
Abstract
We investigated the role of vascular oxidative stress in the mechanisms of the impairment in cerebrovascular regulation produced by the amyloid-β peptide (Aβ). In particular, we sought to provide evidence of vascular oxidative stress in mice overexpressing the amyloid precursor protein (APP) and to determine whether the Aβ-induced attenuation in functional hyperemia is mediated by free radical overproduction. Oxidative/nitrosative stress was assessed by 3-nitrotyrosine immunoreactivity, while free radical production was determined in cerebral microvessels by hydroethidine microfluorography. To study functional hyperemia the somatosensory cortex was activated by whisker stimulation while local blood flow was monitored by laser-Doppler flowmetry. It was found that APP mice show signs of oxidative/nitrosative stress in pial and intracerebral blood vessels well before they develop oxidative stress in neurons and glia or amyloid plaques. Treatment of cerebral microvessels isolated from wild-type mice with Aβ (1 μM) increased free radical production as assessed by the hydroethidine technique. The Aβ-induced attenuation of the increase in somatosensory cortex blood flow produced by whisker stimulation was prevented by treatment with the free radical scavengers MnTBAP or tiron. These data provide evidence that in APP mice vascular oxidative stress precedes the development of parenchymal oxidative stress, and that Aβ-produced vascular reactive oxygen species are involved in the attendant attenuation in functional hyperemia. Thus, vascular oxidative stress is an early event in the course of the brain dysfunction produced by APP overexpression and Aβ, and, as such, could be the target of early therapeutic interventions based on antioxidants.
Keywords
There is increasing evidence that amyloid-β peptides (Aβ) contribute to the pathophysiology of Alzheimer's disease (AD), the most common form of dementia in the elderly. Aβ is cleaved from the amyloid precursor protein (APP) by the proteases β- and γ-secretases (Suh and Checler 2002) and accumulates extracellularly in the brain parenchyma (amyloid plaques) and in cerebral blood vessels (amyloid angiopathy), pathological hallmarks of AD (Braak and Braak 1991). Transgenic mice overexpressing APP have elevated brain levels of Aβ and exhibit neuropathological, cognitive and cerebral metabolic alterations suggestive of AD (Niwa et al. 2002b; Wong et al. 2001). Importantly, immunization of mice overexpressing APP against the Aβ peptide clears amyloid plaques from the brain and leads to cognitive improvement (Janus et al. 2000; Kotilinek et al. 2002; Morgan et al. 2000; Schenk et al. 1999). Therefore, it is widely accepted that Aβ is a critical pathogenic factor in AD (Selkoe and Schenk 2003).
The mechanisms by which Aβ peptides alter brain function have not been clearly defined. Recent evidence suggests that Aβ has profound cerebrovascular actions that could potentially contribute to its pathogenic effect. APP mice have a profound impairment of cerebrovascular regulation that is fully developed at a time when amyloid plaques and amyloid angiopathy are not present (Iadecola et al. 1999; Zhang et al. 1997). Thus, the response of cerebral blood vessels to endothelium-dependent vasodilators, such as acetylcholine, is markedly attenuated (Iadecola et al. 1999). Furthermore, cerebrovascular autoregulation, the ability of cerebral blood vessels to keep cerebral blood flow (CBF) stable in the face of changes in arterial pressure, and functional hyperemia, the increase in CBF produced by synaptic activity, are profoundly impaired in APP mice (Niwa et al. 2002a; Niwa et al. 2000c). The cerebrovascular alterations observed in APP mice can be reproduced by topical application of Aβ 1–40 to the cerebral cortex of normal mice (Niwa et al. 2000b; Niwa et al. 2001; Niwa et al. 2000c), indicating that this peptide is responsible for the dysfunction.
The impairment in endothelium-dependent vasodilation is prevented by free radical scavengers and is not observed in transgenic mice overexpressing both APP and the free radical scavenging enzyme Cu-Zn-superoxide dismutase (Iadecola et al. 1999; Niwa et al. 2000b). Furthermore, a mutated form of Aβ that does not generate reactive oxygen species (ROS) is devoid of cerebrovascular effects (Niwa et al. 2000b). These observations indicate that ROS play a role in the failure of endothelium-dependent relaxation produced by Aβ. However, several critical questions remain to be answered. For example, it has not been established whether oxidative stress is present in APP mice at the age when cerebrovascular dysfunction exists, and if so, whether such oxidative stress is restricted to cerebral blood vessels or also involves brain cells. Furthermore, it is known whether the Aβ-induced attenuation in functional hyperemia, a critical homeostatic mechanism of the cerebral circulation, is also related to overproduction of vascular ROS. In the present study, markers of ROS and agents that scavenge free radicals were used to address some of these issues. It was found that: (a) oxidative stress is present in cerebral vessels of APP mice when there is no evidence of amyloid plaques or oxidative stress in the brain parenchyma; (b) exogenous Aβ 1–40 stimulates ROS production in isolated cerebral microvessels of normal mice, and (c) the attenuation of functional hyperemia produced by Aβ is prevented by free radical scavengers. These findings, collectively, provide strong evidence that vascular oxidative stress is an early event in the pathobiology of APP mice and that ROS are critical factors in the potentially deleterious impairment of functional hyperemia associated with overproduction of Aβ.
METHODS
Methods for surgical preparation of mice, for topical application of drugs, for monitoring CBF using laser-Doppler flowmetry, and for immunocytochemistry have been described in detail in previous publications (Forster et al. 1999; Niwa et al. 2000a) and are briefly summarized below.
Mice
Studies with nitrotyrosine immunocytochemistry were conducted in Tg2576 mice overexpressing Swedish mutant APP695 on a mixed C57BL/6J-SJL/J background (age: 4–7 or 12–14 months) (Hsiao et al. 1996). Age matched APP-negative littermates served as controls. C57BL/6J mice obtained from Jackson Laboratories (Bar Harbor, Maine) were used in experiments in which the Aβ peptide was tested.
General surgical procedures
Mice were anesthetized with isoflurane (Induction: 5%; Maintenance: 1–2%). One of the femoral arteries was cannulated for recording of mean arterial pressure (MAP) and collection of blood samples. The trachea was intubated and mice were artificially ventilated with an oxygen-nitrogen mixture. The O2 concentration in the mixture was adjusted to provide an arterial pO2 (paO2) of 120–140 mmHg (Table 1). Rectal temperature was maintained at 37°C using a thermostatically controlled rectal probe connected to a heating pad. End-tidal CO2, monitored by a CO2 analyzer (Capstar-100, CWE Inc.), was maintained at 2.6–2.7% (Niwa et al. 2000a). After surgery, isoflurane was gradually discontinued and anesthesia was maintained with urethane (750 mg/kg; i.p.) and chloralose (50 mg/kg; i.p.). Throughout the experiment, the level of anesthesia was monitored by testing corneal reflexes and motor responses to tail pinch. To minimize confounding effects of anesthesia on vascular reactivity, the time interval between administration of urethane-chloralose and testing of CBF responses was kept consistent among the different groups of mice studied.
Mean arterial pressure and blood gases in the mice in which CBF was studied
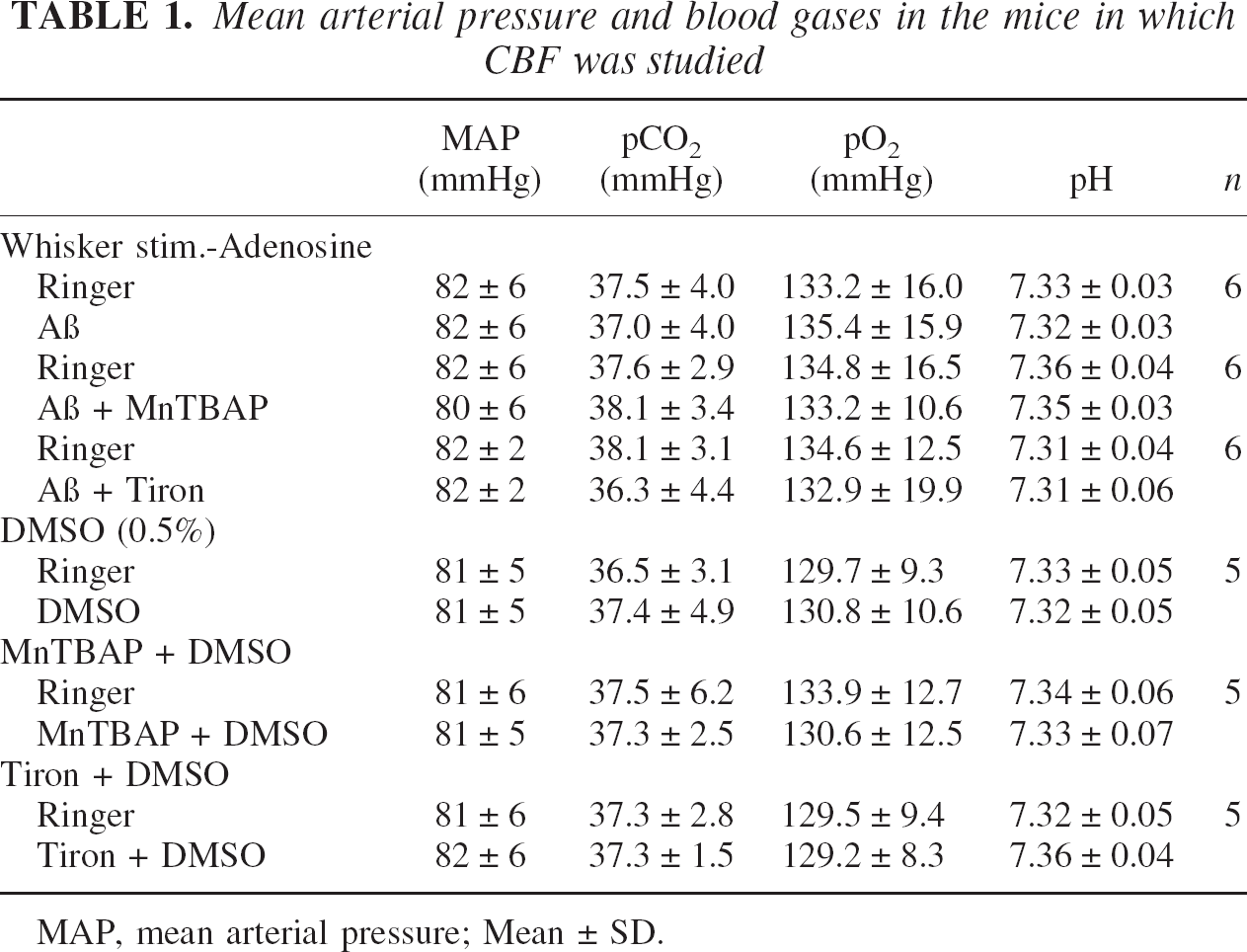
MAP, mean arterial pressure; Mean ± SD.
Monitoring of cerebral blood flow
A small craniotomy (2X2 mm) was performed to expose the parietal cortex, the dura was removed, and the site was superfused with a modified Ringer solution (37°C; pH: 7.3–7.4) (see (Iadecola 1992) for composition) (Niwa et al. 2000a; Zhang et al. 1997). CBF was continuously monitored at the site of superfusion with a laser-Doppler probe (Vasamedic, St. Paul, MN, USA) positioned stereotaxically on the cortical surface and connected to a computerized data acquisition system. CBF values were expressed as percent increase relative to the resting level. Zero values for CBF were obtained after the heart was stopped by an overdose of isoflurane at the end of the experiment. Although laser-Doppler flowmetry is not quantitative, it monitors relative changes in CBF quite accurately (Iadecola 1997).
Cerebral microvessel preparation and measurement of ROS
The cerebral cortices of three C57BL/6J mice/experiment were dissected in Medium 199, pooled and homogenized (Arneric et al. 1988; Galea et al. 1998). The homogenate was mixed with an equal volume of 30% Dextran (15 mL) and centrifuged at 4500 g for 10 minutes at 4°C. The top fatty layer was removed by aspiration, and the solution was decanted and mixed again and recentrifuged. The combined pellets were resuspended in Medium 199 and collected by centrifugation at 900 g for 10 minutes at 4°C. The pellet was resuspended and the solution containing microvessels, red blood cells, and cellular debris was passed through a 40-μm nylon mesh. Microvessels retained on this mesh were washed with 300 mL HBSS (Hank's Balanced Salt Solution). Microvessels were eluted from the mesh with 50 mL HBSS w/o Phenol Red and collected by centrifugation at 900 g for 10 minutes at 4°C. Microvessels were resuspended in HBSS and seeded on TissueTek chamber slides and transferred to a humidified incubator (37°C, 5% CO2). Purity of preparations, assessed by phase contrast microscopy as well as immunolabeling for endothelial, glial and smooth muscle cell markers (see section 5 on immunohistochemistry), was usually greater than 95%. This procedure allows to isolate a range of vessels spanning from capillaries (<10 μm diameter) to small arterioles/venules (50–100 μm diameter). After letting vessels attach for 45 minutes the medium was changed to assay medium (DMEM w/o Phenol Red, 0.5% FBS, 2 μM hydroethidine) and allowed to equilibrate for 16 hours before experiments. ROS production was assessed by the hydroethidine method. Hydroethidine (Dihydroethidium) is cell permeable and is oxidized to become the fluorophore ethidium bromide which intercalates in double stranded DNA and exhibits peak absorbance at 520 nm and an emission maximum at 600 nm (Tarpey and Fridovich 2001). To examine superoxide production with hydroethidine, brain microvessels are incubated in assay medium containing Aβ 1–40 (1 μM) or vehicle for 1 hour. Vessels were then washed and fixed with 3.7% Paraformaldehyde (w/v in Phosphate Buffered Saline) (PBS) for 20 minutes on ice. Nuclei are counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 100 ng/ml for 2 minutes). Fluorescent images of control and Aβ-stimulated vessels were captured with a Nikon E800 microscope equipped with a computer controlled 12 bit digital mono-chrome camera (Coolsnap, Roper Scientific, Trenton, NJ, USA). Ethidium fluorescence was excited at 488 nm and emission captured using a 610 nm long pass filter. Similarly, nuclear DAPI fluorescence was acquired with a filter set that allows for excitation at 350 nm and transmits at 470 nm. For each vessel-fragment an image in the DAPI and ethidium channel was captured. The images were post-processed with IPLab software package (Scanalytics, Fairfax, VA, USA). Briefly, nuclei were identified according to their DAPI signal and the ROI (Region of Interest) was defined accordingly. Planimetric ROI data were transferred to the image acquired with the ethidium filter set. After background subtraction of the camera dark current, pixel intensities of nuclear ethidium signals were quantified. Usually, 20–40 vessel fragments with a total of 500–1000 nuclei were analyzed for each treatment group. The ROI selection and quantification procedure was semi-automated using IPLab's scripting language for unbiased quantification of ethidium signals.
Immunohistochemistry
Immunohistochemical techniques in brain sections were identical to those previously described (Forster et al. 1999; Nogawa et al. 1998). Anesthetized APP mice (pentobarbital, 100 mg/kg i.p.) were perfused through the heart with 4% paraformaldehyde. Brains were embedded in paraffin, sectioned (thickness 5 μm), and processed further as previously described (Nogawa et al. 1998). The following antibodies were used: (a) 4G8, a mouse monoclonal antibody reactive to residue 17–24 of human Aβ used as a marker of amyloid plaques (1:3000; gift of Dr. K. Hsiao Ashe); (b) 3-nitrotyrosine antibody, a marker of peroxynitrite (1:1000; Upstate Biotechnology, Lake Placid, NY, USA). In certain conditions associated with inflammation, tyrosine nitration can also be promoted by nitrite (Eiserich et al. 1998). However, in the absence of florid inflammation this is unlikely to be a relevant factor affecting the specificity of nitrotyrosine as a marker of peroxynitrite. Following incubation with the appropriate secondary antibody, the immunocomplex was visualized using diaminobenzidine as a chromogen. The specificity of the stain was tested by processing sections without the primary antibody and after incubation of the primary antibody with an excess of 3-nitrotyrosine or the 4G8 peptide (see ref. (Forster et al. 1999)).
For immunocytochemical characterization of microvessels, samples were prepared as described above and fixed in Ethanol/Acidic Acid (v/v 95:5) for one minute at room temperature. Slides were blocked with 3% bovine serum albumin (BSA) in PBS. Vessels were double-stained for CD105 (endoglin) as a endothelial marker (anti-mouse CD105 rat monoclonal antibody, clone MJ7/18) and alpha-actin as a marker for vascular smooth muscle cells (anti alpha-actin mouse monoclonal antibody, clone 1A4). Primary antibodies were applied in 1% BSA in PBS for 16 hours at 4°C. After washing the slides three times in PBS, binding of the respective antibodies was revealed with FITC-labeled rabbit anti-rat (Vector Laboratories) and TexasRed-labeled rabbit anti-mouse (Jackson Immunochemicals) antibodies in 1% BSA in PBS for 1 hour at room temperature. Slides were washed twice in PBS and nuclei were counterstained with DAPI. Slides were mounted in Vectashield (Vector Laboratories) and coverslipped. Fluorescent images were acquired with a Nikon E800 microscope equipped with a computer controlled digital monochrome camera (Coolsnap, Roper Scientific, Trenton, NJ, USA) using DAPI, FITC and TexasRed compatible filter sets. Grayscale images were pseudo colored and color channels were merged. Nuclei (DAPI) appear blue, endothelial cells (endoglin) green and smooth muscle cells red (alpha-actin).
Protocols for CBF experiments
Effect of Aβ 1–40 on the increase in CBF produced by somatosensory activation or adenosine
After stabilization of MAP and blood gases (Table 1), the whiskers were activated for 60 seconds and the increase in CBF in the contralateral somatosensory cortex was recorded. After testing responses during Ringer superfusion, the superfusion solution was changed to Ringer containing Aβ 1–40 (5 μM). This concentration of Aβ was previously found to produce maximal cerebrovascular effects (Niwa et al. 2000b; Niwa et al. 2000c). As in previous studies (Niwa et al. 2000b; Niwa et al. 2000c), to minimize aggregation of the peptide during the experiment Aβ was freshly solubilized in DMSO and then diluted in normal Ringer. The final DMSO concentration was less than 0.5%. The CBF response to whisker stimulation was tested after 30–40 min of Aβ superfusion. This time interval was selected on the basis of preliminary experiments in which the time-course of the cerebrovascular effects of Aβ was investigated (Niwa et al. 2000b; Niwa et al. 2000c). In some studies the reactivity of CBF to topical superfusion of adenosine (400 μM) was tested before and after Aβ1–40 application. The Aβ concentration in AD is likely to be smaller than that described to have cerebrovascular effects or to be neurotoxic in vitro (Mattson 1997; Neve and Robakis 1998). However, the Aβ concentration achieved within the cerebral cortex in our study is likely to be less than that topically applied, and, as such, closer to that found in AD. Larger concentrations may be needed because, unlike AD in which the peptide accumulates over years, Aβ was applied only for a relatively short period of time.
Effect of superoxide scavengers on the cerebrovascular actions of Aβ1–40
The cranial window was superfused with Ringer and the effect of whisker stimulation on somatosensory cortex CBF was tested. The superfusion solution was then changed to Ringer containing Aβ 1–40 (5 μM) and either manganic(I-II)meso-tetrakis(4-benzoic acid) porphyrin (MnTBAP) (Porphyrin Products, Logan, UT; 100 μM; n=6), or 4,5-dihydroxy-1,3-benzene disulfonic acid (tiron; Sigma; 10 mM; n=6). The effect of whisker stimulation on CBF was tested 30 min later. MnTBAP and tiron are cell-permanent and scavenge superoxide both extracellularly and intracellularly (Didion and Faraci 2002; Faulkner et al. 1994; Klann 1998). MnTBAP also scavenges hydrogen peroxide (Day et al. 1997). In some experiments (n=5/group), the effect of DMSO 0.5% (Aβ vehicle), tiron, or MnTBAP on the cerebrovascular response to whisker stimulation, or adenosine were studied.
Data analysis
Data in text and figures are expressed as means ± SD. Two-group comparisons were analyzed by the two-tailed t-test for dependent or independent samples, as appropriate. Multiple comparisons were evaluated by the analysis of variance and Tukey's test. Probability values of less than 0.05 were considered statistically significant.
RESULTS
Vascular oxidative/nitrosative stress in APP mice
First, we sought to provide evidence of oxidative stress in cerebral blood vessels of APP mice. In APP-negative mice, no evidence of 3-nitrotyrosine immunoreactivity was observed in pial arterioles supplying the somatosensory cortex (Fig. 1A) (n=5). In contrast, in 4–7 month-old APP+ mice (n=4), abundant immunoreactivity was observed in these pial vessels (Fig. 1B). 3-nitrotyrosine immunoreactivity was localized to endothelial cells and to the adventitial layer, comprised mainly of meningeal cells (Peters et al. 1991), but not to the smooth muscle layer (Fig. 1B). In 4–7 month-old APP+ mice (n=4), 3-nitrotyrosine immunoreactivity was also present in smaller intraparenchymal vessels (Fig. 2A). As in previous studies (Hsiao et al. 1996; Kawarabayashi et al. 2001), no evidence of Aβ deposition, assessed by the 4G8 antibody, was observed in parenchyma and blood vessels at this age (Fig. 2B). As a positive control, 3-nitrotyrosine and 4G8 immunoreactivity were also examined in 12–14 month-old APP+ mice (n=6). In these mice, Aβ deposition in amyloid plaques was also observed in areas in which 3-nitrotyrosine was present in blood vessels (Fig. 2C, D). Furthermore, 3-nitrotyrosine immunoreactivity was found in neurons (Fig. 2E) and amyloid deposition was observed in cerebral blood vessels (Fig. 2F).
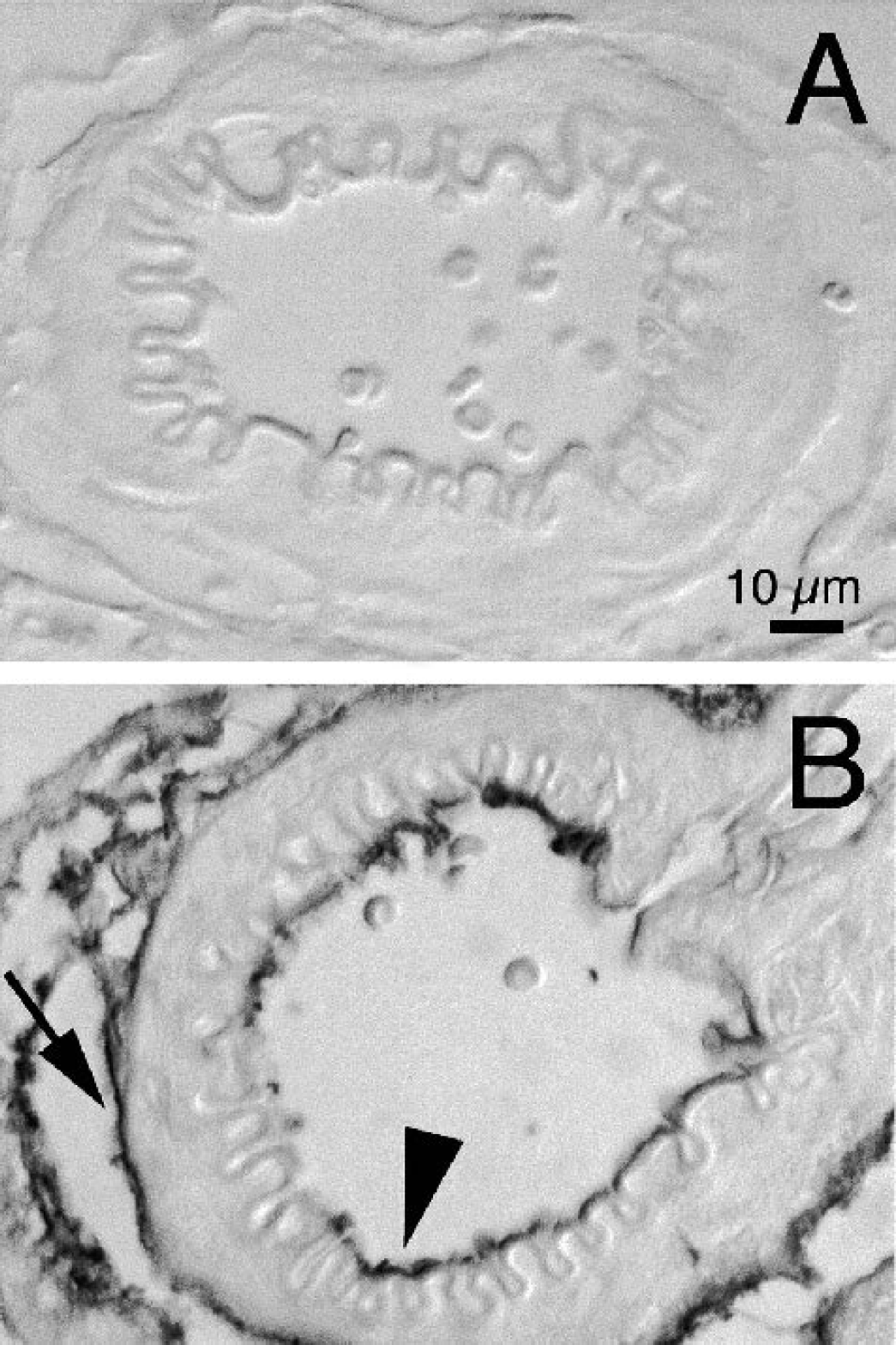
3-nitrotyrosine immunoreactivity in somatosensory cortex pial arterioles of APP negative
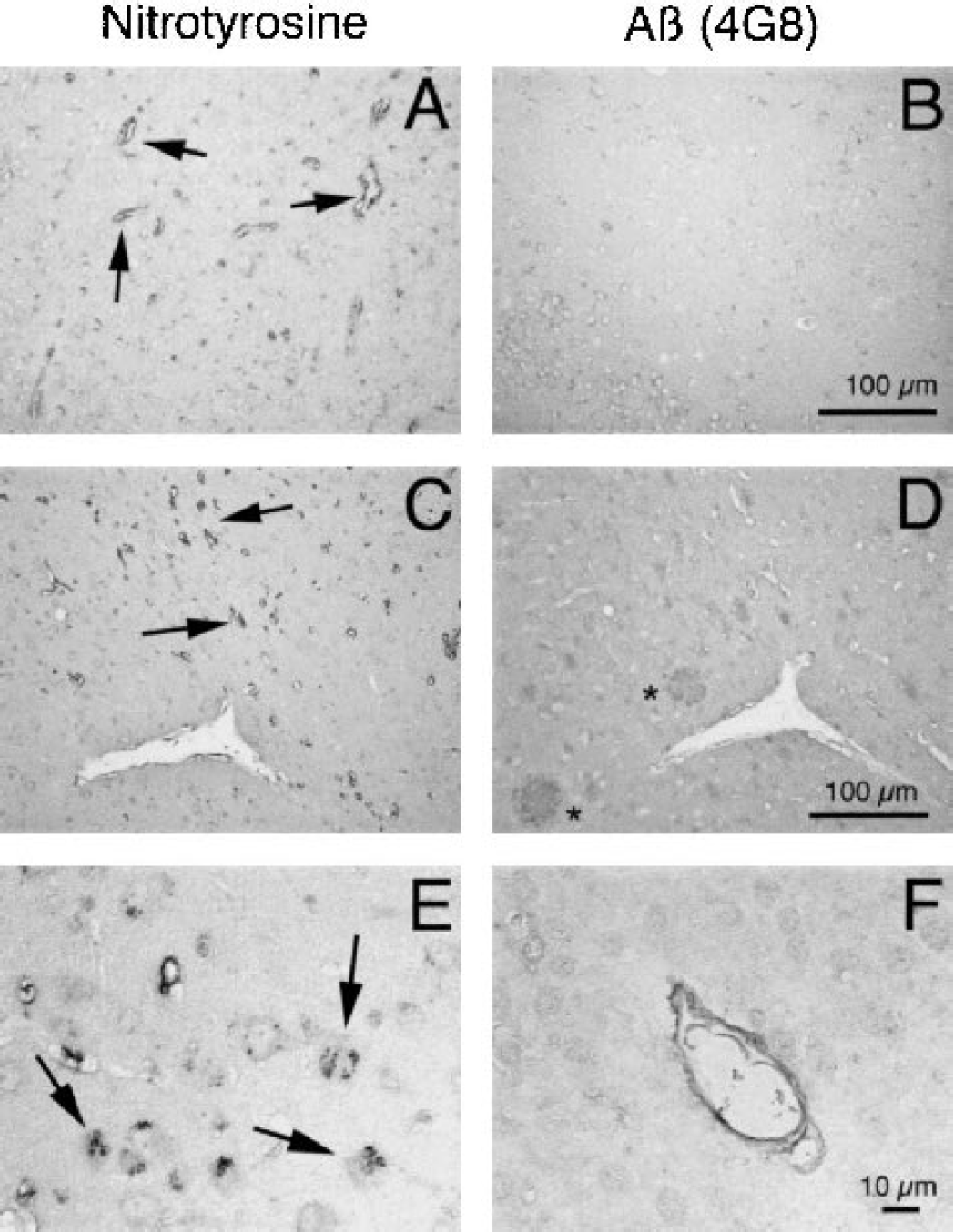
3-nitrotyrosine
Aβ 1–40 increases ROS production in isolated cerebral microvessels
Immunohistochemical characterization of the cerebral microvessels indicates that they include vessels with a smooth muscle coat, most likely arterioles, and capillaries as well (Fig. 3A, B). To determine whether Aβ evoked vascular ROS production, the microvessels were treated with Aβ1–40 (1 μM) and ROS were detected using the hydroethidine method. Treatment with Aβ increased the fluorescence signal in the nuclei of vascular cells, an effect that was blocked by the free radical scavenger MnTBAP (Fig. 3). These data indicate that Aβ 1–40 increases ROS production in cerebral microvessels.
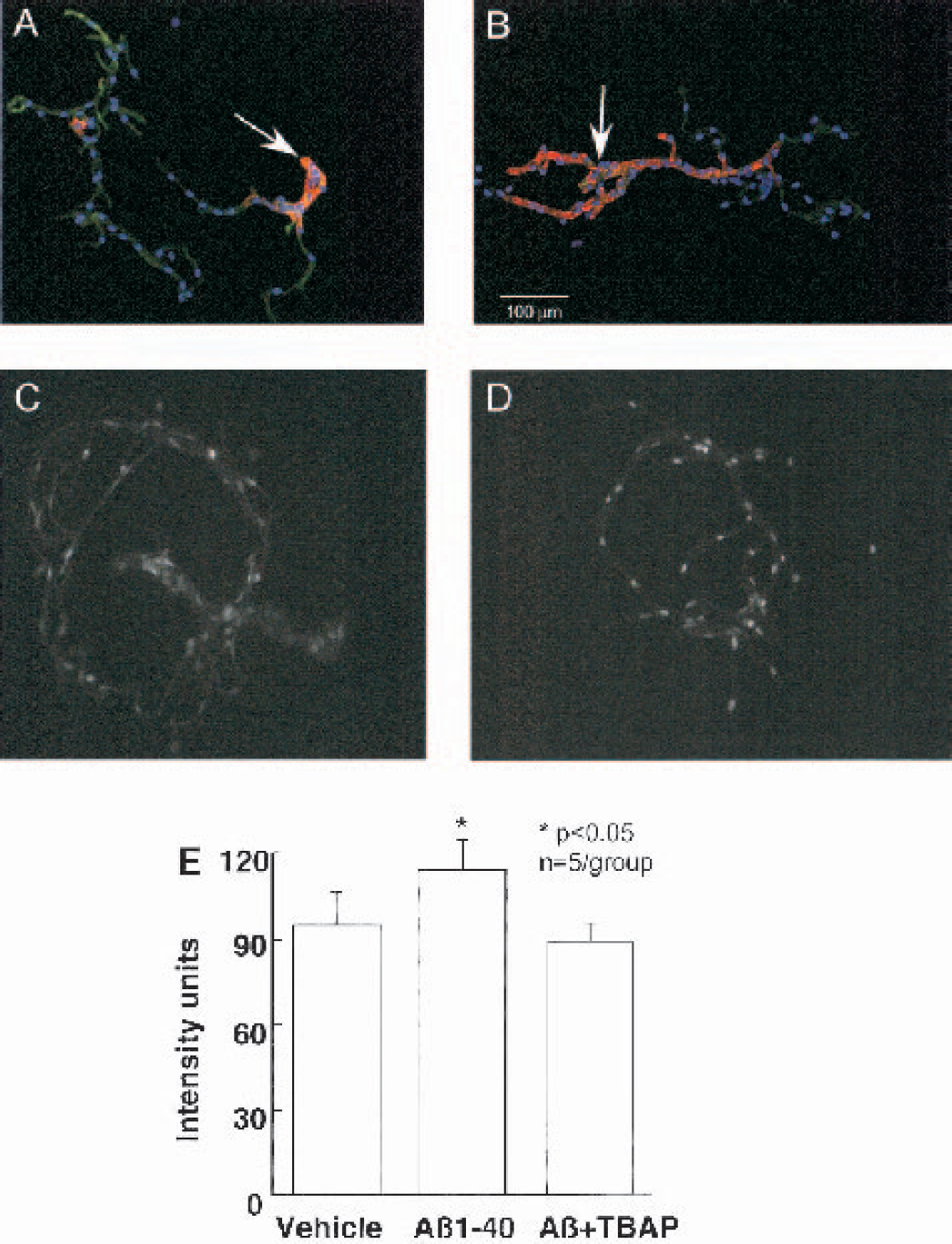
ROS production, assessed by the hydroethidine method, in isolated cerebral microvessels exposed to Aβ1–40.
Free radical scavengers prevent the Aβ-induced attenuation of functional hyperemia
We then sought to establish whether ROS are involved in the attenuation of functional hyperemia produced by topical neocortical application of Aβ. During superfusion with Ringer solution whisker stimulation increased CBF in the somatosensory cortex by 20–30% (Fig. 4B) (n=6). Superfusion with Aβ 1–40 (5 μM) reduced resting CBF and attenuated the increase in CBF produced by whisker stimulation (Fig. 4A, B) (p<0.05; n=6). These effects were not observed if Aβ 1–40 was topically applied in the presence of the free radical scavengers MnTBAP (Fig. 4A, B) or tiron (Fig. 4A, B). The increase in CBF produced by adenosine was not affected by Aβ 1–40, with or without the free radical scavengers (Fig. 4C). DMSO (vehicle for Aβ), MnTBAP or tiron did not alter the increase in CBF produced by whisker stimulation (Fig. 5; n=5/group).
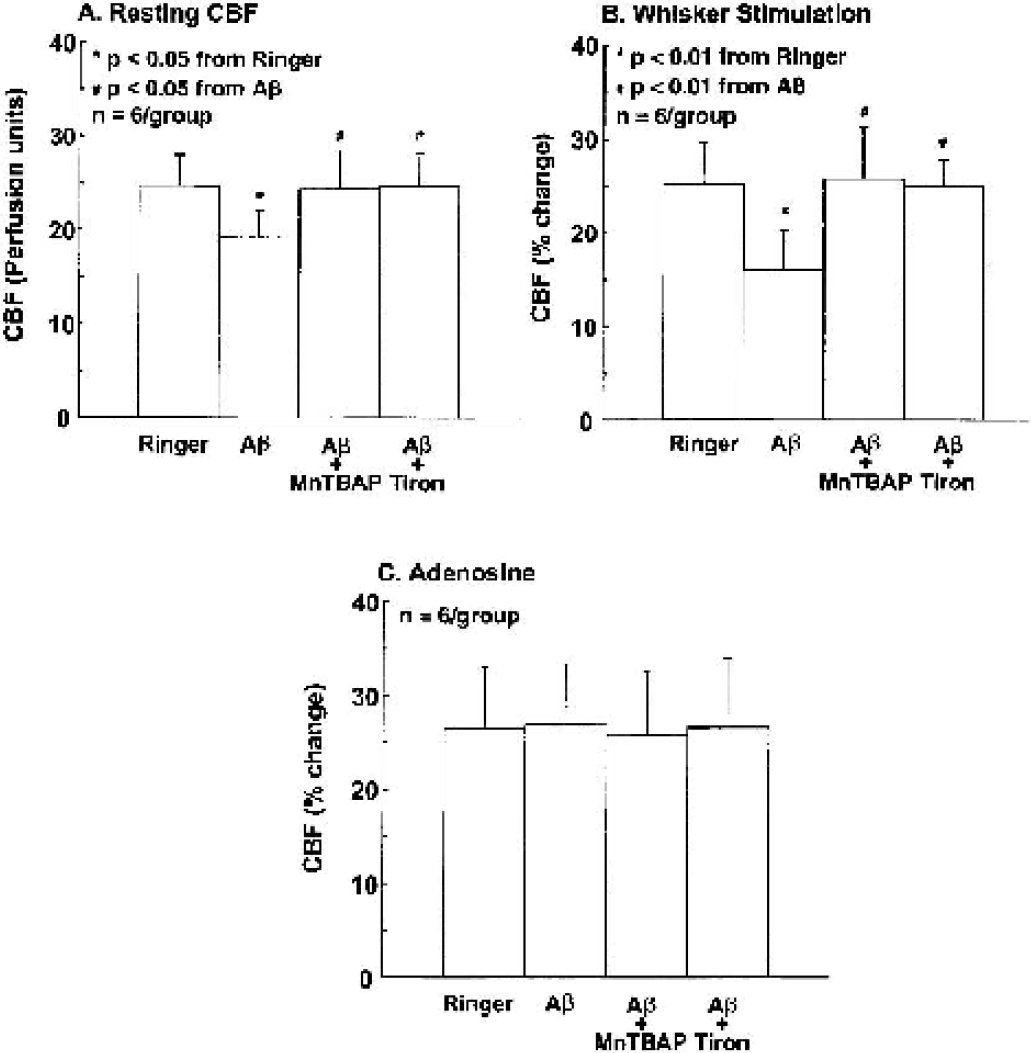
Effect of Aβ1–40 with or without the free radical scavengers MnTBAP (100 μM) or tiron (10 mM) on resting CBF
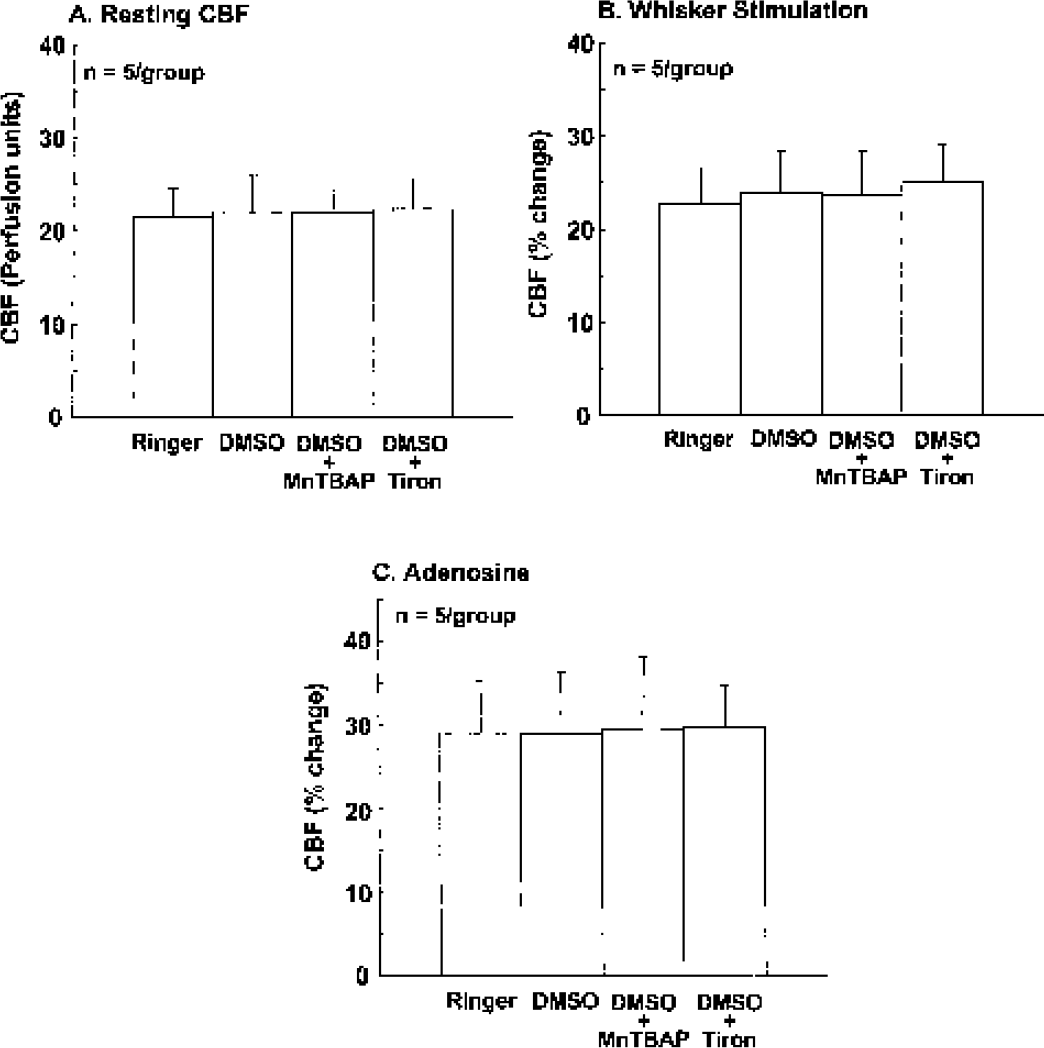
Effect of DMSO (Aβ vehicle, 0.5%), DMSO+MnTBAP (100 μM) and DMSO+tiron (10 mM) on resting CBF
DISCUSSION
We investigated whether cerebrovascular oxidative stress is present in APP mice at an age when they have cerebrovascular dysfunction and, if so, whether exogenous Aβ 1–40 could stimulate ROS production in normal cerebral vessels. Furthermore, we used free radical scavengers to determine whether the attenuation of functional hyperemia produced by Aβ is related to ROS. We found that 3-nitrotyrosine, a marker of oxidative/nitrosative stress, is present in cerebral blood vessels, but not in the brain parenchyma, of “young” APP mice (age 4–7 months). Treatment of isolated cerebral microvessels of normal mice with Aβ 1–40 increases ROS production, an effect blocked by the free radical scavenger MnTBAP. Furthermore, free radical scavengers prevent the attenuation in functional hyperemia produced by Aβ 1–40. These findings, collectively, provide strong evidence that Aβ induces vascular ROS production and that the attenuation of functional hyperemia is related to oxidative stress.
The effect of MnTBAP or tiron on Aβ-induced vascular dysfunction cannot result from changes in MAP and blood gases, because these variables were carefully controlled and did not differ among the groups of mice studied. Similarly, the findings cannot be attributed to a deterioration of the experimental preparation resulting in a progressive and nonspecific impairment in cerebrovascular reactivity, because the CBF response to adenosine was not reduced over the same period of time. Furthermore, the effect of MnTBAP or tiron cannot be caused by a nonspecific enhancement of resting CBF and of functional hyperemia. We have shown here that, in the absence of Aβ, MnTBAP and tiron do not influence resting CBF and its reactivity to whisker stimulation. Therefore, the findings of the present study cannot be attributed to instability of the experimental preparation or to nonspecific cerebrovascular effects of the pharmacological agents used.
One of the new findings of the present study is that in APP mice oxidative/nitrosative stress was observed in cerebral blood vessels, but not in the brain parenchyma, at a time (age 4–7 months) when there was no deposition of Aβ in brain parenchyma or blood vessels. In older mice (12–14 months), however, amyloid plaques were noted and 3-nitrotyrosine immunoreactivity was found in neurons. These observations suggest that vascular oxidative stress is an early event in the pathogenic cascade induced by Aβ in APP mice, and are in agreement with previous results demonstrating that in these mice the cerebrovascular dysfunction precedes the onset of cognitive impairment (Iadecola et al. 1999; Niwa et al. 2000c; Westerman et al. 2002). The cerebrovascular dysfunction is likely to increase the susceptibility of the brain to injury. For example, APP mice have larger brain lesions following middle cerebral artery occlusion, an effect in part due to Aβ-induced cerebrovascular dysfunction (Zhang et al. 1997). However, other factors, such as microglial inflammation, are also likely to play a role (Koistinaho et al. 2002).
It is of interest that in resistance arterioles 3-nitrotyrosine immunoreactivity was observed only in the endothelial cell layer and in the adventitia. This finding is consistent with the hypothesis that endothelial cells and adventitial cells are major sources of Aβ-induced ROS in the vessel wall. A similar pattern of vascular ROS production has been observed in other conditions, such as angiotensin II-induced hypertension or diabetes (Lund et al. 2000; Pagano et al. 1997), in which there is increased production of free radicals in blood vessels. On the other hand, because 3-nitrotyrosine reflects mainly protein nitration by peroxynitrite, the product of the reaction of NO with superoxide, the finding also suggests that the endothelium and the adventitia are major sources of NO. It is unknown whether vascular NO production is upregulated in APP mice. However, upregulation of iNOS has been reported both in Alzheimer patients and in APP mice, and aberrant expression of eNOS has been reported in the vessels of Alzheimer patients (de la Monte et al. 2000; Luth et al. 2001; Vodovotz et al. 1996). Therefore, vascular iNOS and eNOS could be sources of the NO reacting with superoxide to form peroxynitrite. As for superoxide, the experiments with ROS production in isolated microvessels indicate that Aβ 1–40 is able to increase ROS production in brain vessels. The enzymatic system responsible for Aβ-induced vascular superoxide production has not been identified. Recent data suggest that NAD(P)H oxidase is a major source of superoxide at the vascular level in the normal state and in several vascular diseases (Lassegue and Clempus 2003). Furthermore, in situations in which substrates and/or co-factors are rate limiting, eNOS could also produce superoxide (Hink et al. 2001). Accordingly, these enzymes could be involved in the mechanisms of superoxide production by Aβ 1–40. Irrespective of the sources of NO and superoxide, our findings provide strong evidence for increased ROS production at the vascular level. These findings raise the possibility that early treatment with free radical scavengers may be beneficial to correct the cerebrovascular dysregulation and brain dysfunction occurring in AD. Consistent with this hypothesis, clinical studies have demonstrated that antioxidants are beneficial in AD (Morris et al. 2002; Sano et al. 1997).
Another new finding of the present study is that free radical scavengers prevent the attenuation in functional hyperemia produced by Aβ 1–40. This observation, in conjunction with our demonstration of Aβ-induced vascular ROS production, provides strong evidence that ROS are responsible for the attenuation in functional hyperemia. Several lines of evidence suggest that the effects of Aβ on functional hyperemia are not related to neuronal dysfunction. For example, in APP mice the CBF increase produced by somatosensory activation is attenuated but the associated elevation in glucose utilization, an index of neural activity, is not reduced (Niwa et al. 2000c). In addition, APP mice at this age do not exhibit evidence of Aβ-induced neuropathological alterations or behavioral abnormalities (Hsiao et al. 1996; Westerman et al. 2002). Furthermore, the reduction in resting CBF produced by Aβ 1–40 in normal mice cannot be attributed to a comparable reduction in local glucose utilization (Niwa et al. 2000b). Therefore, the attenuation in functional hyperemia is likely to be related to ROS production in blood vessels. We have previously demonstrated that the Aβ-induced impairment of endothelium-dependent relaxation is also mediated by ROS. This observation is in agreement with reports in the cerebral and systemic circulations indicating that oxidative stress alters endothelium-dependent vascular responses (Cai et al. 2000; Didion et al. 2002). Our finding that functional hyperemia is attenuated by vascular ROS production provides, for the first time, evidence that this critical mechanisms of cerebrovascular regulation is highly susceptible to vascular oxidative stress.
The mechanisms by which vascular ROS attenuate functional hyperemia have not been elucidated in full. The evidence to date suggests that the increase in CBF is mediated by multiple agents released from neurons and/or glia, including adenosine, NO, arachidonic acid metabolites, and neuropeptides (Ko et al. 1990; Niwa et al. 2000a; Northington et al. 1992; Peng et al. 2002; Yaksh et al. 1987; Zonta et al. 2003). There are several possible ways in which ROS could attenuate functional hyperemia. First, ROS could impair neural activity and attenuate the release of vasoactive factor from the activated brain. This hypothesis, however, seems unlikely because, as discussed in the previous paragraph, the neural activity evoked by somatosensory stimulation is not altered in APP mice. Another possibility is that ROS impair the reactivity of cerebral vascular smooth muscles to the vasodilator factors released by neural activity. This possibility can also be ruled out because the cerebrovascular reactivity to exogenous NO donors, adenosine, and hypercapnia are not altered by Aβ (Niwa et al. 2000b). A more likely scenario is that ROS remove or inactivate some of the endogenous vasoactive factors released by neural activity. For example, in systemic vessels ROS inactivate NO via formation of peroxynitrite and attenuate NO-dependent vascular responses (Cai et al. 2000). Furthermore, peroxynitrite inhibits prostacylin synthase and P450 epoxygenase (Oyekan 2002; Zou et al. 1999), enzymes that produce vasodilators in cerebral vessels. Therefore, these factors could also be responsible for the Aβ-induced attenuation of the increase in CBF produced by somatosensory activation. Our observation that peroxynitrite is formed in cerebral resistance arterioles of the somatosensory cortex supports this hypothesis. However, further studies are needed to establish this point more firmly.
CONCLUSION
In conclusion, we have provided evidence that vascular oxidative stress is an early event in the pathogenic cascade triggered by APP overexpression and Aβ. Furthermore, exogenous Aβ 1–40 induces ROS production in cerebral microvessels and attenuates functional hyperemia, effects that are prevented by free radical scavengers. These findings, in concert with the previous demonstration that the effect of Aβ cannot be attributed to diminished neuronal activation, indicate that vascular oxidative stress is responsible for the attenuation in functional hyperemia. These observations provide further support to the use of antioxidants in early AD and other conditions associated with increased brain Aβ.