
Abstract
Transgenic mice overexpressing the amyloid precursor protein (APP) have a profound impairment in endothelium-dependent cerebrovascular responses that is counteracted by the superoxide scavenger superoxide dismutase (SOD). The authors investigated whether the amyloid-β peptide (Aβ) is responsible for the cerebrovascular effects of APP overexpression. Cerebral blood flow (CBF) was monitored by a laser—Doppler flowmeter in anesthetized-ventilated mice equipped with a cranial window. Superfusion of Aβ1–40 on the neocortex reduced resting CBF in a dose-dependent fashion (−29% ± 7% at 5 μmol/L) and attenuated the increase in CBF produced by the endothelium-dependent vasodilators acetylcholine (−41% ± 8%), bradykinin (−39% ± 9%), and the calcium ionophore A23187 (−37% ± 5%). Aβ1–40 did not influence the CBF increases produced by the endothelium-independent vasodilators S-nitroso-N-acetylpenicillamine and hypercapnia. In contrast, Aβ1–42 did not attenuate resting CBF or the CBF increases produced by endothelium-dependent vasodilators. Cerebrovascular effects of Aβ1–40 were reversed by the superoxide scavengers SOD or MnTBAP. Furthermore, substitution of methionine 35 with norleucine, a mutation that blocks the ability of Aβ to generate reactive oxygen species, abolished Aβ1–40 vasoactivity. The authors conclude that Aβ1–40, but not Aβ1–42, reproduces the cerebrovascular alterations observed in APP transgenics, Thus, Aβ1–40 could play a role in the cerebrovascular alterations observed in Alzheimer's dementia.
Keywords
There is substantial evidence that the amyloid precursor protein (APP) and peptides derived from its processing are involved in the pathogenesis of Alzheimer's dementia (AD) (Selkoe, 1999). Although mutations of the APP gene are linked to familial AD, the amyloid-β peptide (Aβ), a fragment of APP, is a major component of neuritic plaques, one of the neuropathologic hallmarks of AD (Levy-Lahad and Bird, 1996; Vinters et al., 1996). Furthermore, overexpression of human APP in transgenic mice increases Aβ concentration in brain and reproduces some of the neuropathologic and behavioral alterations observed in patients with AD (Price and Sisodia, 1998). Collectively, these findings suggest that peptides derived from the proteolytic processing of APP play a crucial role in the mechanisms of AD.
The mechanisms by which APP and Aβ exert their pathogenic effects have not been fully elucidated (Mattson, 1997). Recent evidence suggests that APP overexpression and Aβ accumulation impair the function of the cerebral circulation (Iadecola et al., 1999; Zhang et al., 1997). Transgenic mice overexpressing mutant or wildtype APP have a selective alteration in cerebrovascular reactivity to acetylcholine (ACh), bradykinin (BK), and the calcium ionophore A23187, agents that act by releasing vasoactive factors from the endothelium (Iadecola et al., 1999). The deficit in endothelium-dependent vasodilation is not observed in mice overexpressing both APP and the superoxide scavenging enzyme superoxide dismutase-1 (SOD; Iadecola et al., 1999). Furthermore, application of exogenous SOD to the cerebral cortex reverses the attenuation in endothelium-dependent vasodilation in APP transgenic mice (Iadecola et al., 1999). These findings suggest that APP overexpression results in profound alterations in cerebrovascular function and that the effect is mediated through production of reactive oxygen species.
However, it has not been established whether the cerebrovascular alterations observed in these transgenic models result from APP itself or from peptides derived from APP processing. Amyloid precursor protein is cleaved by secretases to produce several peptides, including Aβ1–40 and Aβ1-42, which are thought to mediate the pathogenic effects of APP overexpression (Sinha et al., 1999; Yan et al., 1999). Therefore, in the current study, the authors investigated the cerebrovascular effects of synthetic Aβ peptides on the cerebral microcirculation of mice. The authors found that superfusion of the cerebral cortex of normal mice with Aβ1–40 attenuates resting cerebral blood flow (CBF) and reduces the increase in CBF produced by endothelium-dependent vasodilators. These effects are reversed by treatment with superoxide scavengers and are abolished by substitution of the methionine residue in position 35 with norleucine, a modification that prevents Aβ1–40 from generating free radicals (Varadarajan et al., 1999). The findings provide evidence that Aβ1–40, through production of reactive oxygen species, mediates the cerebrovascular dysfunction induced by APP overexpression, and raise the possibility that this peptide is also responsible for the cerebrovascular alterations observed in patients with AD.
MATERIALS AND METHODS
Methods for surgical preparation of mice, for topical application of drugs, for monitoring CBF using laser—Doppler flowmetry, and for measuring cerebral glucose utilization (CGU) using quantitative autoradiography have been described in detail in previous publications (Iadecola et al., 1999; Niwa et al., 2000a; Zhang et al., 1997) and are summarized briefly below.
General surgical procedures
Studies were conducted in 75 C57BL/6J mice (aged 2 to 3 months, weighing 20 to 30 g) obtained from Jackson Laboratories (Bar Harbor, Maine, U.S.A.). Mice were anesthetized with halothane in 100% O2 (induction: 5%; maintenance: 1% to 2%). Trachea was intubated and mice were artificially ventilated with an oxygen—nitrogen mixture. The O2 concentration in the mixture was adjusted to provide a Pa
Mean arterial pressure and blood gases in mice
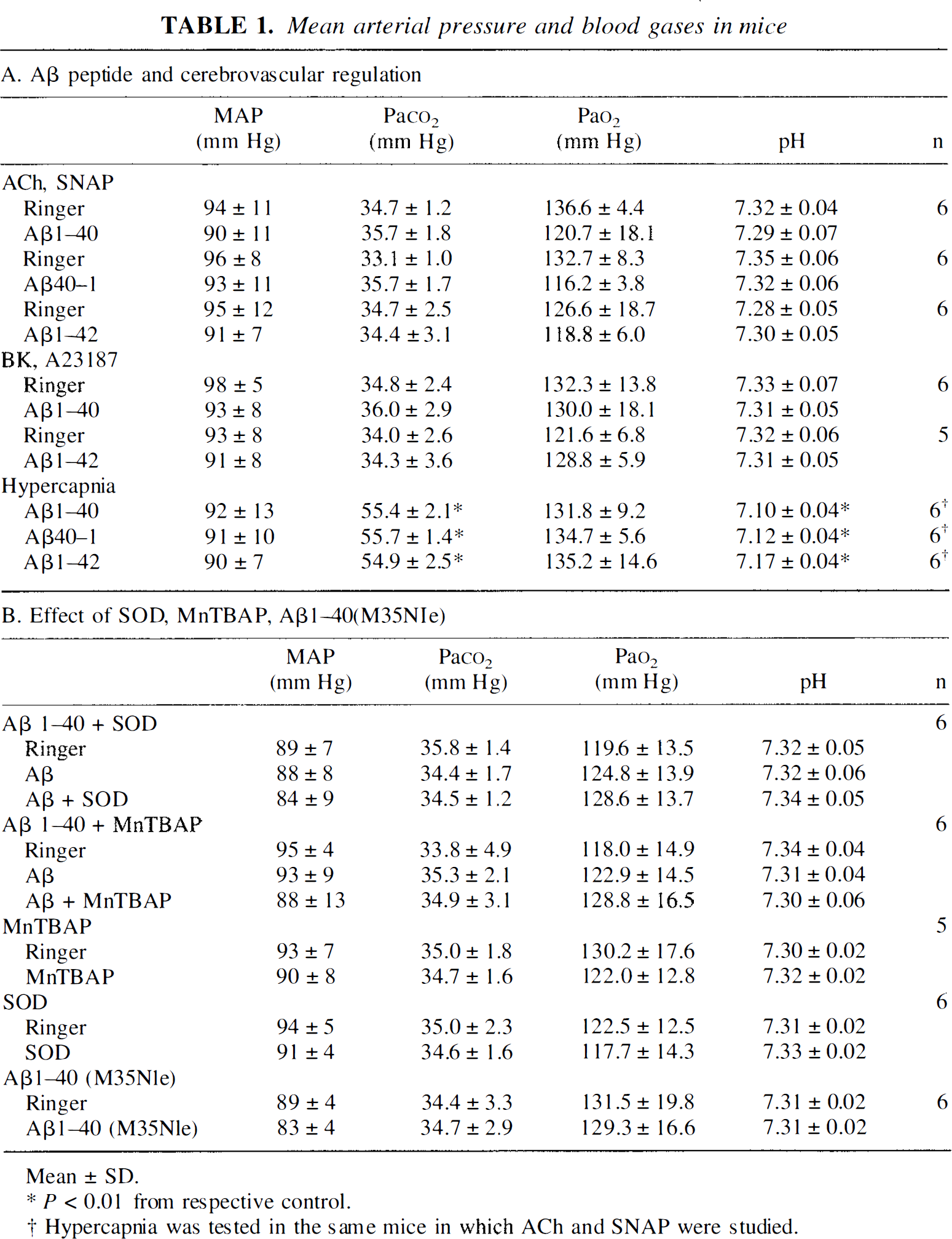
Mean ± SD.
P < 0.01 from respective control.
Hypercapnia was tested in the same mice in which ACh and SNAP were studied.
Monitoring of cerebral blood flow
A small craniotomy (2 × 2 mm) was performed to expose the parietal cortex, the dura was removed, and the site was superfused with Ringer solution (37°C; pH: 7.3 to 7.4) (Niwa et al., 2000a; Zhang et al., 1997). Cerebral blood flow was monitored continuously at the site of superfusion with a laser—Doppler probe (Vasamedic, St. Paul, MN, U.S.A.) positioned stereotaxically on the cortical surface. Cerebral blood flow values were expressed as a percentage increase relative to the resting level. Zero values for CBF were obtained after the heart was stopped by an overdose of halothane at the end of the experiment. Although laser—Doppler flowmetry is not quantitative, it monitors relative changes in CBF quite accurately (Dirnagl et al., 1989; Iadecola and Reis, 1990).
Cerebral glucose utilization
Cerebral glucose utilization was determined using a modification of the 14C-2-deoxyglucose (2-DG) method (Meibach et al., 1980; Niwa et al., 2000a; Sokoloff et al., 1977). Mice were anesthetized and surgically prepared as described for the CBF experiments. One of the femoral arteries was cannulated and used for recording of MAP and collection of blood samples. 14C-labeled 2-DG (New England Nuclear, Boston, MA, U.S.A.) (20 μCi/100 g in 1 mL 0.9% NaCl) was injected intraperitoneally, and approximately 60 μL arterial blood was collected 1, 5, 7, 10, 15, 20, 25, 35, and 45 minutes later. One blood sample was collected before injection of 2-DG. These sampling times were selected in preliminary experiments to accurately resolve the arterial concentration time-course of the tracer. Blood samples were centrifuged and stored on ice. Techniques for determination of tissue 2-DG concentration by quantitative autoradiography have been described previously (Niwa et al., 2000a), but were only summarized. Brains were removed rapidly and frozen in isopentane cooled to −30°C. Serial sections (20 μm) were cut through the brain using a cryostat (Hacker-Bright, model OTF, Fairfield, NJ, U.S.A.), mounted on glass slides, and apposed to x-ray film (Sterling Diagnostic Imaging, Newark, DE, U.S.A.) together with calibrated 14C standards (Niwa et al., 2000a). Fourteen days later the film was developed and the optical density (OD) of regions of interest was determined bilaterally on four adjacent sections using a computerized image analyzer (MCID system; Imaging Research). Optical density was transformed in 14C concentration (nCi/g) using the standards on the film (Niwa et al., 2000a). Radioactivity (nCi/g) of plasma samples was determined by liquid scintillation counting (Niwa et al., 2000a). Plasma glucose was measured using a glucose analyzer (Beckman). Cerebral glucose utilization (μmol/100 g−1 min−1) was calculated from the OD of the regions of interest and the arterial time course of 2-DG using the equation developed by Sokoloff and colleagues (1977). Physiologic parameters in the mice used for CGU measurement (10 minutes after 2-DG injection) were as follows; Ringer—MAP = 94 ± 9 mm Hg; Pa
Experimental protocols
Effect of neocortical application of Aβ1–40 on local cerebral glucose utilization
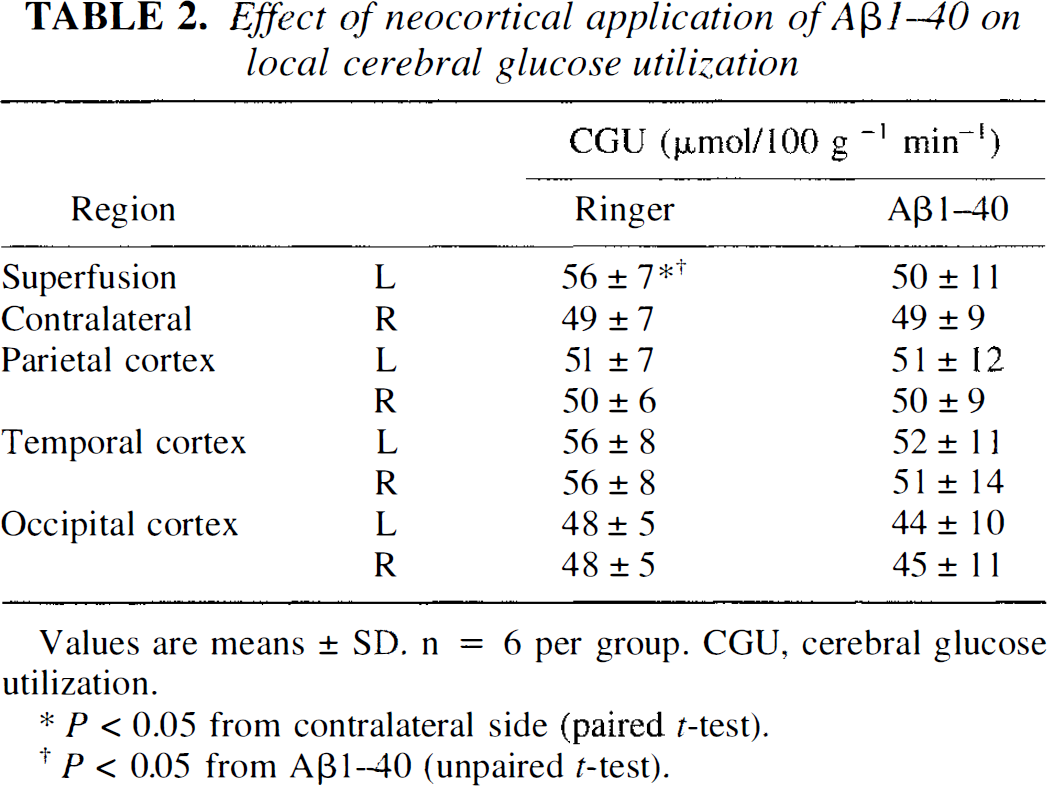
Values are means ± SD. n = 6 per group. CGU, cerebral glucose utilization.
p < 0.05 from contralateral side (paired t-test).
p 0.05 from Aβ1-40 (unpaired t-test).
Data analysis
Data in text and figures are expressed as means ± SD. Two-group comparisons were analyzed by the two-tailed t-test for dependent or independent samples, as appropriate. Multiple comparisons were evaluated by the analysis of variance and Tukey's test. Probability values <0.05 were considered statistically significant.
RESULTS
Effect of Aβ on resting cerebral blood flow and on cerebrovascular reactivity to endothelium-dependent and -independent vasodilators
Cerebral blood flow was stable during superfusion with normal Ringer. However, superfusion with Aβ1–40 (0.01 to 10 μmol/L) reduced resting CBF in a dose-dependent manner (Fig. 1). Reduction at 5 μmol/L averaged 29% ± 7% and was not different from the reduction observed at 10 μmol/L (Fig. 1). Superfusion with Aβ1–40 (5 μmol/L) attenuated the increase in CBF produced by the endothelium-dependent vasodilators ACh (−41% ± 8%; P < 0.05), BK (−39% ± 9%; P < 0.05), and A23187 (−37% ± 5%; P < 0.05) (Figs. 1 and 2). In contrast, Aβ1–40 did not influence the increase in CBF produced by SNAP (100 to 500 μmol/L) or hypercapnia (Pa
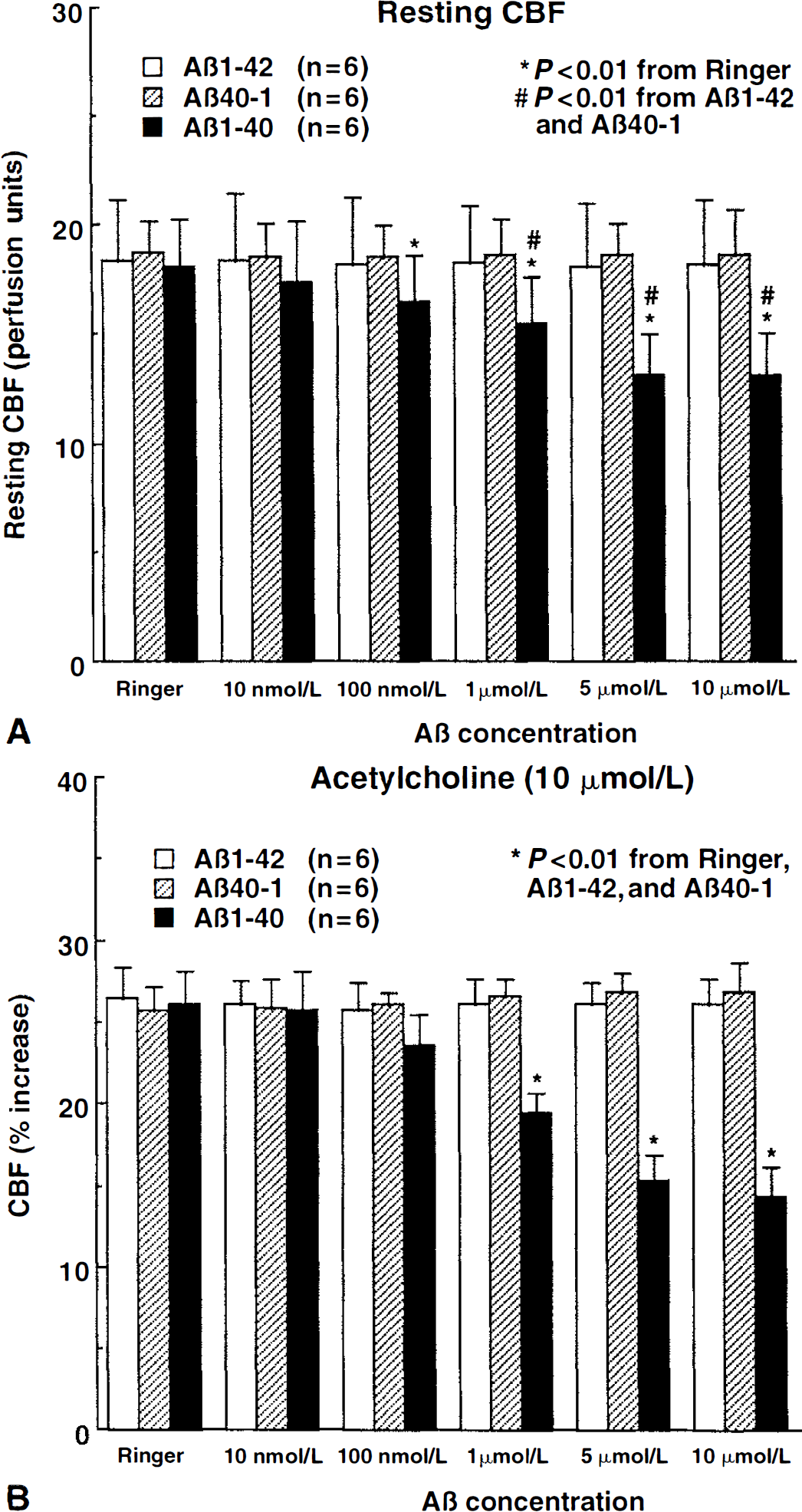
Effect of topical superfusion of Aβ1–40, Aβ40–1, and Aβ1–42 on resting cerebral blood flow (CBF)
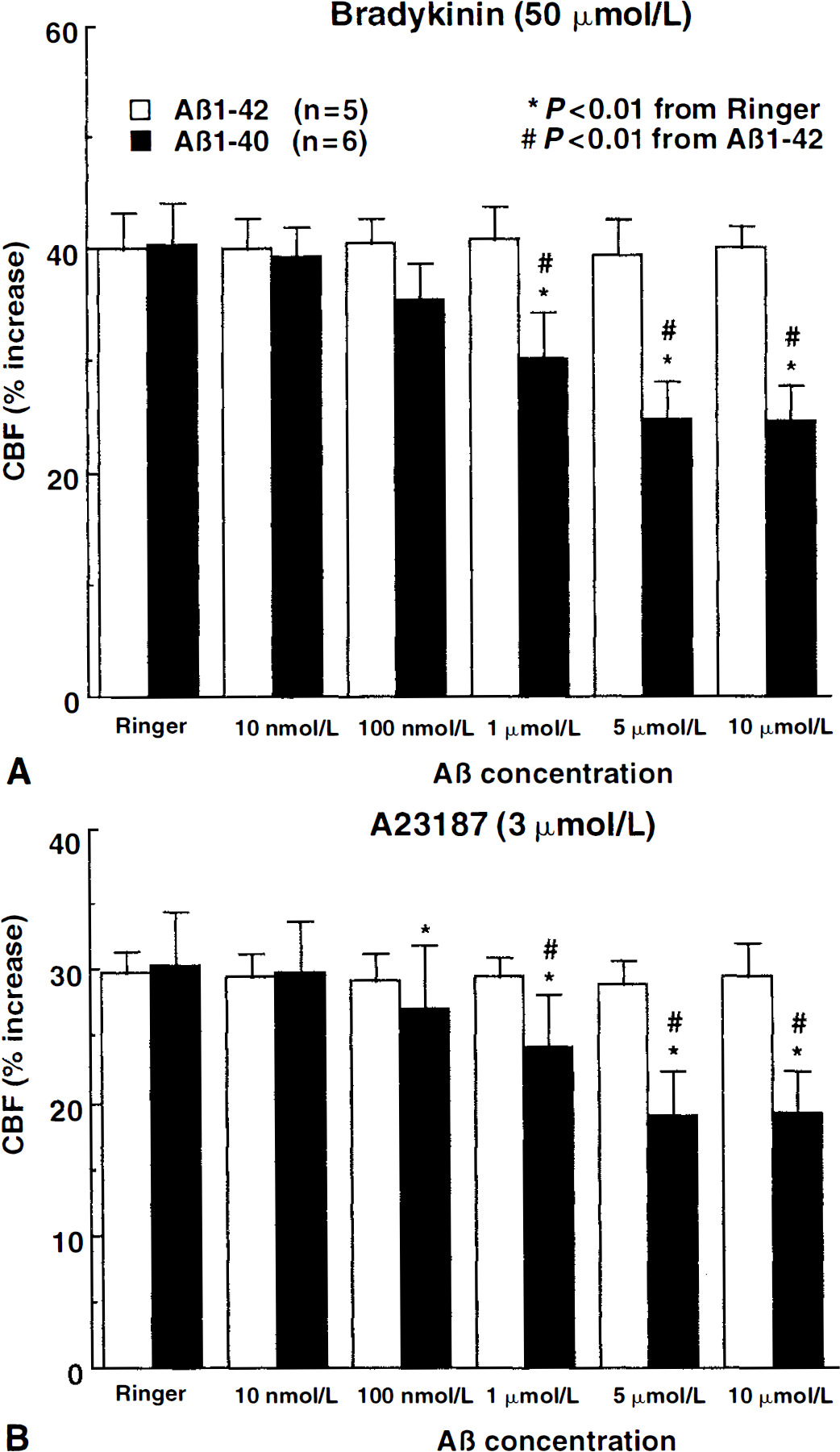
Effect of topical superfusion of Aβ1–40 and Aβ1–42 on the increase in cerebral blood flow (CBF) produced by bradykinin
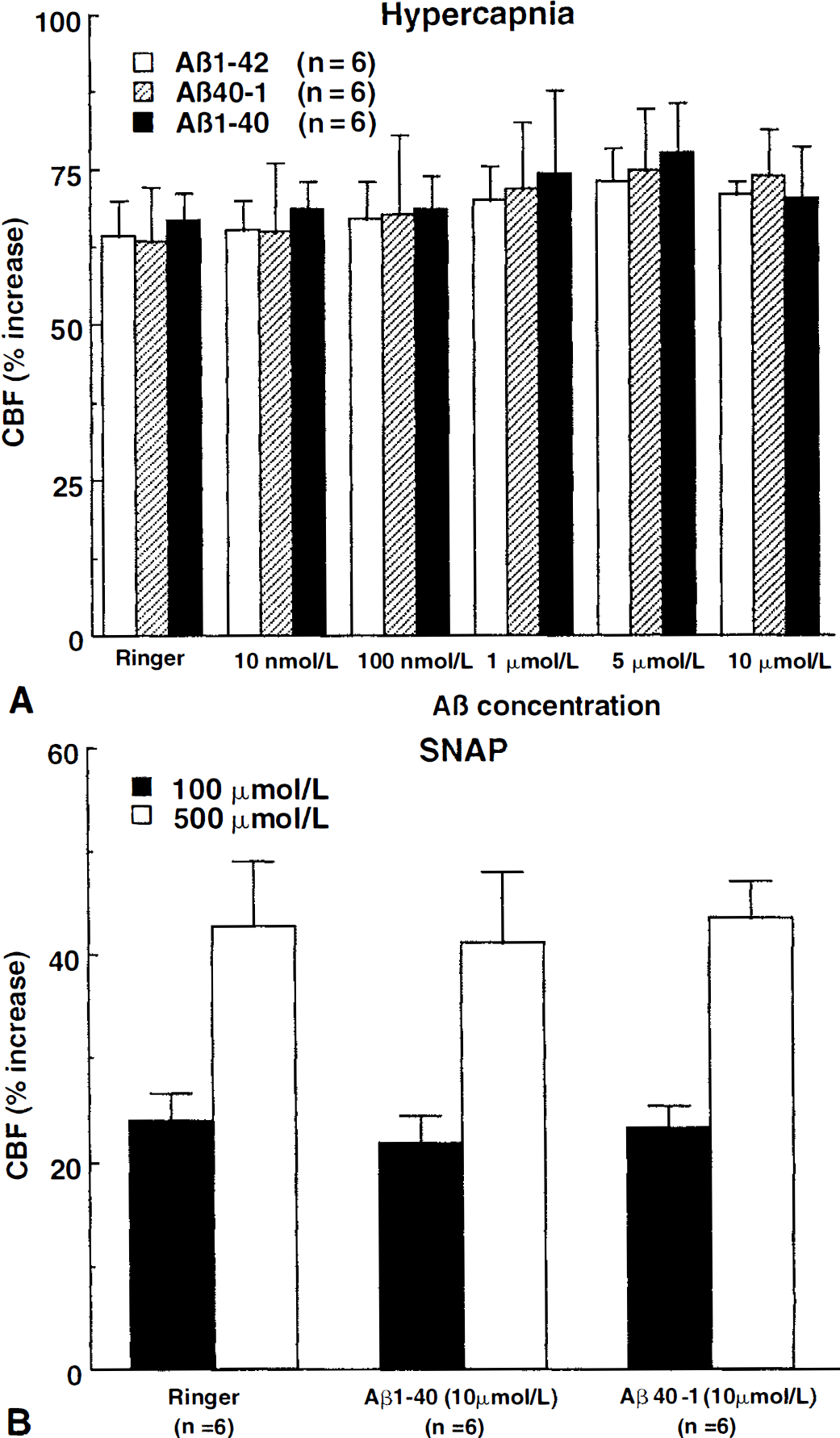
Effect of topical superfusion of Aβ1–40, Aβ1–42, or Aβ40–1 on the increase in cerebral blood flow (CBF) produced by hypercapnia
Effect of Aβ on cerebral glucose utilization
Cerebral blood flow is closely related to CGU, and changes in brain energy metabolism can influence cerebrovascular reactivity (Donegan et al., 1985). To determine whether the effects of Aβ1–40 on resting CBF and its reactivity to endothelium-dependent vasodilators were secondary to alterations in CGU, the effect of topical cortical superfusion with Aβ1–40 on CGU was studied. As previously reported (Iadecola and Xu, 1994), CGU in the exposed cortex was slightly greater than that of the contralateral side during Ringer superfusion (Table 2). However, during Aβ1–40 superfusion, CGU in the cortex exposed to the peptide was not different from that in the contralateral homotopic cortical area (Table 2).
Effect of SOD and MnTBAP on cerebrovascular actions of Aβ
In mice overexpressing APP and Aβ, the alterations in endothelium-dependent responses are counteracted by overexpression of the superoxide-scavenging enzyme SOD (Iadecola et al., 1999). Therefore, the authors investigated whether the cerebrovascular effects of Aβ1–40 also are reversed by the superoxide scavengers SOD or MnTBAP. During Ringer superfusion, Aβ1–40 (5 μmol/L) attenuated resting CBF and vascular response to ACh (Fig. 4). Superfusion with SOD (100 to 500 U/mL) or MnTBAP (25 to 100 μmol/L) counteracted the attenuation in a dose-dependent manner (Fig. 4). MnTBAP was more effective than SOD in restoring resting CBF and reactivity to Ach (Fig. 4), a finding that can be attributed to the smaller molecular weight and better brain penetration of MnTBAP (Day et al., 1995; Faulkner et al., 1994; Klann, 1998). In the absence of Aβ1–40, SOD (500 U/mL; n = 6) or MnTBAP (100 μmol/L; n = 5) did not influence resting CBF (before SOD: 19.4 ± 4.1, After: 19.5 ± 4.1 perfusion units; before MnTBAP: 18.5 ± 5.1; After: 18.5 ± 5.4 perfusion units; P > 0.05) or the increase in CBF produced by ACh (before SOD: 25% ± 3%, after: 25% ± 3%; before MnTBAP: 27% ± 3%, after: 26% ± 2%; P > 0.05).
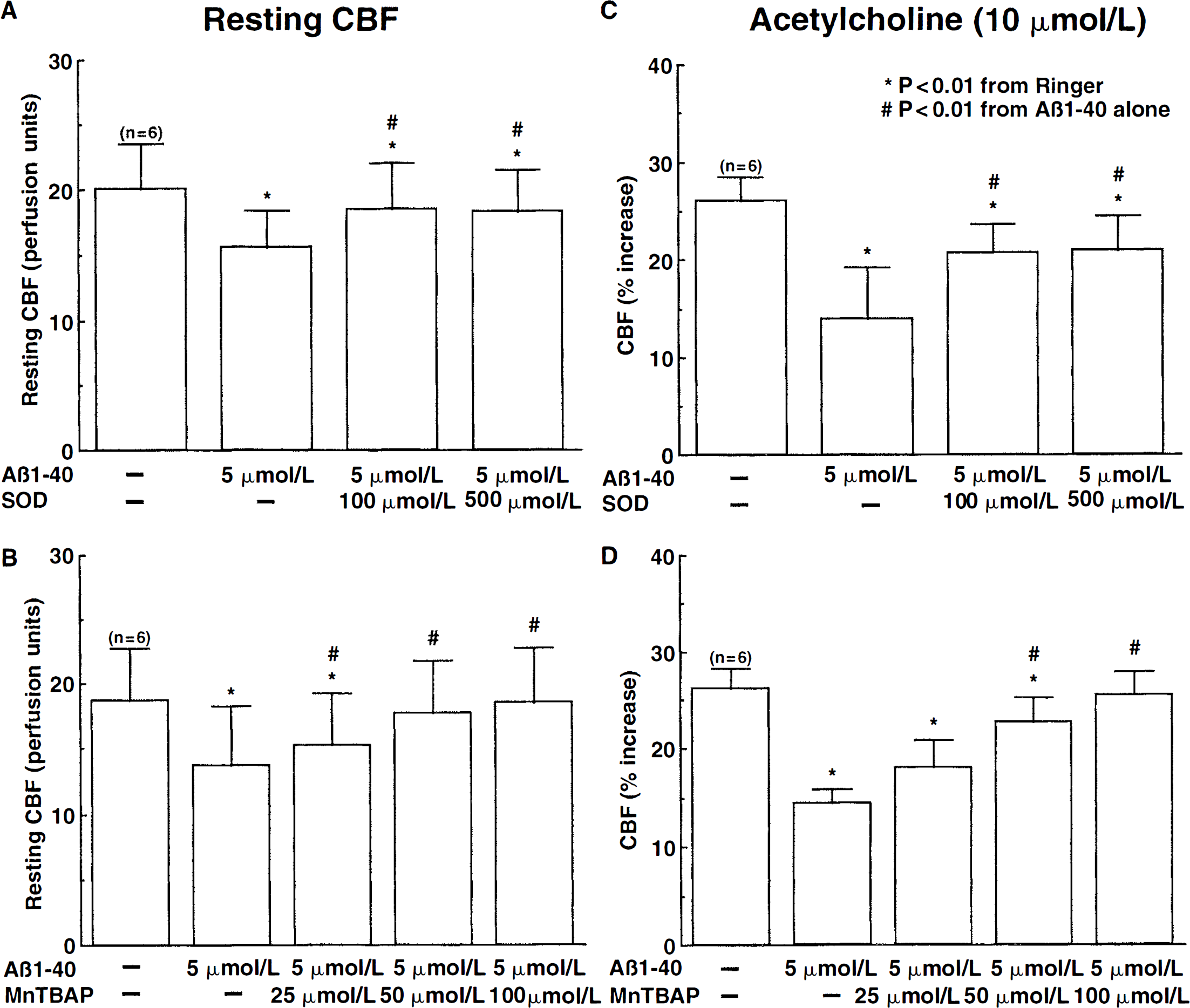
Effect of the free radical scavengers superoxide dismutase (SOD) and MnTBAP on the effect of Aβ1–40 on resting cerebral blood flow (CBF) (
Effect of Aβ1–40(M35Nle) on cerebral blood flow
Substitution of methionine 35 with the structurally-similar aminoacid norleucine attenuates the ability of Aβ to generate reactive oxygen species (Varadarajan et al., 1999). Therefore, the authors used Aβ1–40(M35Nle) to provide additional evidence in support of the hypothesis that the vascular effects of Aβ1–40 are mediated by reactive oxygen species. Aβ1–40(M35Nle) (S μmol/L) did not reduce resting CBF (before: 17.4 ± 3.2; after: 17.3 ± 3.6 perfusion units; P > 0.05; n = 6) and did not alter the increase in CBF produced by endothelium-dependent (ACh, BK, A23187) or endothelium-independent vasodilators (SNAP) (P>0.05; Fig. 5).
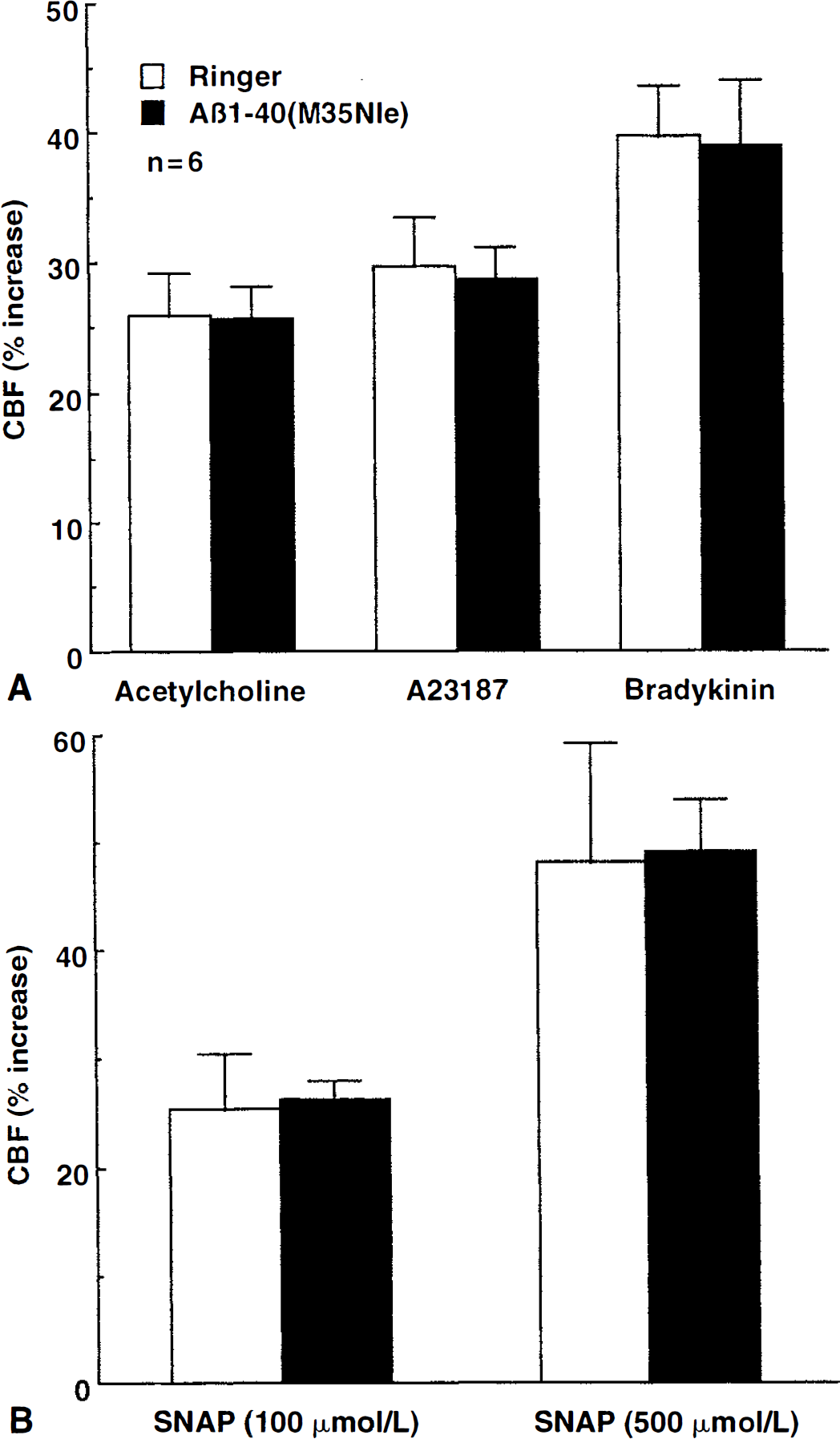
Effect of Aβ1–40(M35Nle) on the increase in cerebral blood flow (CBF) produced by the endothelium-dependent vasodilators, acetylcholine, A23187, and bradykinin
DISCUSSION
The authors sought to determine whether the alterations in endothelium-dependent relaxation observed in transgenic mice overexpressing APP result from APP itself or from peptides derived from its processing. Using a cranial window preparation in mice, the authors found that topical superfusion of the neocortex with Aβ1–40 attenuates the increase in CBF produced by the endothelium-dependent vasodilators ACh, BK, and A23187. The increase in CBF produced by hypercapnia or by the nitric oxide (NO) donor SNAP, vasodilator stimuli that do not act through the endothelium, was not affected. Aβ1–42 was devoid of cerebrovascular effects in this preparation. Impairment in endothelium-dependent responses produced by Aβ1–40 was completely reversed by SOD or MnTBAP, agents that scavenge superoxide. Furthermore, Aβ1–40(M35Nle), a mutated form of Aβ that does not generate reactive oxygen species, was devoid of cerebrovascular effects. These observations demonstrate that Aβ1–40 impairs the ability of cerebral endothelial cells to produce vasodilation and that such impairment is mediated by production of reactive oxygen species. Therefore, Aβ1–40 reproduces in full the cerebrovascular alterations observed in APP mice. Although the cerebrovascular effects of APP itself were not tested, the data support the hypothesis that the cerebrovascular alterations resulting from APP overexpression in transgenic mice are mediated by Aβ1–40.
ACh, BK, and A23187 dilate cerebral blood vessels through different endothelium-dependent mechanisms (Faraci, 1992; Faraci and Heistad, 1998). ACh acts by activating endothelial muscarinic receptors and releasing endothelial NO (Sobey and Faraci, 1997; Wei et al., 1992). BK activates endothelial BK receptors and produces vasodilation by releasing cyclooxygenase products (Mayhan, 1996; Rosenblum, 1987; Sobey et al., 1997), whereas A23187 is thought to act by increasing calcium in endothelial cells through receptor-independent mechanisms (Rosenblum et al., 1989; Rosenblum and Nelson, 1988). The fact that Aβ1–40 attenuates responses to ACh, BK, and A23187 suggests that this peptide produces a global failure in endothelium-dependent vasodilation rather than an alteration of a specific receptor or vasodilator mechanism.
Cerebrovascular effects of Aβ cannot result from changes in MAP and blood gases because these variables were carefully controlled and did not differ among groups. Similarly, the effects of Aβ1–40 cannot be attributed to a deterioration of the experimental preparation resulting in a progressive and nonspecific impairment in cerebrovascular reactivity because the CBF response to endothelium-independent vasodilators was not reduced over the same period of time. Furthermore, Aβ1–42 and Aβ40–1, which were tested using a timing similar to that of Aβ1–40, failed to alter resting CBF and to attenuate responses to ACh, BK, or A23187. Reversal by SOD or MnTBAP of the reduction in CBF and vascular responses Aβ cannot be caused by a nonspecific enhancement of resting CBF and of vasodilator responses. The authors have shown here that SOD and MnTBAP do not influence resting CBF and its reactivity to ACh. Similarly, hydrogen peroxide produced by SOD is unlikely to be responsible for the reversal of the endothelial alteration because this radical is toxic to endothelial cells (Day et al., 1997) and would be expected to exacerbate, rather than ameliorate, endothelial dysfunction. Furthermore, MnTBAP, an antioxidant that scavenges both superoxide and hydrogen peroxide (Day, et al., 1997), yielded results identical to SOD. Therefore, the findings of the current study cannot be attributed to instability of the experimental preparation or to nonspecific cerebrovascular effect of the pharmacologic agents used.
Cerebral blood flow is closely related to CGU, a variable reflecting neural activity. Changes in brain energy metabolism can have profound effects on cerebrovascular reactivity (Donegan et al., 1985). Therefore, the authors investigated whether Aβ1–40 affects CGU. The authors found that Ringer superfusion produces a small but significant increase in CGU in the exposed cortex (Iadecola and Xu, 1994), and that such an increase is abolished by Aβ superfusion. These data provide evidence that Aβ reduces cerebral energy metabolism. However, the small magnitude of the effect suggests that it is unlikely that the Aβ-induced attenuation in vascular reactivity is because of metabolic depression.
The authors found that Aβ–42 does not attenuate the increase in CBF produced by endothelium-dependent vasodilators. Aβ1–42 has a tendency to aggregate into fibrils, and the state of aggregation can influence its biologic effects (Pike et al., 1991, 1995; Weldon et al., 1998). In the authors' experiments, Aβ1–42 was studied immediately after solubilization, and the possibility that Aβ1–42 in a different aggregated state is more vasoactive cannot be ruled out. Furthermore, the authors have not examined whether Aβ1–42 enhances cerebral vasoconstriction, as has been reported for the isolated rat aorta (Crawford et al., 1998), and whether there is an interaction between the biologic activities of Aβ1–40 and Aβ1–42. It is surprising, however, that despite the fact that both Aβ1–40 and Aβ1–42 produce reactive oxygen species, only Aβ1–40 is highly vasoactive. Perhaps Aβ1–40 is more likely to generate free radicals in vascular cell. Further studies will be required to address this issue.
The observation that treatment with SOD or MnTBAP reversed the Aβ–induced impairment of endothelium-dependent responses indicates that reactive oxygen species are involved in the mechanisms of endothelial cell dysfunction. Consistent with this hypothesis, Aβl–40(M35Nle), a mutated form of Aβ that does not produce free radicals, does not influence endothelium-dependent vascular responses. Collectively, these findings provide strong evidence for an involvement of reactive oxygen species in the cerebrovascular effects of Aβ1–40. Although the data suggest that superoxide may be involved, the specific free radical mediating the vascular action of Aβ remains to be firmly established. In this context, it is of interest that Aβ did not attenuate the increase in CBF produced by the NO donor SNAP. Superoxide produced by Aβ could have reacted with NO generated by SNAP to form peroxynitrite (Beckman et al., 1990), resulting in an attenuation of SNAPs vasodilatator potency. One possible explanation for this finding is that the concentrations of SNAP used were sufficiently high to mask such superoxide-induced loss of NO.
The site of oxygen radical production and the mechanisms by which Aβ-induced reactive oxygen species alter the function of endothelial cells remain to be elucidated. Similar alterations in endothelium-dependent vascular responses are seen in hypertension, diabetes, and atherosclerosis—conditions in which there is increased production of reactive oxygen species at the vascular level (Harrison, 1997; Kontos, 1985; Schmidt et al., 1999). Therefore, it is possible that the free radicals responsible for endothelial cell dysfunction are also produced by vascular cells, for example, by the NAD(P)H oxidase system or by activation of RAGE receptors (Bianca et al., 1999; Meda et al., 1996; Yan et al., 1996). As for the mechanism of action, there is evidence that free radicals alter calcium homeostasis in endothelial cells and impair responses, such as release of vasoactive agents, initiated by calcium mobilization (Blanc et al., 1997; Mattson et al., 1993, 1997). However, it remains to be determined whether these alterations observed in vitro occur also in vivo in the experimental preparation.
Experiments in isolated cerebral arteries suggest that Aβ1–40 and Aβ–42 impair endothelium-dependent responses, an effect because of free radical—mediated endothelial cell destruction (Crawford et al., 1998; Thomas et al., 1996). In the current study, the authors demonstrated that the impairment in endothelium-dependent vasodilation produced by Aβ1–40 superfusion was completely reversed by treatment with superoxide scavengers. This observation suggested that the cerebrovascular alterations initiated by Aβ1–40, like those observed in APP mice (Iadecola et al., 1999), were caused by a reversible dysfunction of endothelial cells and not by endothelial cell destruction. Therefore, the mechanisms of Aβ-induced cerebrovascular impairment in vivo seem to be different from those in isolated arteries.
It is well known that AD is associated with morphologic alterations of cerebral blood vessels and with an impairment in the regulation of the cerebral circulation (Kalaria, 1999; de la Tone, 1999). Resting CBF is reduced in the early stages of AD, suggesting that the vascular dysfunction may be an early event that precedes major neuropathologic changes (Friedland, 1990). Recent findings suggest that Aβ-induced vascular alterations may have important implications for brain function. For example, the increase in somatosensory cortex CBF produced by vibrissal stimulation is attenuated in mice overexpressing APP, an effect with a magnitude proportional to brain Aβ concentration (Niwa et al., 2000b). Furthermore, the increase in CBF induced by functional activation is attenuated in asymptomatic patients at high risk for AD, identified on the basis of their ApoE genotype and family history (Smith et al., 1999). However, Aβ concentrations in the brains of patients with AD or APP mice are generally less (Hsiao et al., 1995; Neve and Robakis, 1998) than those described to have cerebrovascular effects in the current study or to be neurotoxic in vitro (Mattson, 1997). Such a discrepancy could be traced to two factors: first, because Aβ was superfused on the cortical surface, the concentration achieved within the substance of the cerebral cortex is likely to be less than that topically applied; second, unlike in APP mice and patients with AD in which the peptide accumulates over months or years, Aβ was applied only for a relatively short period of time in the current study. Therefore, it would be of interest to determine whether lower concentrations of synthetic Aβ applied over longer periods are vasoactive.
In conclusion, the authors demonstrated that exposure of the normal mouse neocortex to Aβ1–40, but not Aβ1–42, produced a profound and selective alteration in the regulation of the cerebral circulation by endothelial cells. Pharmacologic evidence and experiments with mutated Aβ suggest that such effect is mediated by Aβ1–40-induced formation of reactive oxygen species, which in turn, result in a reversible dysfunction of endothelial cells. The authors concluded that Aβ1–40 reproduces in full the cerebrovascular dysfunction observed in mice overexpressing mutated APP and that such impairment could be responsible for the alterations in CBF reported in patients with asymptomatic or early AD.
Footnotes
Acknowledgments
The authors thank Deborah Kabes and Tim Murphy for editorial assistance.