation of striatal damage in ovariectomized females given acute or chronic progesterone treatment after transient MCAO. However, these animals did not have circulating estrogen because of the ovariectomy. Therefore, this may have been a contributing factor to the increase in infarct volume considering the positive results from estrogen treatment in this model.
Progesterone has been the predominant reproductive hormone studied in TBI. Roof and colleagues (1992; 1993; 1994) reported in several studies that progesterone treatment after TBI is neuroprotective. Post-treatment with progesterone (Roof et al., 1992) after cortical impact injury (CCI) or pretreatment by increasing endogenous levels with pseudopregnancy (Roof et al., 1992) prior to CCI have demonstrated decreases in cerebral edema compared to untreated controls or normal cycling females and males, respectively. Roof and colleagues (1994) also showed an improvement in cognitive recovery and a reduction in neuronal degeneration within the thalamus in male rats treated with progesterone compared to sham. Furthermore, females have been shown to be less impaired on the Morris water maze task compared to males after entorhinal cortex lesions (Roof et al., 1993). Other investigators have assessed the influence of other reproductive steroids following injury (for review see Roof and Hall, 2000). Emerson et al. (1993) demonstrated an improvement in males pretreated with 17β-estradiol compared to females on measures of biochemical and neurological outcome after TBI. However, these animals were given a fairly high dose of 17β-estradiol compared to the more recent ischemia studies described above. Furthermore, although Attella et al. (1987) have shown an improvement in cognitive outcome when trauma was induced during pseudopregnancy compared to proestrous females, they suggested that this difference was primarily due to reductions in vasopressin because of the increased actions of progesterone during pseudopregnancy. Hormonal levels were not determined in that study and it is unknown what the exact levels of the various gonadal hormones were in that particular experiment. Roof and Hall (2001) reported smaller reductions in cerebral blood flow following TBI with 17β-estradiol treatment in female rats compared to males. Recently, a decrease in contusion volume after TBI was shown in intact females versus males or ovariectomized females (Bramlett and Dietrich, 2001).
Based on the positive results that hormones can provide neuroprotection after cerebral ischemia or brain trauma, both in vitro and in vivo, studies are investigating possible mechanisms of action, some of which have already been discussed, which influence the outcome measurements. These include cerebral blood flow (Alkayed et al., 1998a; Hurn et al., 1995; Roof and Hall, 2001), leukocyte adhesion (Santizo et al, 2000), edema formation (Roof et al., 1992; Roof et al., 1993; Roof et al., 1994), antioxidant production (Ayres et al., 1998; Bruce-Keller et al., 2000; Culmsee et al., 1999; Sawada et al., 1998), inflammation (Hunt et al., 1997; Ray et al., 1997; Salem et al., 2000; St Clair, 1997; Vegeto et al., 2001), excitotoxicity (Singer et al., 1996; Smith et al., 1987; Weaver et al., 1997), apoptosis (Alkayed et al., 1998b; Alkayed et al., 2001), β amyloid (Goodman et al., 1996; Shi et al., 1998), and growth factor regulation (Gollapudi and Oblinger, 1999; Toran-Allerand, 1996). Hormones have the ability to target multiple pathways that are involved in injury. This area of research is only recently being explored and will provide novel treatment strategies for the future in cerebral ischemia and CNS trauma.
Genetic Aspects of Stroke and Head Trauma
The identification of genetic components of stroke and neurodegenerative disease linked to TBI is critical to the understanding, diagnosis, and treatment of these injuries (Carr et al., 2002; Longhi et al., 2001). While risk and environmental factors can be identified and controlled, genetic determinants of disease, in addition to conferring an enhanced vulnerability to other neurodegenerative disorders, pose a challenging problem for investigative studies. Strong evidence for a genetic component in human stroke comes from twin and family studies (Brass et al., 1992; Bak et al., 2002; Liao et al., 1997). In a study of 300,000 subjects, risk factors such as smoking, a history of diabetes, hypertension, and coronary heart disease, did not alter stroke occurrence in people who had stroke events in previous generations of their family (Liao et al., 1997). Conflicting results regarding candidate gene studies for stroke emphasize the complexity of this multifactorial, complex disorder. Genes encoding for endothelial nitric oxide (eNOS), Apolipoprotein E, as well as candidate genes in the area of homeostasis and thrombosis have been proposed (Carter et al., 1998; Kessler et al., 1997; Markus et al., 1997).
Experimentally, various rat strains, such as spontaneously hypertensive stroke-prone rats (SHRSP) that develop spontaneous cerebral infarction or hemorrhage have been developed (Okamoto et al., 1974). Also, genetically engineered mice that typically over-express or have a specific gene deleted (knock-out) have been developed to study the cause-and-effect relationships in specific injury pathways (Chan et al., 1995). These include the use of mice targeting antioxidant processes, adhesion molecules, interleukins, matrix metalloproteases (MMPs), apoptotic genes, and nitric oxide (Carr et al., 2002).
In TBI research, studies using genetically engineered animals have also been conducted (Longhi et al., 2001; Uryu et al., 2003). Of additional importance to this discussion is the use of genetically engineered mice to investigate the underlying relationships between TBI and neurodegenerative diseases (Longhi et al., 2001). For example, patients expressing the E4 allele of apolipoprotein (APOE) have been reported to increase vulnerability and worse outcomes after brain trauma (Teasdale et al., 1997; Lichtman et al., 2000). Knockout APOE mice subjected to weight-drop brain injury were found to have exacerbated memory impairment and motor deficits, when compared to wild-type controls. In mouse studies, 10-fold over-expression of mutant APP leads to the development of plaques at 6 months of age, with TBI exacerbating cognitive impairments and hippocampal CA3 pathology compared with littermates (Smith et al., 1998). In addition to Alzheimer's disease, TBI may also be a risk factor for other neurodegenerative diseases, including Parkinson's disease, characterized by intracellular filament occlusions in neurons (Hubble et al., 1993). Genetically engineered mice that develop neurofilament-rich inclusions have larger lesion cavities, increased apoptotic cell death, and greater cytoskeletal damage, compared with wild-type mice after TBI (Tu et al., 1997). In addition, a recent TBI study by Uryu and colleagues (2003) investigated age-dependent synuclein pathology in mice. Compared to aged wild type mice, α-synuclein knockouts failed to demonstrate α-synuclein immunoreactivity in vulnerable areas following TBI. These research directions should help elucidate the contributions of environmental and genetic factors and vulnerability patterns to injury, as well as susceptibility to neurodegenerative diseases. Thus, the continued use of genetically engineered mice in experimental studies of ischemia and TBI should provide a clearer mechanistic basis for the development of therapies for the prevention and treatment of both injury types.
Neuroprotective Strategies
As emphasized in the previous sections, ischemic and traumatic insults share many of the same injury processes that are considered important in cell death mechanisms (Fig. 2). Although the location and severity of the primary insult are important factors determining the impact of individual injury processes, excitotoxicity, calcium-mediated events, free radicals, mitochondrial damage, inflammation, and apoptosis are commonly discussed as possible targets for therapeutic interventions. Indeed, much progress has been made developing novel pharmaceutical therapies to treat these injuries including glutamate receptor antagonists, calcium channel blockers, radical scavengers, as well as anti-inflammatory and anti-apoptotic agents (Vink and Nimmo, 2002; Chauhan et al., 2003; Fisher, 1999). Despite the identification of these processes and the development of novel pharmacological agents, clinical trials in stroke and TBI have met with little success (Doppenberg and Bullock, 1997; Gladstone et al., 2002). Various reasons are given for the failures of clinical trials based on solid preclinical data. These include therapeutic window considerations, drug dosing and delivery, proper targets for drug therapy, and flaws in the design of clinical studies (Grotta, 2002; Lees, 2002).
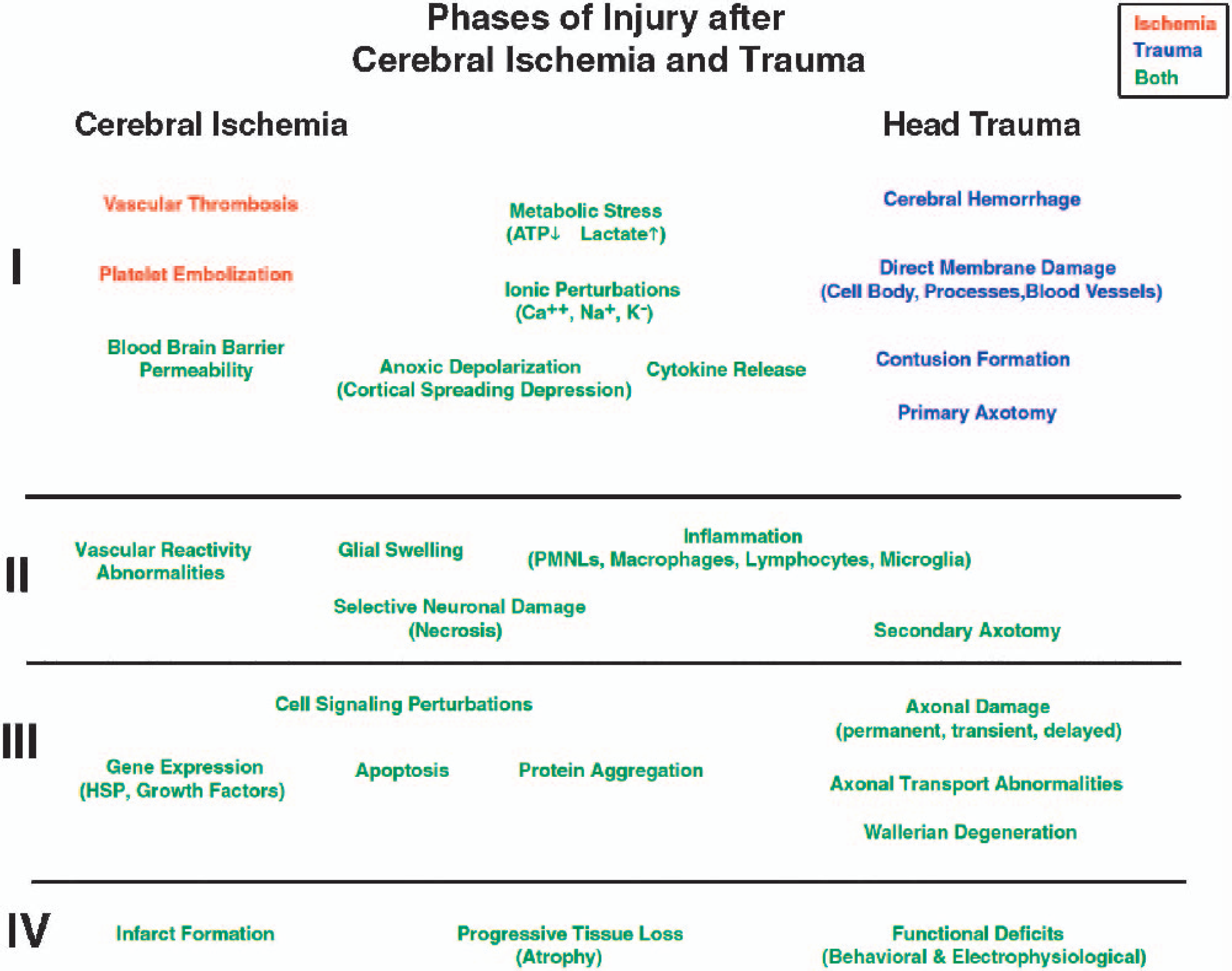
Flow chart summarizing various pathophysiological events involved in cerebral ischemic and traumatic insults. Four phases of injury are indicated that divide the spectrum of processes into immediate, acute, subacute, and ultimate outcome. As indicated, similar pathomechanisms are considered to participate in the structural and functional abnormalities associated with cerebral ischemia and trauma. Degrees of involvement as well as temporal profiles of these events are important in determining whether they play a dominant role in cell death and injury. As in any type of injury, injury severity and the presence of secondary injury mechanisms impact on the robustness of these pathological events. It should be emphasized that there are generally more similarities in terms of pathogenesis between cerebral ischemia and trauma than there are differences.
The relevance of animal models of brain injury to the clinical condition is also frequently discussed (Faden, 2002; Chauhan et al., 2003). Reproducible animal models of cerebral ischemia, stroke, and TBI have been developed to conduct hypothesis-driven research that is pertinent to the clinical condition. Thus, the importance of specific pathomechanisms underlying neural cell death is critically investigated. Likewise, these models are useful in testing novel therapeutic agents and may provide data that can be translated into the clinical arena. However, before such clinical studies are conducted, preclinical data must be critically evaluated in terms of clinical practice and patient treatment. This includes clarification of what specific patient populations relate most closely to the animal model on which the preclinical data was based. Also, the dosing strategy used in the preclinical data must be critically followed in the clinical investigations, including how soon to start treatment as well as duration of treatment. Importantly, clinically relevant outcome measures have to be emphasized in the preclinical experiments. This is an important concern since many neuroprotective strategies have reported cell counts or infarct volumes at relatively short survival periods, while limited functional or behavioral data to support improved outcome is provided. Also, limitations of the therapeutic window have frequently not been incorporated into the design of the clinical trial (Table 2).
Recently, the first demonstration of a neuroprotectant translated from the laboratory to man has been published. Mild therapeutic hypothermia following successful cardiac resuscitation was reported to improve outcome in three recent independent clinical studies (Hypothermia After Cardiac Arrest, HACA Study Group, 2002; Bernard et al., 2002; Hachimi-Idrissi et al., 2001). These clinical studies were the outgrowth of a large amount of experimental data that demonstrated hypothermic protection in models of cardiac arrest, cerebral ischemia, and traumatic brain injury (Busto et al., 1989; Clifton et al., 1991; Dietrich et al., 1994; Koizumi and Povlishock, 1998; Leonov et al., 1990; Safar et al., 1996). An important advantage of hypothermic therapy is that it targets most of the pathomechanisms currently felt to participate in the pathogenesis of ischemic and traumatic brain injury (Dietrich et al., 1996b). This is an important concept, especially when discussing necrotic vs. apoptotic cell death mechanisms, since modulation of one death pathway may lead to enhanced cell death through other pathways (Pohl et al., 1999; Glazner et al., 2000). Thus, a lesson from preclinical and clinical data regarding neuroprotective strategies is that multiple injury mechanisms should be targeted for successful improvements in outcome. Although some clinical evidence for benefit from therapeutic hypothermia and other ischemic and traumatic brain injuries has been obtained (Marion et al., 1997; Clifton et al., 1993; Polderman et al., 2002; Schwab et al., 1998), more experimental data are required to answer major scientific questions regarding therapeutic hypothermia, including therapeutic window, level and duration of hypothermia, rewarming phase, and gender considerations.
Epidemiology of Stroke and Traumatic Brain Injury (USA statistics)

Concluding Remarks
Ischemic and traumatic brain injury result from the interaction of complex pathophysiological processes that are activated by ischemic or traumatic events (Fig. 2). In both injury settings, areas of risk are present that may be salvaged by specific treatment strategies. Although each of these pathophysiological mechanisms are targets for therapeutic interventions, the complex interaction of these pathomechanisms may make it difficult for targeted pharmacological agents to protect the brain long-term and improve behavioral outcome. Also tissue responses to different injury severities and types (i.e. ischemic vs traumatic) may differ and therefore complicate treatment strategies not tailored to individual cases. We are presently at a crossroad regarding translating preclinical data to specific patient populations. Thus, continued investigations of how ischemia and traumatic pathomechanisms interact and what possible combination therapies should be advanced continue to be important in this research arena. A resurgence of therapeutic hypothermia has occurred over the last several years and has recently provided the first clinical evidence for a neuroprotective benefit in cardiac arrest patients. Continuing research along this line, in combination with pharmacological interventions may provide better treatments for these large populations of individuals.
Footnotes
Acknowledgements
The authors would like to thank Charlaine Rowlette and Georgina Escobar for editorial assistance.