
Abstract
Preservation of endothelial functions with low-dose nitric oxide (NO) and inhibition of excessive production of NO from inducible NO synthase (iNOS) is a potential therapeutic approach for acute stroke. Based on this hypothesis, an NO modulator, S-nitrosoglutathione (GSNO) was used, which provided neuroprotection in a rat model of focal cerebral ischemia. Administration of GSNO after the onset of ischemia reduced infarction and improved cerebral blood flow. To understand the mechanism of protection, the involvement of inflammation in ischemic brain injury was examined. Treatment with GSNO reduced the expression of tumor necrosis factor-α, interleukin-1β, and iNOS; inhibited the activation of microglia/macrophage (ED1, CD11-b); and downregulated the expression of leukocyte function-associated antigen-1 and intercellular adhesion molecule-1 in the ischemic brain. The number of apoptotic cells (including neurons) and the activity of caspase-3 were also decreased after GSNO treatment. Further, the antiinflammatory effect of GSNO on expression of iNOS and activation of NF-κB machinery in rat primary astrocytes and in the murine microglial cell line BV2 was tested. Cytokine-mediated expression of iNOS and activation of NF-κB were inhibited by GSNO treatment. That GSNO protects the brain against ischemia/reperfusion injury by modulating NO systems, resulting in a reduction in inflammation and neuronal cell death was documented by the results.
Introduction
Stroke is the third leading cause of death in the US and is associated with serious long-term physical and cognitive disabilities, especially in elderly patients (Feigin et al, 2003; Sarti et al, 2000). Nearly 85% of strokes are ischemic, and under these conditions neurons in the ischemic center (core) quickly undergo necrosis because of ATP depletion and ionic failure (Barber et al, 2003). The core is surrounded by a ring-like penumbra (Leker and Shohami, 2002), which during a stroke is electrically silent but still has significant blood flow. Delayed oxygenation/reperfusion and apoptosis contribute to the vulnerability of this area to inflammation and free-radical attack. The focal cerebral ischemia model using the middle cerebral artery occlusion (MCAO) technique is widely accepted as being closer to the clinical manifestations of stroke in humans (Ginsberg and Busto, 1989). To date there are no clinically effective therapies for brain damage because of ischemic stroke. The attenuation of neuronal death by drugs that protect endothelial functions and downregulate the inflammatory process may provide effective therapy against ischemia/reperfusion (I/R) injury.
Nitrosative stress has been implicated in cerebral ischemia (Bolanos and Almeida, 1999; Suzuki et al, 2002; Zhu et al, 2002). The major reactive nitrogen species, nitric oxide (NO), is a multimodal molecule with complex actions in stroke (Willmot and Bath, 2003). Endothelial NO synthase (eNOS)-derived NO modulates endothelium integrity and invokes antiinflammatory properties as well as antiapoptotic activity (Li and Forstermann, 2000). It also exerts its beneficial effects by maintaining cerebral blood flow (CBF), inhibiting platelet aggregation and reducing leukocyte adhesion (Cirino et al, 2003). In fact, eNOS-deficient transgenic mice show larger infarct volumes compared with controls after MCAO (Huang et al, 1996). At an early time-point of ischemic insult, NO protects the brain by increasing CBF (Zhang and Iadecola, 1993; Zhang et al, 1994) and prevents apoptosis partly by inhibiting active caspases via S-nitrosylation (Mannick et al, 1999) and by inhibiting p50 subunit binding of NF-κB to DNA via S-nitrosylation (Marshall and Stamler, 2001). In contrast to eNOS, the activities of neuronal NO synthase (nNOS) and inducible NO synthase (iNOS) are broadly deleterious and their inhibition as well as gene deficiencies are neuroprotective in stroke (Huang et al, 1994; Zhao et al, 2000). Transient ischemic periods and the expression of proinflammatory cytokines, including tumor necrosis factor-α, (TNF-α), induce the expression of iNOS in various populations of cells including macrophages, neutrophils, and reactive microglia (Iadecola, 1997; Iadecola et al, 1995; Zhao et al, 2003; Zhu et al, 2002). Nitric oxide derived from iNOS is involved in the induction of apoptosis and cellular damage (Burney et al, 1999; Chung et al, 2001; Kim et al, 1999; Stewart and Heales, 2003). These multifactorial protective and deleterious mechanisms of NO suggest that intervention with NO modulation may be most effective when delivered at the appropriate time and in an optimum concentration either alone or in a suitable combination with neuroprotective agents.
Attention has been focused recently on the role of S-nitrosylation (Foster et al, 2003) by S-nitrosothiol (RSNO) including S-nitrosoglutathione (GSNO) in storage and transport of NO (Hogg, 2002; Janero, 2000; Richardson, 2002). S-Nitrosoglutathione is a physiological metabolite of glutathione (GSH) and NO (Megson, 2000; Schrammel et al, 2003), and is involved in several pharmacological activities (Chiueh, 2002) and signaling (Choi et al, 2000; Gu et al, 2002; Lane et al, 2001; Stamler et al, 1997). It is present in micromolar concentrations in the rat brain (Kluge et al, 1997) and is severalfold more potent than GSH against oxidative stress caused by peroxynitrite (ONOO−) (Rauhala et al, 1998). Unlike other classes of NO donors, GSNO is a stable compound and does not decompose spontaneously, and requires additional agents or enzymes including GSNO reductase or thioredoxin system (Steffen et al, 2001; Zeng et al, 2001) for its metabolism. It reduces the frequency of embolic signals (Kaposzta et al, 2002a, 2002b; Molloy et al, 1998) and can reverse acute vasoconstriction, preventing ischemic brain injury after subarachnoid hemorrhage (Sehba et al, 1999). Administration of GSNO has been shown to suppress iNOS induction and enhance eNOS expression in pedicle vessels, resulting in blood perfusion and a higher flap survival after I/R injury (Kuo et al, 2004a, 2004b).
The selectivity and regulation among the NO synthase (NOS) isoforms has been recognized as highly critical for therapeutic purposes. Therefore, the focus of this study is the modulation of inflammation and integrity of endothelium by GSNO to achieve the protection of the brain in a rat model of experimental stroke. We found that treatment of rats with GSNO after the onset of cerebral ischemia reduced brain infarction, improved neurological deficit, increased CBF, limited apoptotic cell death, reduced endothelial dysfunction, blocked infiltration of macrophages, downregulated expression of cytokines, and inhibited induction of iNOS. An in vitro study of GSNO treatment of astrocytes and BV2 cells showing inhibition of iNOS induction and NF-κB activation documents the role of GSNO in downregulation of inflammation. Collectively, these studies establish that GSNO protects the brain by improving CBF and reducing inflammation in a rat model of experimental stroke.
Materials and methods
S-Nitrosoglutathione was purchased from World Precision Instruments (Sarasota, FL, USA). Labeled 3H-glutathione (glycine −2-3H) of specific activity 20–50 Ci/mmol was obtained from American Radiolabeled Inc. (St Louis, MO, USA). DMEM (4.5 g glucose/L), RPMI 1640 medium, fetal bovine serum, and Hanks balanced salt solution were from Life Technologies (Grand Island, NY, USA). All other chemicals and reagents used were purchased from Sigma (St Louis, MO, USA) unless stated otherwise.
Experimental Animals
A total of 90 male Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 250 to 275 g were used in this study. All animals received humane care in compliance with the Medical University of South Carolina's guidance and the National Research Council's criteria for humane care as outlined in Guide for the Care and Use of Laboratory Animals.
Experimental Design and Administration of Drugs
All animal procedures were approved by the Medical University of South Carolina Animal Review Committee and were in accordance with the guidelines for animal use published by the National Institute of Health. The animals were divided into three groups: (i) control (sham-operated) group (Sham, n=25); (ii) ischemia (20 minutes) and reperfusion (24 hours) group (Vehicle, n=25); and (iii) GSNO treatment group (GSNO, n=40). In the treatment group, GSNO (1 mg/kg body weight) solution in saline (∼250 μL) was slowly infused by jugular vein cannulation at the time of reperfusion. The rats in the ischemia (vehicle) and control (sham) groups were administered the same volume of normal saline instead of GSNO.
Focal Cerebral Ischemia Model
Rats were fasted overnight but allowed free access to water before the experiments. Rats were anesthetized with an intraperitoneal injection of xylazine (10 mg/kg body weight) and an intramuscular injection of ketamine hydrochloride (100 mg/kg). A rectal temperature probe was introduced, and a heating pad maintained the body temperature at 37°C±0.5°C. Right middle cerebral artery (MCA) was occluded as described by Longa et al (1989). Briefly, under a dissecting microscope, the right common carotid artery was first exposed and the occipital branch of the external carotid artery (ECA) was coagulated by electrocautery. A 4-0 monofilament nylon (Harvard Apparatus, MA, USA) coated with poly-
Measurement of Physiological Variables
The physiological variables were measured before, during MCAO and at 30 mins of reperfusion. Regional CBF was monitored using laser Doppler flow meter (Perimed Inc., Sweden and Oxford Optronix Ltd, Oxford, UK). Animals under anesthesia were placed in a stereotaxic apparatus and a needle probe was placed at bregma with the following coordinates (anterioposterior, −1.0 mm; lateral, −4.0 mm). Measurements were recorded continuously before and during MCAO, and up to 3 hours after reperfusion with the probe in place continuously. However, animals were reanesthetized once within the recording period. Body temperature was monitored by a rectal probe and maintained at 37°C±0.5°C by a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). Cranial temperature and mean arterial blood pressure were measured by HSE Plugsys TAM-D and TCAM (Harvard Apparatus), respectively. Blood gases and blood pH were measured by pH/blood gas analyzer iSTAT (Heska, Fort Collins, CO, USA). Heart rate was monitored by Standard Needle Electrodes (Model No. MLA1203, AD Instruments, Colorado Springs, CO, USA) connected to a bioamplifier (ML-132, AD Instruments) linked to a Powerlab system (PowerLab/8 SP—AD Instruments).
Evaluation of Ischemic Infarct and Neurological Score
A 2,3,5-triphenyltetrazolium chloride (TTC) staining technique was used for the evaluation of ischemic infarct followed by image acquisition by computer. Briefly, after an overdose of pentobarbital, the rats were killed by decapitation after 24 hours of reperfusion. The brains were quickly removed and placed in ice-cold saline for 5 mins. Six serial sections from each brain were cut at 2-mm intervals from the frontal pole by Rodent Brain Matrix (ASI Instrument Inc., Warren, MI, USA). The sections were incubated in 2% TTC (Sigma) and dissolved in saline for 15 mins at 37°C. The stained brain sections were stored in 10% formalin and refrigerated at 4°C for further processing and storage. Coronal sections (2 mm) were placed on a flat-bed color scanner (HP scan jet 5400 C) connected to a computer. The infarct area, outlined in white, was acquired by image-analysis software (Photoshop 4.0 Adobe System) and measured by NIH image software. Neurological evaluation was performed by an observer masked from the identity of the group. Neurological deficits were assessed at 30 mins, 1 hour, 24 hours, and 72 hours after reperfusion (before killing) and scored as follows: 0, no observable neurological deficit (normal); 1, failure to extend left forepaw on lifting the whole body by tail (mild); 2, circling to the contralateral side (moderate); 3, leaning to the contralateral side at rest or no spontaneous motor activity (severe). The animals not showing paralysis at 1 hour time-points after MCAO were excluded from the study as described in our earlier publication (Sekhon et al, 2003). We judged that in rats with no paralysis, the reduction of blood flow was not sufficient to produce infarction of adequate size, hence those animals likely had no significant neurological deficit.
Measurement of Membrane Permeability of S-Nitrosoglutathione
To examine the ability of GSNO to cross the blood–brain barrier (BBB), an in vitro model of BBB, consisting of a coculture of mouse brain capillary endothelial cells in transwell (0.4 μmol/L pore size) and rat astrocytes (6-well plate), was used as described earlier (Dehouck et al, 1990). 3H-labeled GSNO (1 μCi of specific activity 20 to 50 Ci/mmol), prepared in our lab from 3H-labeled GSH on a microscale following the method described by Al-Sa'doni et al (2000), was added to the culture-inserts medium in contact with endothelial cells at a dose of 100 μmol/L (nonlabeled GSNO) at 37°C. The permeability of GSNO was measured by counting in scintillation liquid in the basal compartment (astrocytes compartment) at 10, 20, 40, and 60 mins. The composition of radioactive compounds in the medium of the lower compartment was analyzed by thin layer chromatography as described for GSH derivatives (Anderson et al, 1985).
Measurement of Caspase-3 Activity in Rat Brain
The caspase-3 enzyme activity assay was performed as described earlier (Haq et al, 2003). Briefly, the reaction mixture contained 50 μg of cytosolic protein prepared from rat brain homogenates and 500 μmol/L Ac-DEVD-AMC (caspase-3 substrate II, fluorogenic; Calbiochem Cat# 235425) in 900 μL of buffer B (100 mmol/L HEPES, pH 7.4; 20% glycerol; and 2 mmol/L dithiothreitol). The enzyme reaction was initiated by adding the substrate at 37°C. The caspase-3-like activity was measured using a spectrofluorometer at an excitation wavelength of 380 nm and an emission wavelength of 460 nm for detecting the shift in fluorescence on cleavage of AMC fluoropore.
Cell Culture
Primary rat astrocytes were prepared from 1 to 3-day-old postnatal Sprague–Dawley rat pups and maintained in DMEM (4.5 g glucose/L) with 10% fetal bovine serum (FBS) and antibiotics. Based on glial fibrillary acidic protein (GFAP) positive immunostaining, astrocytes were determined to be greater than 95% pure. BV2, a murine microglia cell line, was provided by Dr Michael McKinney of Mayo Clinic (Jacksonville, FL, USA) and maintained in DMEM (4.5 g glucose/L) supplemented with 10% FBS and antibiotics. Cytotoxic effects of treatments were determined by measuring the metabolic activity of cells with 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and lactate dehydrogenase (LDH) release assay.
Western Blot Analysis
Fresh or frozen brain tissue or cultured cells were used for the Western blot analysis. Tissues (brain) were homogenized in an ice-cold buffer containing 20 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L Na pyrophosphate, 1 mmol/L vanadate and 1 g leupeptin. The homogenates (160 μg protein each) were treated with cold acetone, vortexed, and stored at −70°C for 4 hours. The samples were centrifuged at 11,500g for 10 mins to precipitate the protein. The dry pellets were then boiled for 5 mins in loading buffer. Equal amounts 40 μg/lane of protein were subjected to SDS-PAGE and transferred to nitrocellulose (Amersham Biosciences, Piscataway, NJ, USA). Samples from the cultured cells for immunoblot were prepared as described earlier (Giri et al, 2002). Briefly, the cells were harvested and then lysed in ice-cold lysis-buffer (50 mmol/L Tris-HCl, pH 7.4, containing 50 mmol/L NaCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 10% glycerol, and protease inhibitor cocktail). The samples were centrifuged at 8,000g for 10 mins. Supernatant (50 μg protein/lane) was analyzed by SDS-PAGE and blotted to nitrocellulose (Amersham). Blots were blocked for 1 hour in 5% nonfat dry milk-TBS-0.1% Tween 20, washed, and then incubated overnight with iNOS antibody (1:1,000) in 5% BSA-TBS-0.1% Tween 20 at 4°C, and then washed. This was followed by incubation for 1 hour with rabbit secondary peroxidase conjugated antibody (1:5,000, Sigma). Immunoreactivity was detected using the enhanced chemiluminescence detection method according to the manufacturer's instructions (Amersham Pharmacia Biotech), and subsequent exposure of the membrane to X-ray film. Densitometric analysis was performed on a Bio-Rad densitometer (Model GS-670, imaging densitometer). Protein concentration was determined using a commercially available protein assay dye (Bradford reagent) from Bio-Rad, Hercules, CA, USA.
Transcriptional Assays
Primary astrocytes or microglial cell line (BV2) were transiently transfected with NF-κB- or iNOS-luciferase reporter gene (1.5 μg/well) with β-galactosidase by lipofectamine-2000 (Invitrogen, Baltimore, MD, USA) for astrocytes and lipofectamine-Plus (Invitrogen) for BV2 cells, according to the manufacturer's instructions, in 12-well plates as described (Giri et al, 2004). For cotransfection studies, cells were transfected with reporter in the presence or absence of p65 and/or p50 expression vector. Total DNA (3 μg/well) was kept constant and pcDNA3 was used to normalize all groups to equal amounts of DNA. Luciferase activity was determined using a luciferase kit (Promega, Madison, WI, USA).
Apoptosis by TUNEL Assay
Apoptosis was detected by TUNEL (TdT-mediated dUTP nick end labeling) assay. Briefly, sections were deparaffinized with xylene and rehydrated through three changes of graded alcohol and incubated in phosphate buffer saline (PBS) for 15 mins at room temperature and then in 20 μg/mL proteinase K for 15 mins at room temperature. The ApopTag plus peroxidase kit (Chemicon, Temecula, CA, USA) was used for detection of apoptosis. Endogenous peroxidase activity in the brain sections was blocked by incubation with 3% H2O2 in PBS for 5 mins followed by incubation for 10 secs with equilibration buffer. The sections were incubated for 60 mins at 37°C with terminal deoxynucleotidyl transferase enzyme (TdT) in reaction buffer. The reaction was terminated by incubation with stop/wash buffer at room temperature. Sections were then incubated with peroxidase-conjugated antidigoxigenin antibody (affinity purified sheep polyclonal) for 30 mins at room temperature and the reaction was developed with diaminobenzidine (DAB) substrate for 4 mins at room temperature. Sections were counterstained with methyl green for 30 secs and mounted in Permount (Fisher Scientific, Fair Lawn, NJ, USA). Double labeling was used to identify TUNEL-positive cells by using the ApopTag fluorescein in situ apoptosis detection kit (Chemican) followed by incubation with neuronal specific enolase antibody (rabbit polyclonal 1:100, Chemicon), and visualized with anti-rabbit Texas Red (1:100, Vector Labs, Burlington, CA, USA).
Immunohistochemistry
Expressions of cytokines (TNF-α and interleukin-1β (IL-1β)), microglia/macrophage markers (ED1 and CD11-b), intercellular adhesion molecule (ICAM-1), leukocyte function-associated antigen (LFA-1), and iNOS were detected by immunohistochemical analysis using specific antibodies. Paraffin-embedded sections from the formalin-fixed brain tissues were stained for TNF-α, IL-1β, iNOS, ED1, CD11-b, LFA-1, and ICAM-1. In brief, the brain tissue sections were deparaffinized, sequentially rehydrated in graded alcohol, and then immersed in PBS (pH 7.4). Slides were then microwaved for 2 mins in antigen unmasking solution (Vector Labs), cooled, and washed 3 times for 2 mins in PBS. Sections were immersed for 25 mins in 3% hydrogen peroxide in distilled water to eliminate endogenous peroxidase activity, then blocked in immunohistochemical grade 1% bovine serum albumin in PBS for 1 hour and diluted goat serum for 30 mins to reduce nonspecific staining. Sections were incubated overnight with mouse monoclonal TNF-α antibody (Bio Source Inc., Camarillo, CA, USA); goat polyclonal IL-1β antibody (Santa Cruz Biotechnology, CA, USA); rabbit polyclonal iNOS antibody (Transduction Labs, CA, USA); ED1 (mouse monoclonal from Bio Source); mouse monoclonal CD11-b antibody (Bio Source Inc.); mouse monoclonal LFA-1 antibody (Phar Mingen, San Diego, CA, USA); and mouse monoclonal ICAM-1 antibody (Phar Mingen). They were then rinsed 3 times for 5 mins in PBS containing 0.1% Tween-20. Secondary biotinylated anti-rabbit or anti-goat or anti-mouse IgG was incubated on slides for 30 mins followed by an avidin–biotin HRP complex (Vectastain ABC-Elite Kit, Vector Labs) with DAB as substrate. The slides were then dehydrated through a graded series of alcohol, mounted in Permount and cover-slipped. All the sections were analyzed using an Olympus microscope and images were captured using a digital video camera controlled by Adobe Photoshop (Adobe Systems, CA, USA).
For immunofluorescent double-labeling, sections were incubated first with anti-iNOS (1:10), followed by macrophage marker ED1 (1:100, mouse monoclonal from Bio Source) or astrocyte marker GFAP (1:100, mouse monoclonal, clone 6F-01 from Accurate, Westbury, NY, USA). Anti-iNOS was visualized using Texas Red conjugated anti-rabbit IgG (1:100, Vector Labs) and ED1 or GFAP using FITC conjugated anti-mouse IgG (1:100, Vector Labs). Rabbit or mouse polyclonal IgG was used as control primary antibody. Sections were also incubated with FITC or Texas Red conjugated IgG without the primary antibody as a negative control. After washing, slides were air-dried and mounted with aqueous mounting media (Vector Labs). Slides were examined for immunofluorescence using an Olympus microscope equipped for epifluorescence with dual wavelength filter and Adobe Photoshop software. Individual color channels (red or green) were separated with Adobe Photoshop software.
Statistical Analysis
All values are expressed as mean±s.d. Comparisons among means of groups were made with a two-tailed Student's t-test for unpaired variables. The Mann–Whitney test was used for comparing two scores (groups). Differences among groups were considered significant when P<0.05.
Results
Effect of GSNO on Reduction of Infarction and on Recovery of Neurological score
All the ischemic animals (vehicle) were subjected to MCAO (20 mins), followed by reperfusion (24 hours). Treated ischemic animals (GSNO) were administered GSNO (1 mg/kg body weight) once just after the onset of ischemia in this study. 2,3,5-Triphenyltetrazolium chloride-stained representative sections (numbers 3 and 4 of a total of 6 sections arranged from cranial to caudal regions) from vehicle and GSNO ischemic brains are presented in Figure 1A. Treatment with GSNO, compared with vehicle, reduced the infarct as measured by the disappearance of significant areas of the brain's white region. The infarct volume (Figure 1B), which was based on all 6 slices from 7 different animals, was found to decrease significantly from 623±21.05 (vehicle) to 220±12.13 (GSNO). There was a significant difference in the neurological scores between vehicle and GSNO animals (Figure 1C). Twenty minutes of MCA occlusion and 24 hours of reperfusion led to a neurological score of 2.71±0.48. S-Nitrosoglutathione animals had an average neurological score of 1.28±0.48. The selection of dose of GSNO (1 mg/kg body weight) is based on maximal brain protection (infarct volume). This dose had no effect on resting arterial blood pressure, intracranial pressure, and other physiologic parameters (Table 1). A transient decrease in blood pressure was observed at 2 mg/kg and higher doses, but not at 1 mg/kg and lower doses. Furthermore, administration of GSNO after the onset of ischemia was associated with significantly increased CBF compared with vehicle (Figure 1D; monitored up to 3 hours after reperfusion). We also monitored the survival of animals up to 7 days after ischemia both in GSNO-treated and untreated groups. While all the GSNO-treated animals (n=7) survived up to 7 days and remained healthy and free from neurological deficits, the untreated animals (n=7) either died or were killed because of poor clinical conditions within 3 days after ischemia.
Physiologic parameters
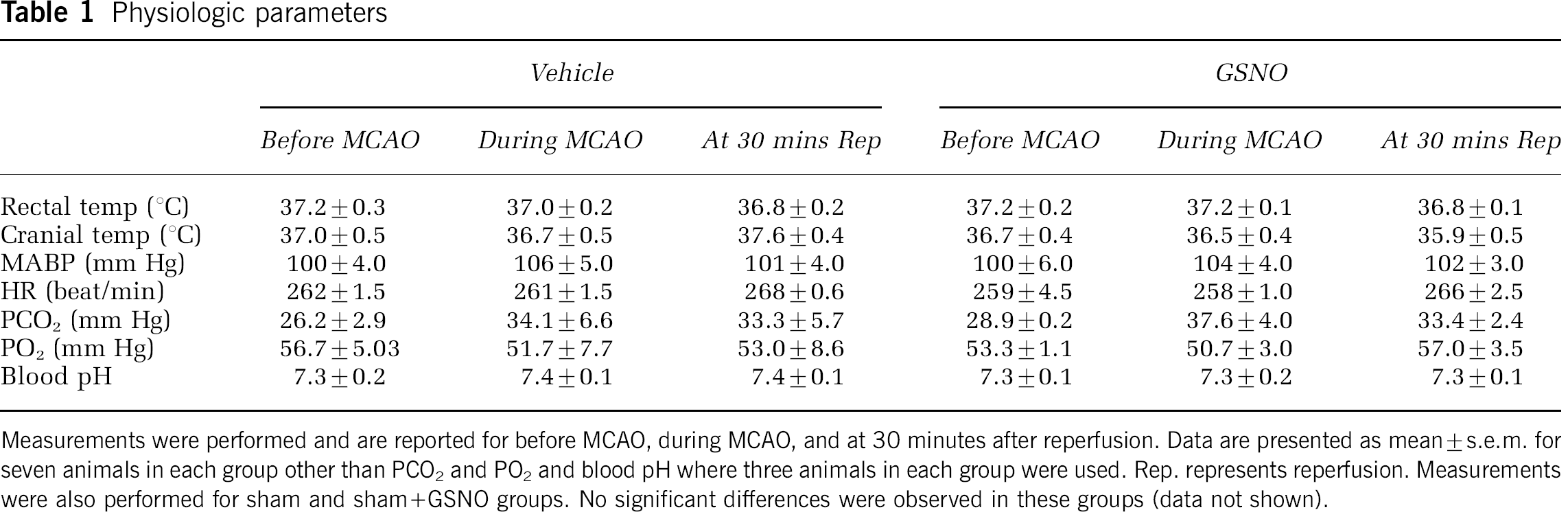
Measurements were performed and are reported for before MCAO, during MCAO, and at 30 minutes after reperfusion. Data are presented as mean±s.e.m. for seven animals in each group other than PCO2 and PO2 and blood pH where three animals in each group were used. Rep. represents reperfusion. Measurements were also performed for sham and sham+GSNO groups. No significant differences were observed in these groups (data not shown).
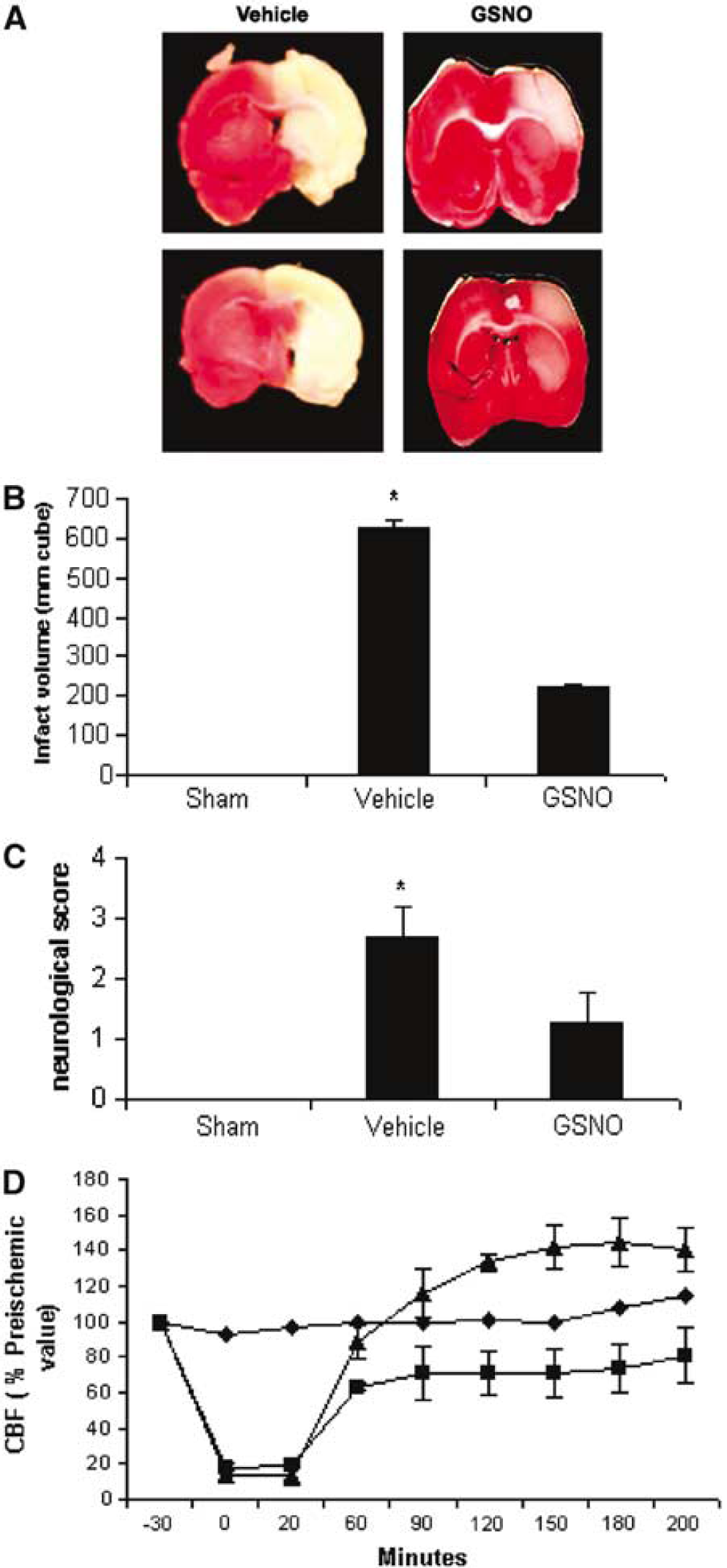
Two representative TTC-stained brain sections (# 3 and # 4 out of the six consecutive sections from cranial to caudate region) corresponding to the largest infarct area from each group (
GSNO Crosses the Endothelial Cells Slowly
Metabolism of GSNO has been described as a complex phenomenon especially in vivo as it is dependent on several factors, both enzymatic and nonenzymatic (Chiueh, 2002; Eiserich et al, 1998; Singh et al, 1996; Zeng et al, 2001). We used a cell culture model of BBB consisting of a coculture of mouse brain capillary endothelial cells (Omidi et al, 2003) and rat astrocytes in vitro (Dehouck et al, 1990; Deplanque et al, 2003) to assess the membrane permeability of GSNO. The permeability (25.2% of total radioactivity, at 60 mins) shows GSNO as a weak permeant molecule, indicating that GSNO may be crossing the BBB at a slow rate. Furthermore, we analyzed the composition of radioactive compounds present in the lower compartment and found that total labeled intact GSNO was only 8.4% of total radioactivity present after 60 mins in the lower compartment. This result does not indicate how much intact GSNO crossed endothelial cells without being metabolized. However, it indicates that some GSNO, as such, crossed the endothelial membrane.
Effect of GSNO on the Induction of Proinflammatory Mediators (Cytokines and iNOS)
The cytokines, TNF-α (Figures 2A–2C) and IL-1β (Figures 2D–2F) were highly expressed after 24 hours of reperfusion in the ipsilateral hemisphere (mainly penumbral cortex) of the ischemic brain, and this expression was significantly reduced in GSNO-treated animals. The expression of iNOS (Figures 2G–2I), detected by immunostaining after 24 hours reperfusion, followed a similar pattern to TNF-α and IL-1β, and treatment with GSNO reduced drastically the number of detectable iNOS-positive cells. The sham-operated group had no iNOS-positive cells present in any examined region of the brain. The presence of expression of iNOS in the ipsilateral hemisphere and its absence in the GSNO-treated ipsilateral hemisphere were also supported by Western blot analysis as shown in Figures 3A and 3B. The expression of iNOS was found mainly in activated microglia/macrophages as the expression of iNOS colocalized with the expression of ED-1 (Figure 4). Inducible NO synthase-expressing macrophages/microglia were present prominently in the cortex region (Figure 4A) as well as in vessels (Figure 4B). The expression of iNOS also colocalized with some of the GFAP expression, a marker for activated astrocytes, thereby indicating the participation of activated astrocytes in iNOS-induced stress (Figure 4C).
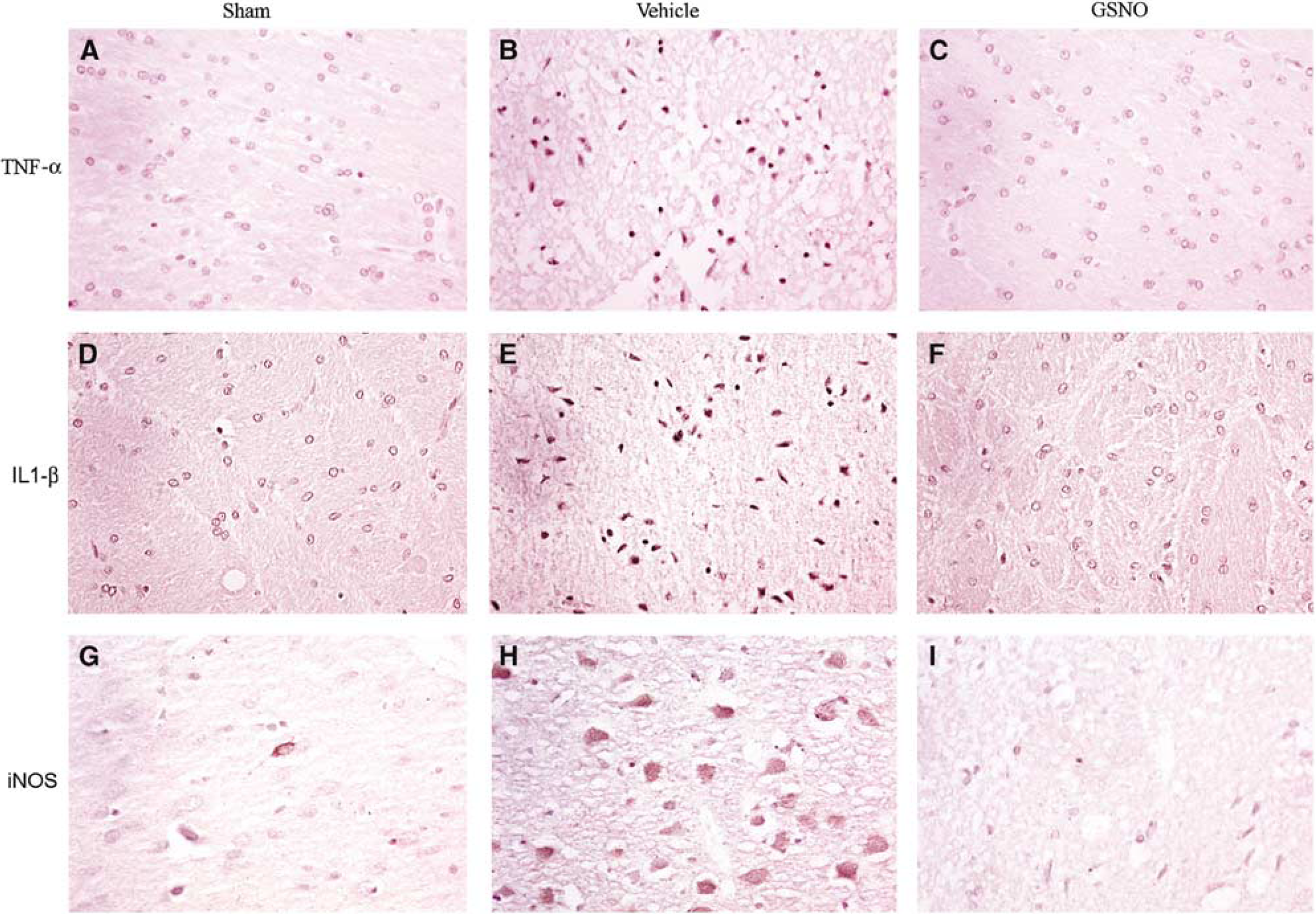
Photomicrographs of immunohistochemistry of rat brain at 24 hours of reperfusion after 20 minutes middle cerebral artery occlusion (MCAO). Enhanced reaction (brown DAB staining) showed higher expression of TNF-α (

Western blot of rat brain at 24 hours of reperfusion after 20 minutes middle cerebral artery occlusion (MCAO). Representative Western blot from three different sets of experiments showed expression of inducible nitric oxide synthase (iNOS) present in ipsilateral hemisphere of untreated animals (vehicle). Sham-operated animals (sham) and treated ischemic animals (S-nitrosoglutathione, GSNO) did not show significant expression of iNOS present in the brain (
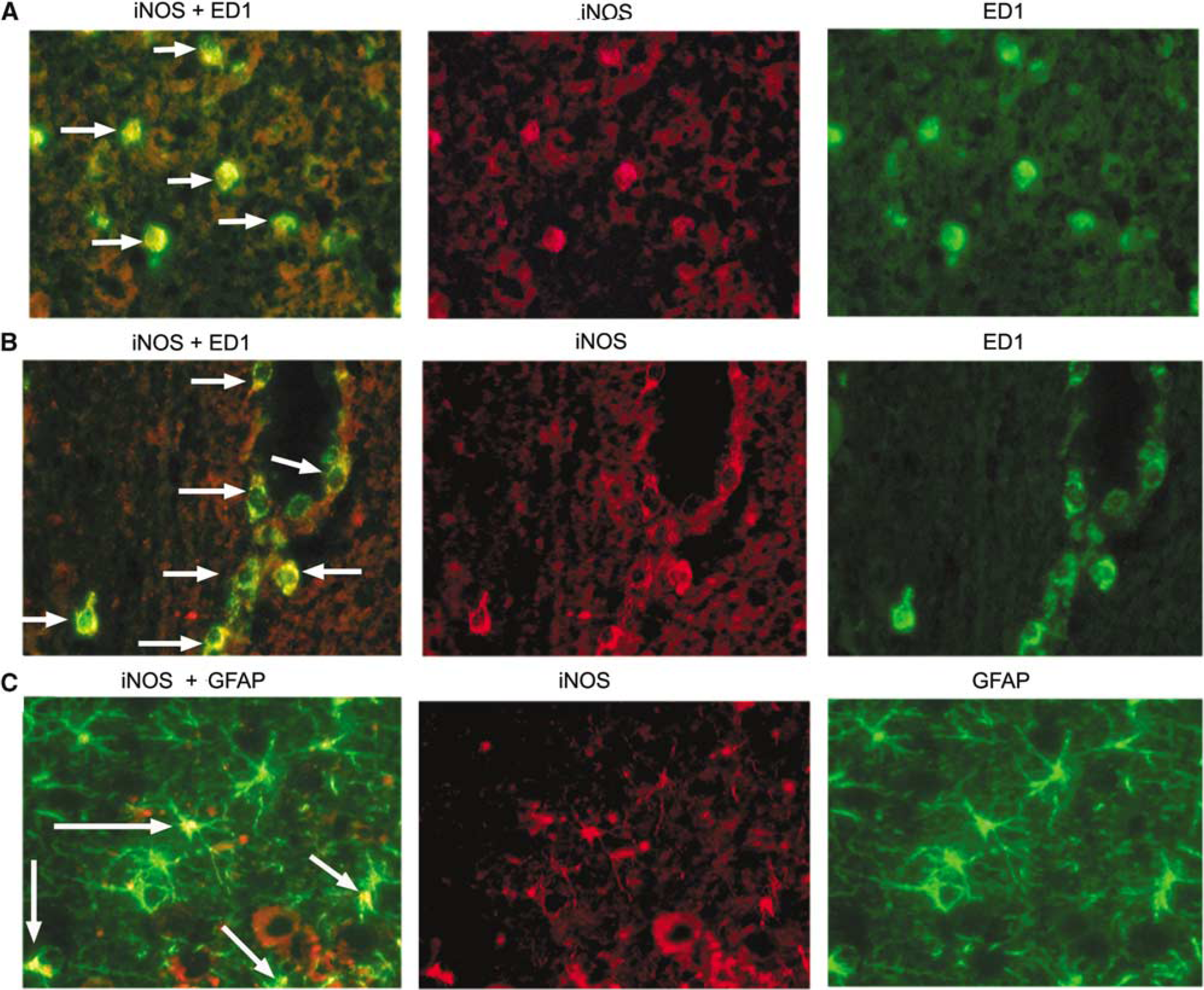
Colocalization of expression of iNOS with ED1 and GFAP at 24 hours of reperfusion after 20 minutes middle cerebral artery occlusion (MCAO). Immunostaining for inducible NO synthase (iNOS) and ED1 in a penumbral section colocalized and appeared as yellowish fluorescence (
Effect of GSNO on the Expression of Activated Microglia/Macrophage, LFA-1 and ICAM-1
Inflammatory mediators, secreted by either infiltrating blood-borne cells or by activated glial cells, enhance oxidative and nitrosative stresses and induce apoptotic cell death. ED1 and CD11-b markers were used to detect cells of monocyte origin, including activated microglia. There were significant specific stainings for ED1 (Figures 5A–5C) and CD11-b (Figures 5D–5F) in the penumbra region of ipsilateral hemisphere (vehicle). Treatment with GSNO reduced the number of ED1 as well as CD11-b-positive cells. In ischemia, the infiltration of monocytes–macrophages requires the expression of endothelial adhesion molecule (ICAM-1) as well as its ligand (LFA-1) on macrophages. We detected the enhanced immunoreactivity of both LFA-1 (Figures 5G–5I) and ICAM-1 (Figures 5J–5L) in vehicle brain. Treatment with GSNO reduced immunoreactivity of both LFA-1 and ICAM-1. The sham-operated brain had no significant staining for these molecules.
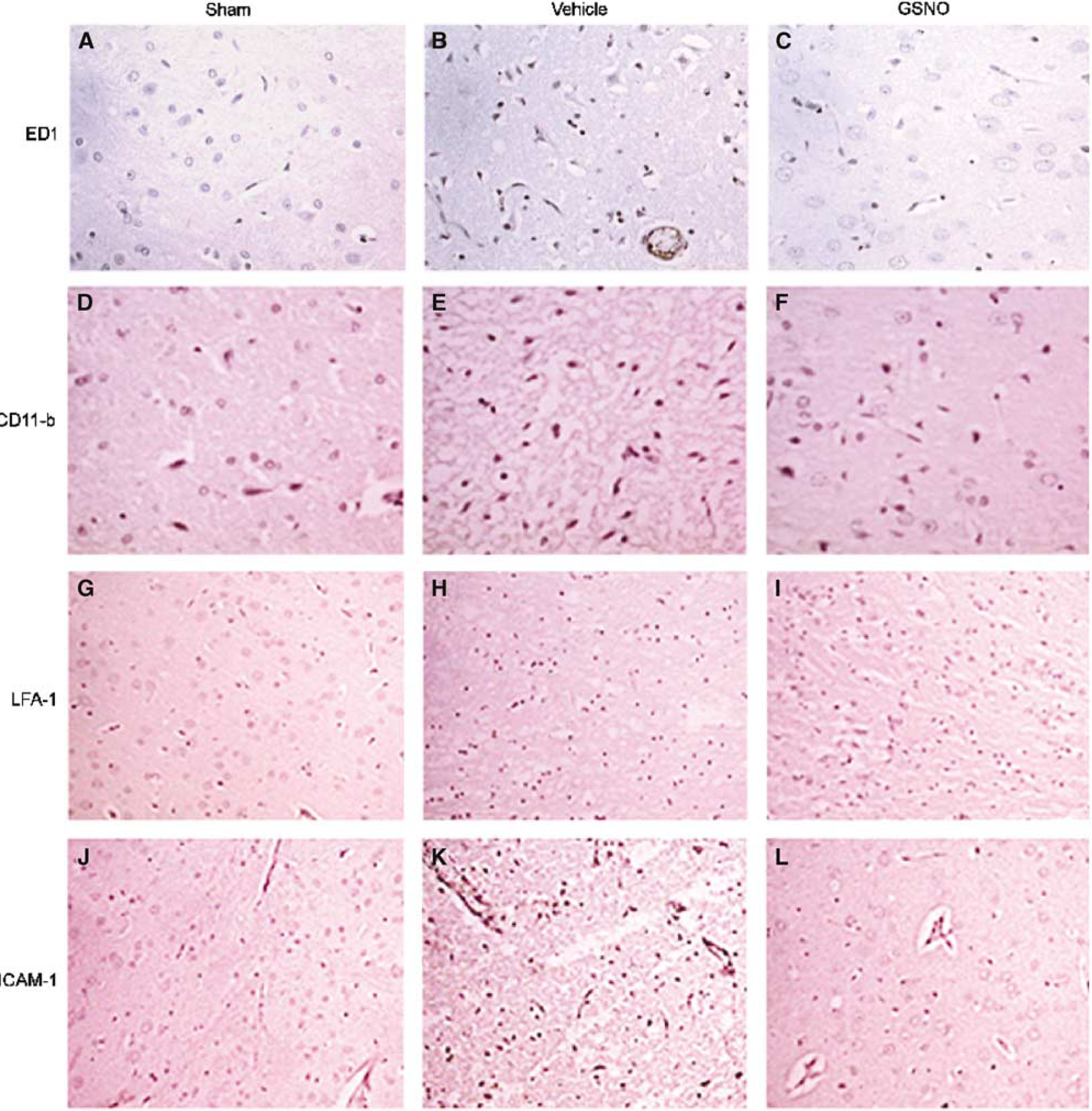
Photomicrographs of immunohistochemistry of rat brain at 24 hours of reperfusion after 20 minutes middle cerebral artery occlusion (MCAO). Expression of ED1 (
Effect of GSNO on Apoptotic Cell Death and Caspase-3 Activity
DNA fragmentation as an indicator of apoptosis was determined by transferase-mediated d-UTP-labeled nick end labeling (TUNEL) assay. DNA fragmentation in the ipsilateral hemisphere, especially around the border of infarct, was increased significantly (Figure 6A, vehicle). S-Nitrosoglutathione treatment (Figure 6A, GSNO) resulted in a decreased number of apoptotic cells. The sham-operated brain (Figure 6A, sham) as well as the contralateral hemisphere (not shown) of the ischemic brain did not show TUNEL-positive cells. The fact that the TUNEL-positive cells were mainly neurons was confirmed by colocalization of the TUNEL staining with the neuron-specific marker-NSE (Figure 6B, TUNEL+NSE). Furthermore, we found the caspase-3 activity significantly increased in the ipsilateral hemisphere (Figure 6C, vehicle) of ischemic brain as compared with the sham-operated brain (Figure 6C, sham), and this activity returned to close to basal level in the GSNO-treated brain (Figure 6C, GSNO).
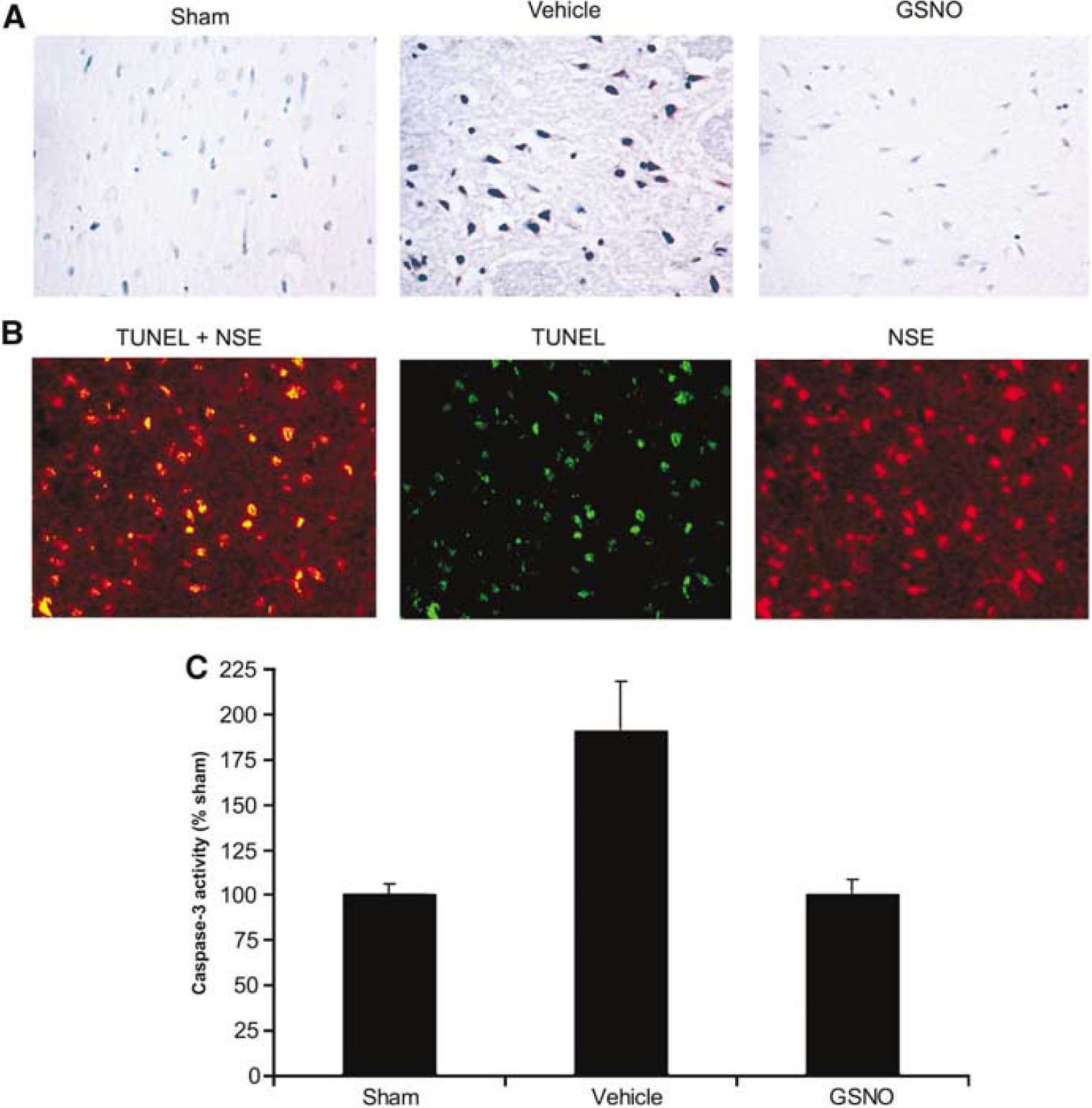
Photomicrographs of immunohistochemistry/colocalization of TUNEL assay with neurons and caspase-3 activity in rat brain at 24 hours of reperfusion after 20 minutes middle cerebral artery occlusion (MCAO). (
Effect of GSNO on LPS/IFNγ-Induced iNOS Expression In Vitro (Rat Primary Astrocytes and Microglia Cell Line BV2)
To understand the mechanism of GSNO-mediated inhibition of iNOS induction in vivo (Figures 2 and 3), we examined the effect of GSNO in vitro on LPS+IFN-γ-stimulated rat primary astrocytes and LPS-stimulated microglia cell line BV2 (microglia are involved in the propagation of inflammation after I/R insult). The cells were treated with either LPS (1 μg/mL) in the case of BV2 cells or LPS (1 μg/mL) +IFN-γ (50 U/mL) for astrocytes, after incubation with different concentrations (0.1 to 2 mmol/L) of GSNO for 30 mins. After 24 hours, the cells were analyzed for iNOS protein by Western blot. Treatment with GSNO inhibited the expression of iNOS both in astrocytes (Figure 7A) and BV2 cells (Figure 7C) in a dose-dependent manner. S-Nitrosoglutathione (1 mmol/L) alone had no effect on iNOS expression. The inhibitory effect of GSNO on cytokine-induced iNOS expression was further confirmed by iNOS luciferase activity assay both in astrocytes (Figure 7B) and BV2 cells (Figure 7D). The higher concentration of GSNO required in our in vitro experiment may be dependent on cell type. A much lower concentration of GSNO (25 to 100 μmol/L) was required in our other studies performed on TNF-α-stimulated mouse endothelial cells for inhibition of endothelial-monocyte adhesion (data not shown).
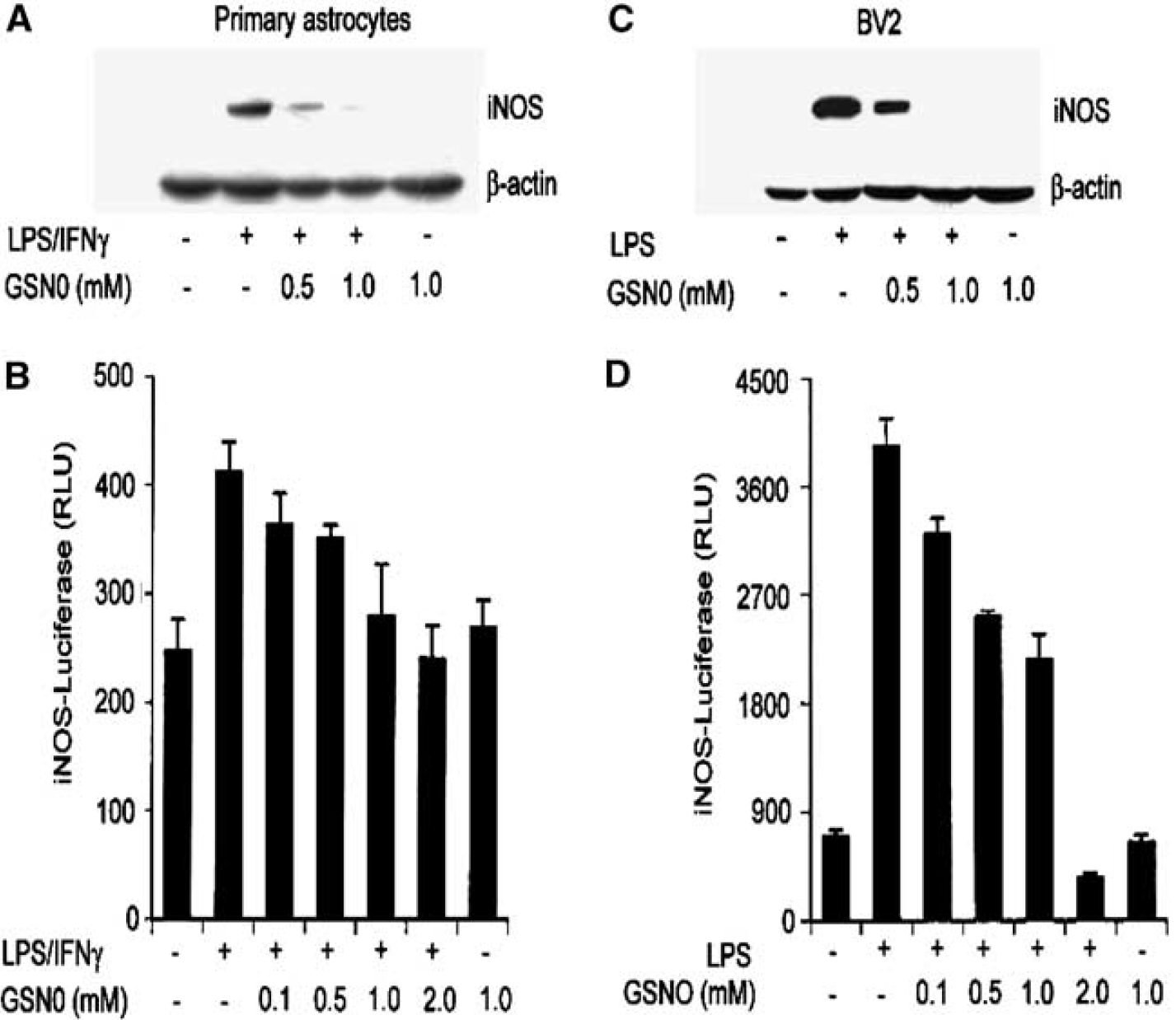
S-nitrosoglutathione (GSNO) inhibits LPS/IFN-γ or LPS-mediated iNOS gene expression in rat primary astrocytes and microglial cell line (BV2). Primary astrocytes and microglial cell BV2 were incubated for 30 minutes with different concentrations of GSNO as indicated, followed by LPS (1 μg/mL) or LPS/IFNγ (1 μg/50 U/mL) treatment for 24 hours. For detection of inducible NO synthase (iNOS) protein expression by immunoblot, cell lysate from astrocytes (
Effect of GSNO on LPS/IFNγ-Induced NF-κB Luciferase Activity In Vitro (Rat Primary Astrocytes and BV2 Cells)
NF-κB/IκB complex in cytoplasm dissociates under stimulus by phosphorylation/degradation of IκB and free NF-κB translocates to the nucleus, binds with DNA, and regulates the transcription of NF-κB-responsive genes including iNOS. We investigated the effect of GSNO treatment in LPS/IFNγ-mediated NF-κB activation and iNOS induction in rat primary astrocytes and BV2 cells. The cells were transfected with the NF-κB-luciferase reporter and luciferase activities were monitored in response to LPS+IFN-γ (astrocytes) or LPS (BV2 cells) challenge. Treatment with different concentrations of GSNO (0.1 to 2 mmol/L) decreased the LPS+IFNγ or LPS-mediated activation of NF-κB luciferase reporter activity in astrocytes and BV2 cells, respectively (Figures 8A, 8D). Further, to investigate the direct effect of GSNO on NF-κB luciferase activity, we cotransfected the expression vector of p65/p50 along with NF-κB luciferase reporter in astrocytes and BV2 cells. After 48 hours of transfection, cells were treated with different concentrations of GSNO followed by LPS or LPS/IFN-γ for 5 hours. S-Nitrosoglutathione treatment inhibited the p65/p50-mediated luciferase activity in primary astrocytes as well as in BV2 cells in a dose-dependent manner (Figures 8B, 8E). S-Nitrosoglutathione also attenuated the p65/p50-mediated iNOS-luciferase activity in these cells (Figures 8C, 8F) indicating that GSNO also mediated its effect directly on NF-κB subunits and modified their ability to bind to DNA for the transcription of iNOS gene expression. These observations document the antiinflammatory properties of GSNO.
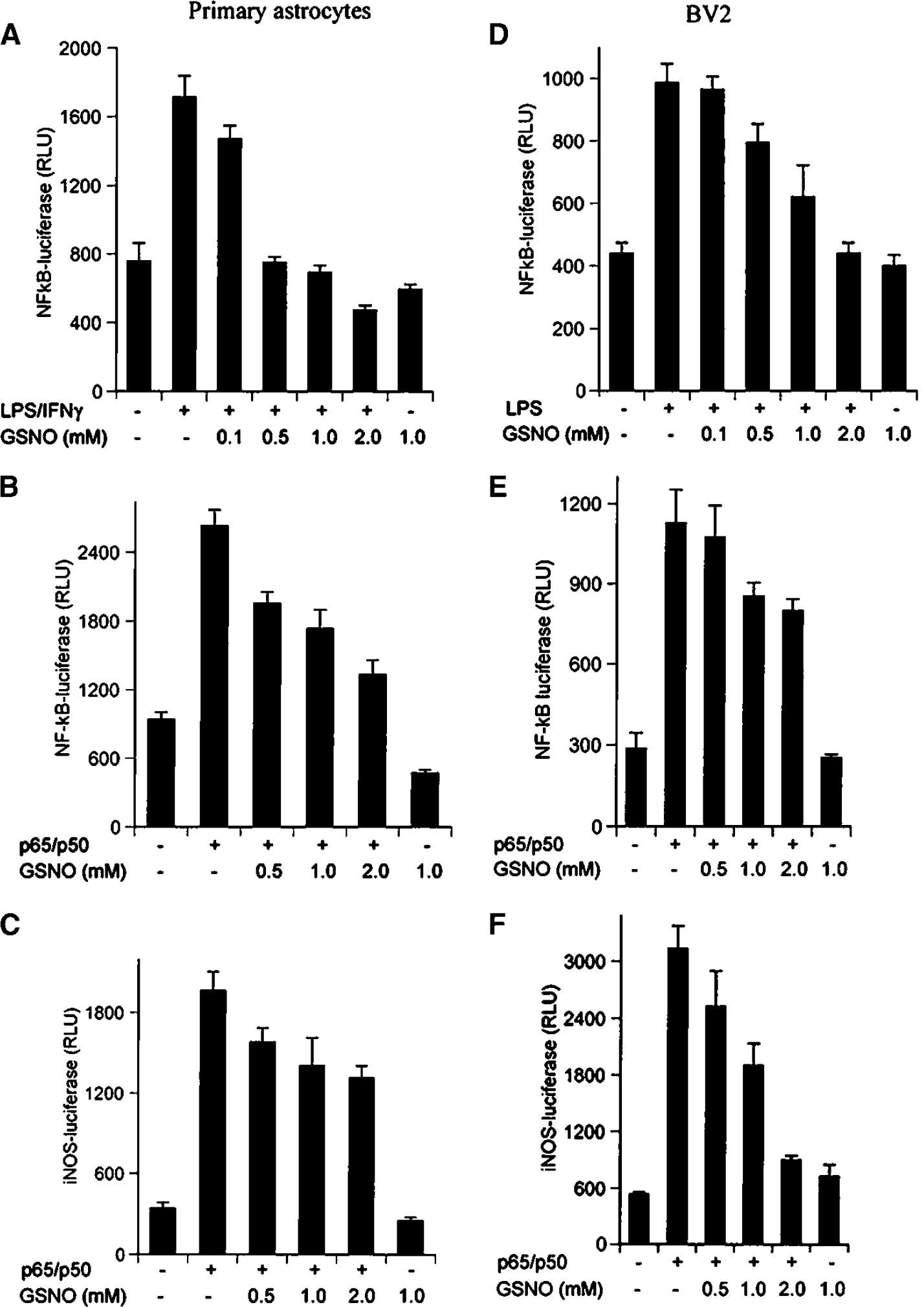
S-nitrosoglutathione (GSNO) inhibits LPS/IFN-γ- or LPS-mediated NF-κB reporter activity in rat primary astrocytes and microglial cell line BV2. (
Discussion
Treatment with GSNO increased CBF (Figure 1D), inhibited expression of TNF-α, IL-1β, and iNOS (Figure 2) and reduced apoptotic cell death via caspase-3 inhibition (Figure 6) in the ipsilateral hemisphere of the brain in a rat model of experimental stroke. This in turn resulted in protection against injury in terms of reduction of infarction (Figure 1A and 1B) and improvement in neurological score (Figure 1C). The conclusion of neuroprotection by GSNO is based on the observations that GSNO treatment inhibited the activation of both endothelial cells and microglia/macrophages (Figure 5) participating in the inflammatory process of stroke. Furthermore, the treatment with GSNO in vitro inhibited the cytokine and/or LPS-induced iNOS expression and cytokine and/or LPS-mediated NF-κB luciferase activity both in rat primary astrocytes and BV2 cells (Figures 7 and 8), indicating that GSNO is an antiinflammatory agent.
Inflammatory components in stroke may be amenable to treatment by antiinflammatory agents (Barone and Feuerstein, 1999) including N-acetylcysteine (Khan et al, 2004; Sekhon et al, 2003). Cerebral ischemia promotes activation of glial cells (resident microglia and astrocytes) and infiltration of blood-borne cells including monocytes (del Zoppo et al, 2000; Garcia et al, 1994; Stoll et al, 1998), leading to inflammation. The infiltration of monocytes is dependent on endothelial function, which is compromised under ischemic conditions (Barone and Feuerstein, 1999). Nitric oxide produced by eNOS in low amounts regulates physiological endothelial functions including CBF and vasodilatation via the guanylyl cyclase (GC)-cGMP pathway and/or by nitrosylation/transnitrosylation of cysteine residues of targeted proteins and small peptides (Tseng et al, 2000). The role of NO seems to be distinct and different in the presence of excessive reactive oxygen species (ROS)/O2− compared with the physiological levels of ROS/O2− (Davis et al, 2001). In the absence of excessive O2−, NO may terminate the deleterious free radicals leading to cellular protection. However, NO may either act in concert with ROS or react with ROS/O2− to produce ONOO− and HO· (Eiserich et al, 1998).
In view of the above documented evidence, we hypothesize that an NO modulator, which mimics the function of eNOS-derived NO and inhibits the inflammatory signals, will act as a neuroprotective and antiinflammatory agent in stroke. S-Nitrosoglutathione is identified as such a modulator of the NO system that protected the brain against stroke injury. It reduced infarction (Figures 1A and 1B), improved neurological score (Figure 1C), and enhanced CBF (Figure 1D). S-Nitrosoglutathione has been shown to be a potent inhibitor of platelet aggregation (Langford et al, 1994) and also reduces embolization in humans (Molloy et al, 1998). Systemic administration of GSNO results in reduction of the thrombosis rate in swine (Vodovotz et al, 2000). It also has the ability to modulate blood vessel tone (Rodriguez et al, 2003) and blood perfusion (Kuo et al, 2004b). S-Nitrosoglutathione has been shown to reverse acute vasoconstriction and prevent ischemic brain injury after subarachnoid hemorrhage (Sehba et al, 1999). It also protects the brain from mitochondrial dysfunction (Ju et al, 2004). In our model, GSNO protected the brain, at least in part, from endothelial dysfunction by mimicking eNOS-derived NO, thereby increasing the basal level of CBF (Figure 1D). It also has the ability to regulate the CBF mainly via nitrosylation/transnitrosylation of targeted proteins (Foster et al, 2003), and likely independent of the cGMP pathway. However, the inhibition of endothelial activation was further documented by a decreased expression of ICAM-1 and ED-1 in the GSNO-treated brain (Figures 5L, 5C) compared with vehicle (Figures 5K, 5B).
To substantiate further that GSNO has an antiinflammatory effect, we treated brain cells (rat primary astrocytes and microglial cell line BV2) in vitro with GSNO and monitored the regulation of iNOS and its transcriptional regulator, NF-κB. The treatment with GSNO inhibited the iNOS expression dose dependently in astrocytes and BV2 cells, respectively (Figures 7A and 7C). Inhibition of iNOS was further confirmed by iNOS-luciferase activity assay (Figures 7B and 7D). To examine whether inhibition of iNOS expression by GSNO was mediated by inhibition of activation of NF-κB, we investigated the role of NF-κB in this process. The treatment with GSNO inhibited the NF-κB-luciferase activity both in LPS+IFN-γ-stimulated astrocytes (Figure 8A) and in LPS-stimulated BV2 cells (Figure 8D) in a dose-dependent manner. The evidence of GSNO-mediated inhibition of LPS and/or cytokine-mediated NF-κB luciferase activity (Figures 8B, 8E) and iNOS luciferase activity (Figures 8C, 8F) was provided by transfecting the cells with p65/p50. These results indicated that GSNO did not inhibit the translocation of p65/p50 to the nucleus but altered the structure of p65/p50 complex, thereby blocking its ability to bind on to DNA. This modification of p65/p50 is likely to be responsible for the absence of NF-κB dependent iNOS gene expression. Inhibition of NF-κB by S-nitrosylation of thiol group of p50 using S-nitrosocysteine has been documented previously in murine macrophages and human respiratory cells (Marshall and Stamler, 2001). Theoretically, at least two targets for inhibition of iNOS induction via NF-κB pathway exist; one in the cytoplasm (possibly the IKK complex) and the other in the nucleus (p65/p50), depending on cell type (Marshall and Stamler, 2002). Inhibition of iNOS expression has been related with neuroprotection in an animal model of experimental stroke. Mice deficient in the iNOS gene show reduction in infarct volumes compared with respective controls (Zhao et al, 2000). In our model, we observed enhanced expression of iNOS (Figure 2H) along with TNF-α (Figure 2B) and IL-1β (Figure 2E) at 24 hours of reperfusion after 20 mins of MCAO. Treatment with GSNO inhibited the expression of iNOS (Figure 2I), TNF-α (Figure 2C), and IL-1β (Figure 2F).
The cell types that express iNOS in the ischemic brain were activated microglia/macrophages (ED-1 and CD11-b positive) present in infarct and peri-infarct areas of the ipsilateral hemisphere (Figures 5B and 5E). Although both ED-1 and CD11-b markers do not differentiate between resident activated microglia and peripheral monocytes–macrophages, this may not alter the interpretation of participation of monocyte-lineage cells in ischemic injury. The infiltration of macrophage/monocytes was corroborated by enhanced expression of adhesion molecule, ICAM-1 (Figure 5H), and its ligand LFA-1 (Figure 5H) in ischemic brain compared with sham-operated and GSNO-treated brains (Figure 5). The activated astrocytes were identified by the enhanced expression of GFAP (a marker of activation of astrocytes) presented in Figure 4. Because normal astrocytic function has been identified as critical for support of neuronal survival in acute stroke (Anderson et al, 2003), the abnormal astrocytic structure including activation is suggested to participate in injury. The possible role of iNOS-derived NO was investigated by the colocalization of expression of iNOS with expressions of both ED-1 (Figures 4A, 4B) and GFAP (Figure 4C). Further, a colocalization of TUNEL with NSE (a neuronal marker, Figure 6B) confirmed that neurons are under apoptotic stress. Activation of caspase-3 is a hallmark of apoptosis in experimental cerebral ischemia (Namura et al, 1998; Robertson et al, 2000). Intervention by antiapoptotic drugs to rescue the cells from apoptosis is one of the primary achievable clinical goals (Waldmeier, 2003). Treatment with GSNO reduced the apoptotic cell death (Figure 6A) and inhibited the activation of caspase-3 as shown in Figure 6C. Caspase-3 remains inactivated in its nitrosylated form (Mohr et al, 1997) and the Fas-induced apoptotic pathway has been shown to activate denitrosylation of caspase-3 (Mannick et al, 1999), leading to its activation. The observed caspase-3 inhibition in our studies indicates that in addition to inactivation of NF-κB, GSNO may mediate its antiapoptotic effects through nitrosylation of caspase-3.
To investigate the role of GSH, a GSNO metabolite, the animals were treated with exogenous GSH under the same conditions as that of GSNO. The treatment with GSH (150 mg/kg) administered either at reperfusion after MCAO or before the onset of ischemia had limited neuroprotective effect (data not shown). This observation indicates that the action of GSNO is based mainly on signaling and posttranslational modifications, most probably via nitrosylation and NO insertion in targeted proteins.
In conclusion, the studies described here document that GSNO provides neuroprotection against injury from focal cerebral ischemia. The inhibition of inflammatory mediators in cell culture studies via subunits p65/p50 of NF-κB clearly indicates that the protection observed in vivo may not depend on protective effects of GSNO on vasculature, circulatory cells and endothelium only, but GSNO may also exert a direct antiinflammatory effect. Furthermore, inhibition of caspase-3 activation and reduction of TUNEL positive cells in vivo by treatment with GSNO document its ability to act as an antiapoptotic drug in I/R injury. Collectively, these observations indicate that GSNO provides protection against ischemic injury by multiple mechanisms. Further therapeutic work with GSNO may involve a detailed mechanistic study and preclinical evaluation before assessment in carefully conceived clinical trials.
Footnotes
Acknowledgements
The authors thank Joyce Bryan for procurement of animals and chemicals used in this study and Carrie Barnes for cutting brain sections for morphological evaluation and immunostaining. Thanks to Jennifer G Schnellmann, PhD, ELS for technical writing assistance and Dr Abdul Aziz for suggestions on statistical evaluations.