We investigated the l-arginine-induced, regional cerebral blood flow (rCBF) enhancement after different durations of transient focal cerebral ischemia in the rat to determine if l-arginine increases rCBF after transient focal cerebral ischemia. Focal ischemia (5 minutes and 20 minutes) followed by 90 minutes of reperfusion was induced in a normotensive rat suture-model. Regional cerebral blood flow in both hemispheres was measured by laser-Doppler-flowmetry. Reactivity of rCBF to l-arginine (300 mg/kg) was measured 45 minutes after reperfusion, and hypercapnia 90 minutes after reperfusion. The effect of d-arginine and pretreatment with the nitric oxide (NO) synthase inhibitor Nω-nitro-l-arginine (l-NA) (10 mg/kg) was examined in additional groups. Hypercapnia and l-arginine increased rCBF in sham operated controls and on the nonischemic hemispheres. d-arginine did not. Twenty-minute long ischemia significantly reduced the response to l-arginine (control side: 115 ± 5.9%; ischemic side: 107 ± 6.1%, n = 7) and hypercapnia, 5 minutes of ischemia did not. Nω-nitro-l-arginine pretreatment partly restored the l-arginine-induced rCBF increase. Thus, rCBF increase caused by l-arginine in the reperfusion period was unaffected by 5 minutes of ischemia, but reduced by 20 minutes of ischemia. The restoration after pretreatment with l-NA may be caused by attenuated production of cytotoxic substances, e.g., NO and related compounds.
Nitric oxide (NO) and NO donors are thought to act neuroprotectively in early stages of cerebral ischemia by improving CBF in the border zones of ischemia. Therefore, NO and NO donors may be of relevance for clinical stroke therapy. However, there have also been many reports on the neurotoxic effects of NO in cerebral ischemia (Dawson et al., 1993; Huang et al., 1994; Iadecola et al., 1994; Yoshida et al., 1994; Dawson, 1995).
l-arginine, the substrate for NO-generation, dilates pial arteries and increases regional cerebral blood flow (rCBF) under physiological conditions (Morikawa et al., 1992b; Morikawa et al., 1994). A nitric oxide-induced rCBF increase in the ischemic region was reported to be attenuated, when the drugs were applied 20 minutes after onset of ischemia (Dalkara et al., 1994; Zhang and Iadecola, 1994). It is unclear if the reduced effect of l-arginine in cerebral ischemia is simply caused by the hypoxic condition or if there is evidence for a dysfunction of vascular smooth muscle response to NO.
The formation of NO from l-arginine is limited by the amount of the substrate level and oxygen requirement (Matheis et al., 1992). Nitric oxide production measured by nitrite concentrations or by a porphyritic microsensor method showed an increase up to 20 or 25 minutes, respectively, and a subsequent decrease to baseline 60 minutes after onset of focal cerebral ischemia (Kader et al., 1993; Malinski et al., 1993). Nitric oxide increased at the beginning of the reperfusion period (Malinski et al., 1993).
The aim of our study was to determine the influence of transient focal cerebral ischemia on the l-arginine-induced rCBF increase in an area of compromised blood flow (25 to 50%) which has the potential to recover (Dirnagl, 1993), and to determine the dependency of the impairment of cerebrovascular reactivity on the duration of ischemia. The rCBF response to l-arginine was measured in the reperfusion period to exclude a lack of oxygen as the limiting factor of constitutive nitric oxide synthase activity. One additional group underwent pretreatment with a high dose of the nitric oxide synthase (NOS)-inhibitor Nω-nitro-l-arginine (l-NA) to study whether NO formation may contribute to the impairment of vascular responses in the reperfusion period.
MATERIALS AND METHODS
Rat model of transient focal cerebral ischemia
Adult male Wistar rats, weighing 260 to 300 g, were anesthetized with thiopental (100 mg/kg intraperitoneally), trache-otomized, and artificially ventilated with an animal ventilator (model 683, Harvard Apparatus, South Natick, MA, U.S.A.). A catheter was inserted into the right femoral artery to continuously measure mean arterial blood pressure and analyze blood gases. The right femoral vein was cannulated to substitute fluid and to apply drugs. The rats were kept at a constant temperature of approximately 37.5°C using a rectal thermometer-controlled heating pad.
Transient focal cerebral ischemia was induced by a modified method of intraluminal vascular occlusion (Longa et al., 1989). Briefly, the left common carotid artery was ligated and cannulated with a polyethylene tube. A 3–0 surgical nylon suture with its tip heated near a flame was inserted through the tube approximately 11 mm from the internal carotid artery/external carotid artery bifurcation toward the intracranial part of the internal carotid artery. After placement of the laser-Doppler probes the suture was advanced 17 to 19 mm from the internal carotid artery/external carotid artery bifurcation until the laser-Doppler flow decreased to ischemic levels. Reperfusion was initiated by pulling back the suture into the lumen of the common carotid artery and allowing recirculation via the circle of Willis. Animals were excluded if there was no ischemic laser-Doppler flow decrease, no sufficient reperfusion or a massive laser-Doppler flow decrease in the contralateral hemisphere after the final insertion of the suture, suggesting hemorrhagic infarction. At the end of the experiment, the base of skull was inspected to exclude hemorrhagic infarction.
Measurement of regional cerebral blood flow
Regional cerebral blood flow was continuously and simultaneously measured by a two channel laser-Doppler flowmeter (model Perimed, Periflux 4001 Master, Järfälla, Sweden) in both hemispheres. For placement of the laser-Doppler probes parts of the parietal bones were gently abraded to a transparent bone layer. The laser-Doppler flow probe was placed over an area free of large cerebral vessels. The measurement area was 5- to 6-mm lateral to midline and 1- to 2-mm caudal to bregma in both hemispheres. Before ischemia was induced, baseline cortical blood flow was monitored until it remained stable for at least 20 minutes. Then CO2 reactivity was tested to prove the integrity of the vascular bed. Another baseline of 30 minutes followed. Regional cerebral blood flow was expressed as percentage of change related to a baseline of 100%. At the end of the experiment, CO2 reactivity was measured a second time. Hypercapnia was produced by mechanically ventilating with 10% CO2, 21% O2, and the balance of N2. Arterial blood gases and pH was analyzed before and after 7 minutes of hypercapnia. CO2 reactivity was considered normal if there was an increase in rCBF of at least 2% per 1 mm Hg increase in Pco2 (Jones et al., 1989).
Materials and experimental groups
We used l-NA, l-arginine, and d-arginine (Sigma Chemicals, Deisenhofen, Germany). Nω-nitro-l-arginine (10 mg/kg body weight), l-arginine (300 mg/kg body weight), and D-arginine (300 mg/kg body weight) were dissolved in 0.9% saline.
Five experimental groups were investigated. Three groups (3, 4, and 5) were subjected to 20 minutes and one group (2) to 5 minutes of transient focal cerebral ischemia. Group 1 comprised sham-operated controls.
sham-operated controls (n = 5),
5 minutes of ischemia/90 minutes of reperfusion-l-arginine (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6).
20 minutes of ischemia/90 minutes of reperfusion-l-arginine (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 7).
20 minutes of ischemia/90 minutes of reperfusion-d-arginine (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6).
l-NA pretreatment (10 mg/kg intravenously/5 min) 20 minutes before ischemia - 20 minutes of ischemia/90 minutes of reperfusion-l-arginine (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6).
Small amounts of arterial blood were withdrawn to avoid an excessive mean arterial blood pressure increase above the upper limit of cerebral autoregulation (∼150 mm Hg) (Dirnagl and Pulsinelli, 1990) after infusion of l-NA.
Statistical analyses were performed by one-way analysis of variance and Student-Newman-Keuls procedure to detect group differences. To detect changes over time within each group a paired t-test was performed. Differences between hemispheres within each group were compared using an unpaired t-test. Data are given as mean ± SD.
RESULTS
Physiological variables
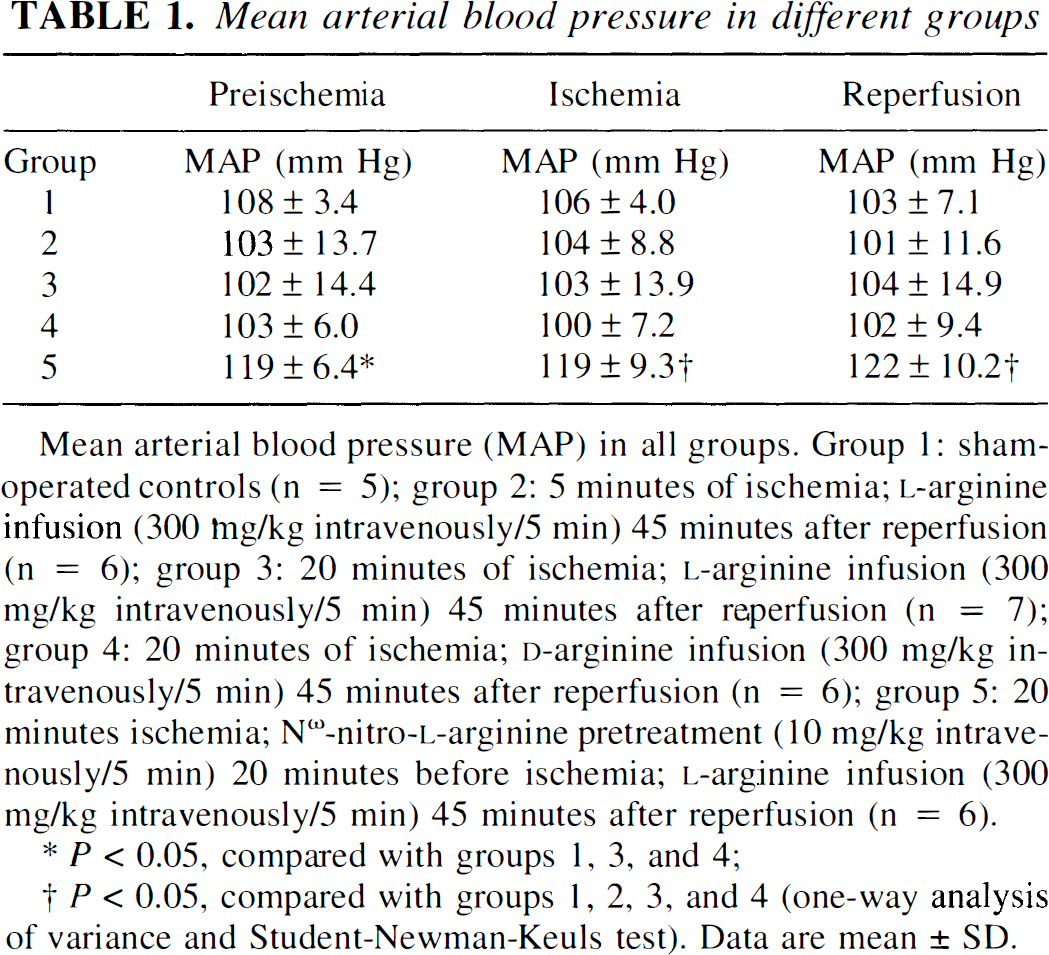
Mean arterial blood pressure, Po2, Pco2, pH, and hematocrit levels were within normal ranges in all experimental groups. In preliminary experiments the administration of l-NA induced an excessive increase in mean arterial pressure up to or even above the upper limit of cerebral autoregulation. Therefore, we modulated mean arterial pressure increase by withdrawing blood. Nevertheless, mean arterial pressure values were significantly higher in l-NA-treated than in untreated animals (Table 1).
Mean arterial blood pressure (MAP) in all groups. Group 1: sham-operated controls (n = 5); group 2: 5 minutes of ischemia; l-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6); group 3: 20 minutes of ischemia; l-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 7); group 4: 20 minutes of ischemia; d-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6); group 5: 20 minutes ischemia; Nω-nitro-l-arginine pretreatment (10 mg/kg intravenously/5 min) 20 minutes before ischemia; l-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6).
P < 0.05, compared with groups 1, 3, and 4;
P < 0.05, compared with groups 1, 2, 3, and 4 (one-way analysis of variance and Student-Newman-Keuls test). Data are mean ± SD.
l-Arginine effect in the reperfusion period
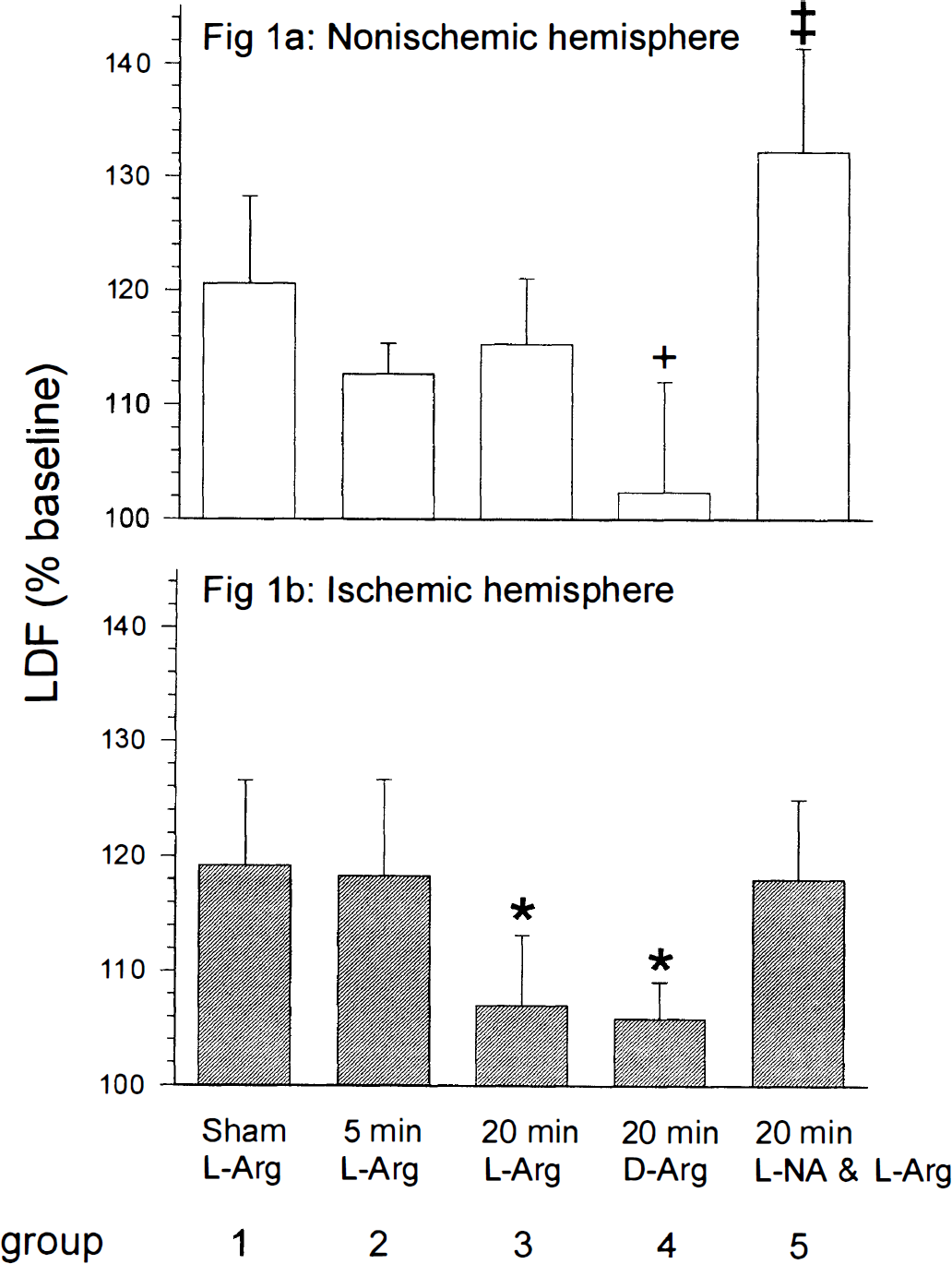
l-Arginine significantly increased rCBF in the nonischemic hemisphere in all groups (Fig. 1). In the ischemic hemisphere, the rCBF response to l-arginine was significantly reduced after 20 minutes, but not after 5 minutes of ischemia. In contrast to l-arginine, the stereoisomeric and normally inactive d-arginine had no effect on rCBF in either ischemic or control hemispheres within 30 minutes of infusion.
. Regional cerebral blood flow (rCBF) responses to l-arginine in the nonischemic and ischemic hemispheres. l-arginine was administered 45 minutes after reperfusion. The rCBF values were recorded 30 minutes after l-arginine infusion. (a) In the nonischemic hemispheres (control) rCBF increase in response to l-arginine administration was apparent in all groups. Nω-nitro-l-arginine (l-NA) pretreatment enhanced the l-arginine-induced rCBF increase (‡ P < 0.05, compared with corresponding hemispheres of groups 1, 2, 3, and 4 using one-way analysis of variance and Student-Newman-Keuls test). d-arginine had no significant effect on rCBF (+ P < 0.05, compared with groups 1, 2, 3, and 5 using one-way analysis of variance and Student-Newman-Keuls test). (b) Twenty minutes, but not 5 minutes of ischemia significantly reduced rCBF response to l-arginine in the ischemic hemisphere (*P < 0.05, compared with groups 1, 2, and 5 using one-way analysis of variance and Student-Newman-Keuls test). There was a significant difference between the ischemic and the nonischemic hemispheres in l-arginine-induced rCBF increase after 20 minutes of ischemia (P < 0.05, using unpaired Student's t-test). No significant difference was observed between rCBF values after administration of either l-arginine or d-arginine. Nω-nitro-l-arginine pretreatment partly restored rCBF response to l-arginine in the ischemic hemisphere, although there was still a significant difference between the ischemic and the nonischemic hemispheres (P < 0.05, using unpaired Student's t-test). Data are mean ± SD. LDF, laser-Doppler flow.
Pretreatment with l-NA partly restored l-arginine-induced rCBF increase in the ischemic hemisphere, and enhanced rCBF response to l-arginine in the nonischemic hemisphere. There was a significant difference in the rCBF change between the ischemic and the nonischemic hemisphere (Fig. 1).
Nω-nitro-l-arginine attenuated hyperemia during early reperfusion, but had no effect on rCBF during ischemia and advanced reperfusion
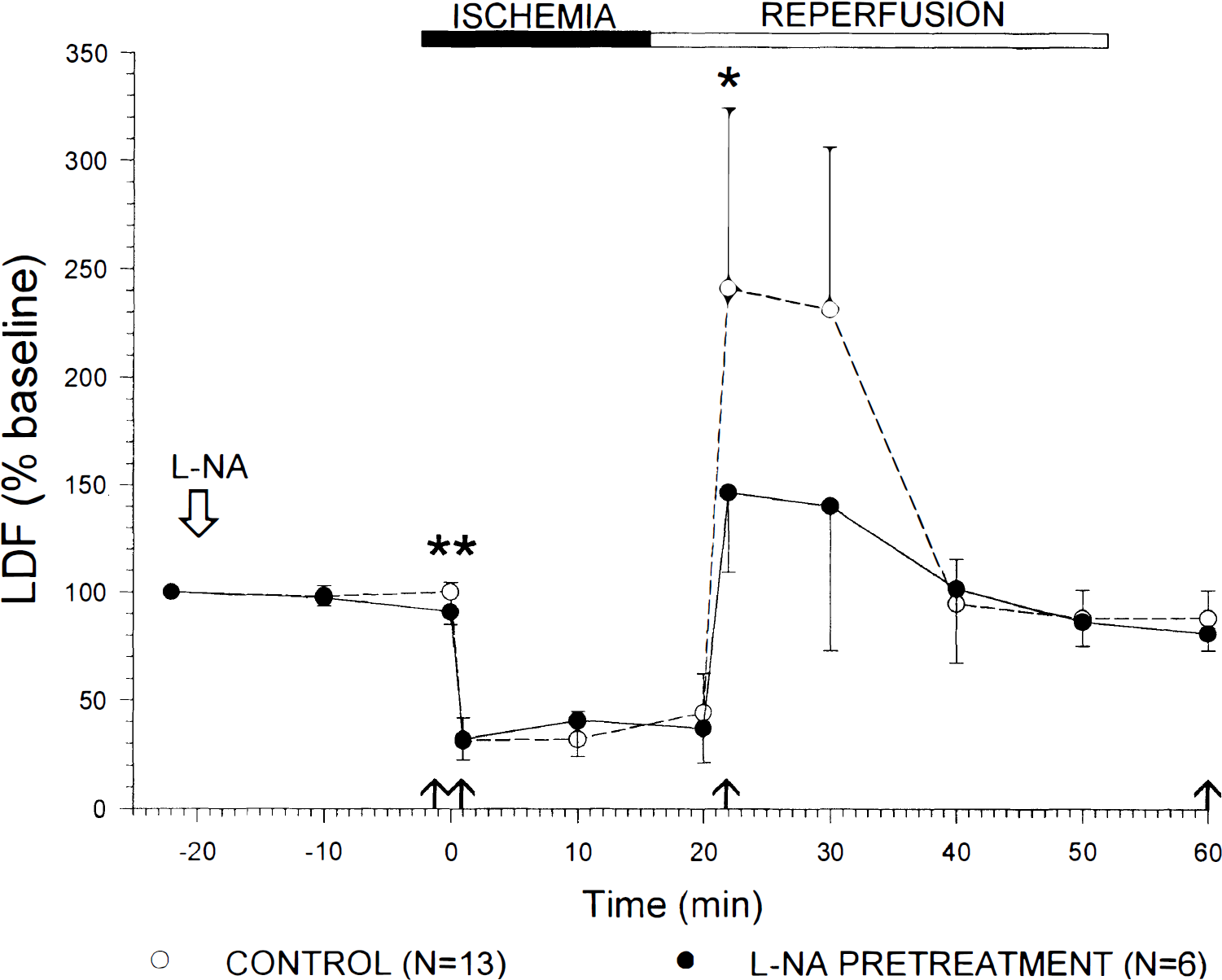
Preischemic rCBF values were significantly decreased in both hemispheres 20 minutes after administration of l-NA (ischemic hemisphere: 91 ± 6%; control hemisphere: 82 ± 9% of baseline). This effect reached a maximum in the control hemisphere 30 minutes after l-NA application (78 ± 9% of baseline), and was still detected until l-arginine was applied. On occlusion of the middle cerebral artery, rCBF decreased to 32 ± 7% in group 2, 32 ± 7% in group 3, and 25 ± 7% in group 4 of the preischemic level without significant differences between these groups. Nω-nitro-l-arginine treatment had no effect on ischemic rCBF values (31 ± 8%; not significant). On reperfusion, hyperperfusion developed (group 2: 180 ± 36%; groups 3 and 4: 251 ± 83%; not significant), followed by mild hypoperfusion (rCBF at 45 minutes of reperfusion, group 2: 73 ± 6%; group 3 and 4: 88 ± 12%). Nω-nitro-l-arginine treatment significantly reduced hyperperfusion (146 ± 37%, P < 0.05 compared with groups 3 and 4; Fig. 2), but did not influence hypoperfusion (81 ± 8%).
Effect of l-nitro-l-arginine (l-NA) pretreatment on rCBF after 20 minutes of transient focal ischemia in l-NA pretreated versus untreated rats. rCBF significantly decreased 20 minutes after administration of l-NA (**; unpaired Student's t-test; P < 0.001). Intraischemic rCBF values are not influenced by l-NA therapy. l-nitro-l-arginine significantly attenuated rCBF values during early reperfusion (*; unpaired Student's t-test; P < 0.05). LDF, laser-Doppler flow. ↑ time points of statistical analysis. Data are mean ± SD.
Influence of transient focal cerebral ischemia on hypercapnic reactivity
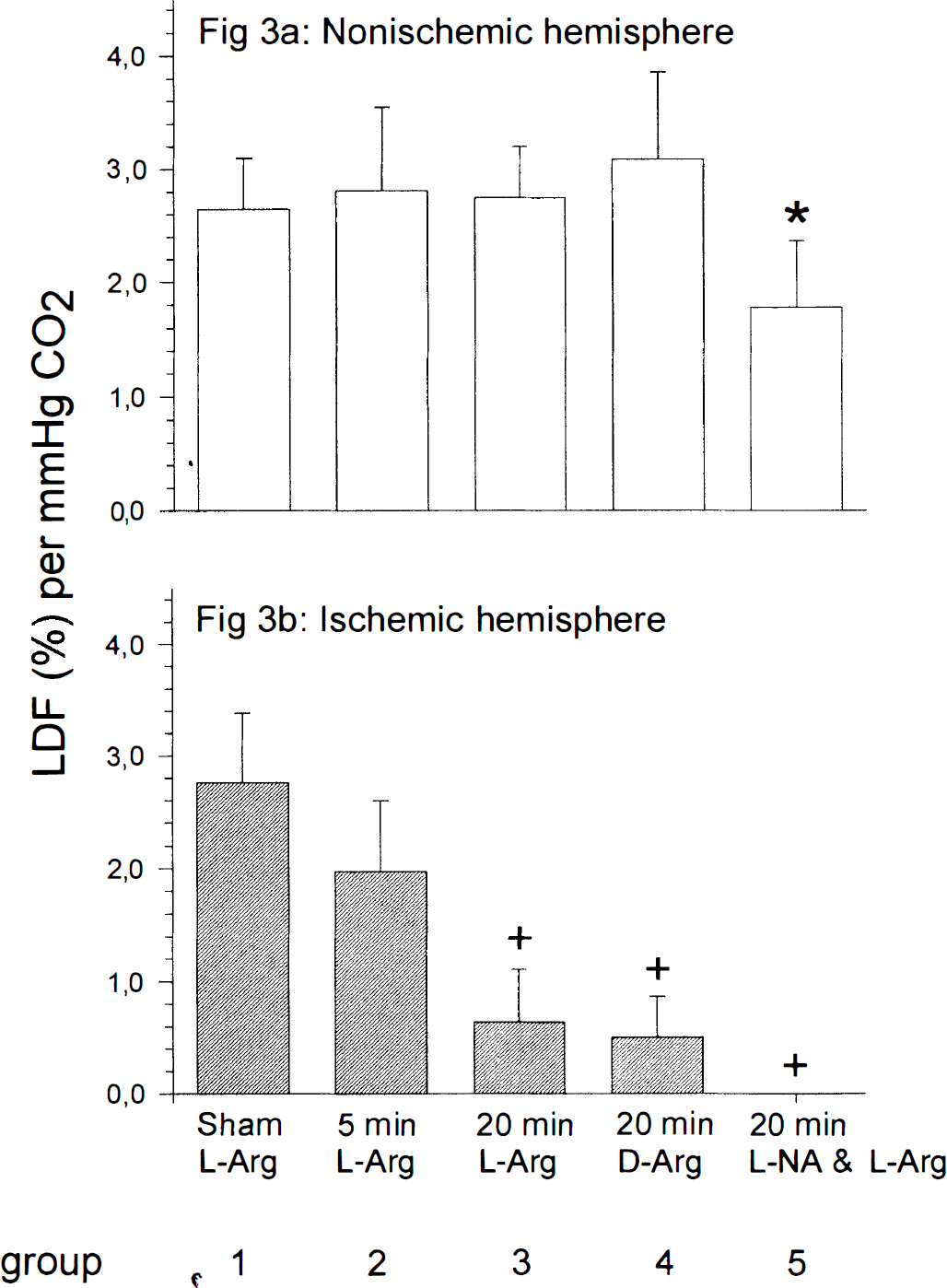
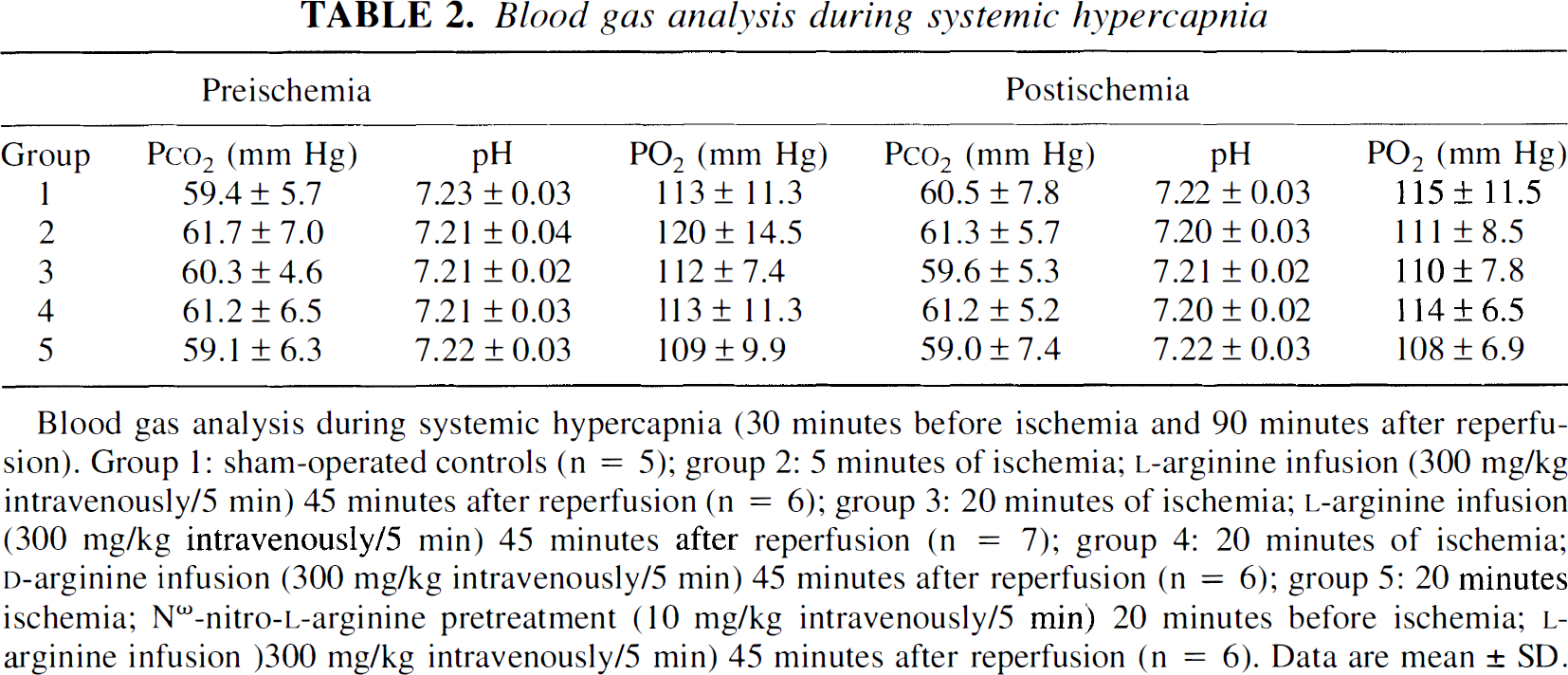
At the beginning of the experiments, hypercapnic reactivity was within normal ranges in both hemispheres in all groups investigated (data not shown). In the ischemic hemisphere, the rCBF response to hypercapnia was significantly reduced after 20 minutes, but not after 5 minutes of ischemia (Fig. 3; Table 2). Neither l-NA nor l-arginine nor d-arginine had a significant effect on the reduction of hypercapnic reactivity. In the nonischemic hemisphere no change of CO2 reactivity was observed, with the exception of group 5 (pretreatment with l-NA), in which CO2 reactivity was slightly reduced (1.78 ± 0.59%).
. CO2 reactivity after transient cerebral ischemia in the nonischemic and ischemic hemispheres. Measurements of hypercapnic reactivity were performed 30 minutes before ischemia and 90 minutes after reperfusion in all groups. (a) In the nonischemic hemispheres, hypercapnic reactivity was within normal range in all groups except of the l-NA pretreated group. Nω-nitro-l-arginine pretreatment caused a slight reduction of hypercapnic reactivity (* P < 0.05, compared with groups 1, 2, 3, and 4 using one-way analysis of variance and Student-Newman-Keuls test). (b) In the ischemic hemispheres 20 minutes but not 5 minutes of ischemia caused a marked reduction of CO2 reactivity (+ P < 0.05, compared with groups 1 and 2 using one-way analysis of variance and Student-Newman-Keuls test). In addition, there was a significant difference in CO2-induced rCBF increase between the nonischemic and the ischemic hemispheres of groups 3, 4, and 5 (P < 0.001, compared with the control hemispheres using unpaired Student's t-test). LDF, laser-Doppler flow. Data are mean ± SD.
Blood gas analysis during systemic hypercapnia
Preischemia
Postischemia
Group
Pco2 (mm Hg)
pH
PO2 (mm Hg)
Pco2 (mm Hg)
pH
PO2 (mm Hg)
1
59.4 ± 5.7
7.23 ± 0.03
113 ± 11.3
60.5 ± 7.8
7.22 ± 0.03
115 ± 11.5
2
61.7 ± 7.0
7.21 ± 0.04
120 ± 14.5
61.3 ± 5.7
7.20 ± 0.03
111 ± 8.5
3
60.3 ± 4.6
7.21 ± 0.02
112 ± 7.4
59.6 ± 5.3
7.21 ± 0.02
110 ± 7.8
4
61.2 ± 6.5
7.21 ± 0.03
113 ± 11.3
61.2 ± 5.2
7.20 ± 0.02
114 ± 6.5
5
59.1 ± 6.3
7.22 ± 0.03
109 ± 9.9
59.0 ± 7.4
7.22 ± 0.03
108 ± 6.9
Blood gas analysis during systemic hypercapnia (30 minutes before ischemia and 90 minutes after reperfusion). Group 1: sham-operated controls (n = 5); group 2: 5 minutes of ischemia; l-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6); group 3: 20 minutes of ischemia; l-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 7); group 4: 20 minutes of ischemia; d-arginine infusion (300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6); group 5: 20 minutes ischemia; Nω-nitro-l-arginine pretreatment (10 mg/kg intravenously/5 min) 20 minutes before ischemia; l-arginine infusion)300 mg/kg intravenously/5 min) 45 minutes after reperfusion (n = 6). Data are mean ± SD.
DISCUSSION
The present study shows that 20 minutes (but not 5 minutes) of transient focal cerebral ischemia followed by 90 minutes of reperfusion diminishes the rCBF increase elicited by l-arginine or hypercapnia. Pretreatment with l-NA attenuated cerebral hyperperfusion in the early reperfusion period. In addition, l-NA partly restored rCBF increase after administration of l-arginine. However, l-NA did not prevent loss of the hypercapnic reactivity of cerebral vessels.
The mode of action of exogenous l-arginine is still under discussion. The fact that endogenous l-arginine is present in amounts that far exceed the Km of NOS suggests that administering l-arginine should not enhance the synthesis of NO. However, previous studies showed that either topical or systemic l-arginine administration may induce an increase in rCBF under physiologic and pathophysiologic conditions (Morikawa et al., 1994). The linkage of the l-arginine effect to the l-arginine/NO pathway was generally shown by the ineffectiveness of d-arginine and/or by the blockade with NOS inhibitors. There are several possible explanations for the so-called “l-arginine paradox” (Forstermann et al., 1994). First, the “functional” Km in intact cells may be higher than that of the purified enzyme. Second, an endogenous inhibitor of NOS may cause an increased requirement of l-arginine (Faraci et al., 1995; Vallance et al., 1992). Third, exogenous l-arginine induces NO release by an indirect mechanism. Arnal et al. reported that l-arginine was able to reverse the inhibitory effect of l-glutamine on receptor-mediated NO release (Arnal et al., 1995).
In experimental stroke models, exogenous l-arginine was found to decrease infarction volume. This protective effect of l-arginine therapy is probably caused by an increase of rCBF in the ischemic penumbra (Morikawa et al., 1992 a,
b
; Morikawa et al., 1994). Thus, l-arginine (30 or 300 mg/kg intravenously), when administered early after onset of ischemia (5 minutes), enhanced ischemic rCBF and reduced infarct volume after permanent middle cerebral artery occlusion in spontaneously hypertensive rats and Sprague-Dawley rats (Morikawa et al., 1992a; Morikawa et al., 1994). Dalkara et al. (1994) found a significantly greater rCBF increase after early rather than delayed l-arginine administration in a spontaneously hypertensive rat model of focal cerebral ischemia. However, after photothrombotic occlusion of the middle cerebral artery, l-arginine did not enhance CBF and did not reduce infarction volume in spontaneously hypertensive rats after early or late application (Prado et al., 1996). The inconsistent findings may be explained by differences in the experimental models. Ischemia is complicated by thrombosis in the photothrombotic stroke model. Tissue exposed to thrombosis combined with ischemia revealed considerably more severe changes than those exposed to mechanically induced ischemia (Dietrich et al., 1989; Prado et al., 1996).
Thrombosis combined with ischemia may aggravate reperfusion injury, possibly by releasing of platelet-derived substances (Dietrich et al., 1988; Wester et al., 1992; Prado et al., 1996). In the mechanically induced middle cerebral artery occlusion model, the beneficial effect of l-arginine therapy seems to depend on the time of drug administration after onset of cerebral ischemia. This suggests that a reduction in NO production within the ischemic endothelium or a decrease of the smooth muscle response to NO accounts for the loss of reaction after longer lasting ischemia (Dalkara et al., 1994). Nitric oxide synthase activity and NO/nitrite concentration within ischemic tissue were measured in rat models of middle cerebral artery occlusion (Kader et al., 1993; Malinski et al., 1993; Zhang et al., 1994). Initiation of ischemia caused NO/nitrite concentrations to increase for up to 20 minutes, to peak at 10 to 20 minutes and to decrease at 60 minutes (Kader et al., 1993; Malinski et al., 1993). Similarly, NOS activity increased approximately 10-fold from baseline levels 10 minutes after occlusion but decreased by 60 minutes after initiation of ischemia (Kader et al., 1993). Reperfusion after 2 hours of ischemia stimulated an increase in NO concentrations to above baseline levels for at least 1 hour (Malinski et al., 1993). In our study, l-NA pretreatment attenuated postischemic hyperemia. In an earlier study, inhibition of NOS with l-NAME decreased early postischemic hyperemia after global cerebral ischemia in piglets (Greenberg et al., 1995). These findings suggest the integrity of NO production and the vascular smooth muscle response to NO during early reperfusion after short-term ischemia.
Because NO increase during reperfusion (Malinski et al., 1993) indicates the integrity of NO synthesis, the inefficacy of l-arginine therapy may be caused by a reduction of the vascular smooth muscle response to NO. The microvascular bed may be damaged by the release of cytotoxic substances, e.g., NO and reactive oxygen species. Similar to NO, superoxide anion is generated during reperfusion (Nelson et al., 1992; Schreiber et al., 1995). The simultaneous production of NO and superoxide may cause formation of cytotoxic peroxynitrite anion (Beckman, 1990; Beckman et al., 1990), which exerts a variety of cytopathic effects, including initiation of membrane lipid peroxidation (Rubbo et al., 1994) and inhibition of mitochondrial electron transport (Radi et al., 1994). The incomplete restoration of the l-arginine/NO-induced rCBF increase by l-NA treatment may be caused by an incomplete reduction in the levels of these cytotoxic substances.
Whereas l-arginine reactivity was partly restored after l-NA pretreatment, hypercapnic vasodilation remained abolished. Similar to the present study, earlier investigations showed a blunted response of CBF to systemic hypercapnia in the core and adjacent areas of ischemia (Jones et al., 1989). The reason for the differential effect of l-NA pretreatment on the response to l-arginine and hypercapnia is unclear. Although previous studies suggest that NO may be a mediator of hypercapnic dilation in the brain (Iadecola and Zhang, 1994; Wang et al., 1994), there are other mechanisms involved in CBF regulation during hypercapnia (Wang et al., 1994). In addition, different isoforms of the NOS may be involved, i.e. the endothelial NOS in l-arginine induced vasodilation and the neuronal NOS in hypercapnia (Dawson, 1994; Iadecola et al., 1994; Wang et al., 1995). The difference in the restoration of vascular responsiveness after reperfusion by l-NA supports the hypothesis that there is a selective vulnerability of vessel wall reactivity after ischemia, rather than a global loss of cell vascular response.
Footnotes
Abbreviations used
References
1.
ArnalJFMünzelTVenemaRCJamesNLBaiCLMitchWEHarrisonDG (1995) Interactions between L-arginine and L-glutamine change endothelial NO production. An effect independent of NO synthase substrate availability. J Clin Invest95:2565–2572
BeckmanJSBeckmanTWChenJMarshallPAFreemanBA (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Acad Natl Sci USA87:1620–1624
4.
DalkaraTMorikawaEPanahianNMoskowitzMA (1994) Blood flow-dependent functional recovery in a rat model of focal cerebral ischemia. Am J Physiol267:H678–H683
5.
DawsonDA (1994) Nitric oxide and focal cerebral ischemia: multiplicity of actions and diverse outcome. Cerebrovasc Brain Metab Rev6:299–324
6.
DawsonVLDawsonTMBartleyDAUhlGRSnyderSH (1993) Mechanism of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci13:2651–2661
7.
DawsonVL (1995) Nitric oxide: role in neurotoxicity. Clin Exp Pharmacol Physiol22:305–308
DietrichWDNakayamaHWatsonBDKanemitsuH (1989) Morphological consequences of early reperfusion following thrombotic or mechanical occlusion of the rat middle cerebral artery. Acta Neuropathol78:605–614
10.
DirnaglU (1993) Cerebral ischemia: the microcirculation as trigger and target. Prog Brain Res96:49–64
11.
DirnaglUPulsinelliW (1990) Autoregulation of cerebral blood flow in experimental focal brain ischemia. J Cereb Blood Flow Metab10:327–336
12.
FaraciFMBrianJEJrHeistadDD (1995) Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol269:H1522–H1527
GreenbergRSHelfaerMAKirschJRTraystmanRJ (1995) Effect of nitric oxide synthase inhibition on postischemic cerebral hyperemia. Am J Physiol38:H341–H347
15.
HuangZHuangPLPanahianNDalkaraTFishmanMCMoskowitzMA (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science265:1883–1885
IadecolaCZhangF (1994) Nitric oxide-dependent and -independent components of cerebrovasodilatation elicited by hypercapnia. Am J Physiol266:R564–R552
18.
JonesSCBoseBFurianAJFrielHTEasleyKAMeredithMPLittleJR (1989) CO2 reactivity and heterogeneity of cerebral blood flow in ischemic, border zone, and normal cortex. Am J Physiol257:H473–H482
19.
KaderAFrazziniVISolomonRATrifilettiRR. (1993) Nitric oxide production during local cerebral ischemia in rats. Stroke24:1709–1716
20.
LongaEZWeinsteinPRCarlsonSCumminsR (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke20:84–91
21.
MalinskiTBaileyFZhangZGChoppM (1993) Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab13:355–358
22.
MatheisGShermanMPBuckbergGDHaybronDMYoungHHIgnarroLJ (1992) Role of L-arginine-nitric-oxide pathway in myocardial reoxygenation injury. Am J Physiol262:H616–H620
23.
MorikawaEHuangZMoskowitzMA (1992a) L-arginine decreases infarct size caused by middle cerebral arterial occlusion in SHR. Am J Physiol263:H1632–H1635
24.
MorikawaERosenblattSMoskowitzMA (1992b) L-arginine dilates rat pial arterioles by nitric oxide-dependent mechanisms and increases blood flow during focal cerebral ischaemia. Br J Pharmacol107:905–907
25.
MorikawaEMoskowitzMAHuangZYoshidaTIrikuraKDalkaraT. (1994) L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke25:429–435
26.
NelsonCWWeiEPPovlishockJTKontosHAMoskowitzMA (1992) Oxygen radicals in cerebral ischemia. Am J Physiol263:H1356–H1362
27.
PradoRWatsonBDZhaoWYaoHBustoRDietrichWDGinsbergMD (1996) L-arginine does not improve cortical perfusion or histopathological outcome in sponaneously hypertensive rats subjected to distal middle cerebral artery photothrombotic occlusion. J Cereb Blood Flow Metab16:612–622
28.
RadiRRodriquezMCastroLTelleriR (1994) Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys308:89–95
29.
RubboHRadiRTrujilloMTelleriRKalyanaramanBBarnesSKirkMFreemanBA (1994) Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem269:26066–26075
30.
SchreiberSJMegowDRaupachAVictorovIVDimaglU (1995) Age-related changes of oxygen free radical production in the rat brain slice after hypoxia: on-line measurement using enhanced chemiluminescence. Brain Res703:227–230
31.
VallancePLeoneACalverACollierJMoncadaS (1992) Accumulation of an endogeneous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet339:572–575
32.
WangQPelligrinoDAPaulsonOBLassenNA (1994) Comparison of the effects of NG-nitro-L-arginine and indomethacin on the hypercapnic cerebral blood flow increase in rats. Brain Res641:257–264
33.
WangQPelligrinoDABaughmanVLKoenigHMAlbrechtRF (1995) The role of neuronal nitric oxide synthase in regulation of cerebral blood flow in normocapnia and hypercapnia in rats. J Cereb Blood Flow Metab15:774–778
34.
WesterPDietrichWDPradoRWatsonBDGlobusMYT (1992) Serotonin release into plasma following common carotid artery thrombosis. Stroke23:870–875
35.
YoshidaTLimmrothVIrikuraKMoskowitzMA (1994) The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab14:924–929
36.
ZhangFIadecolaC (1994) Reduction of focal cerebral ischemic damage by delayed treatment with nitric oxide donors. J Cereb Blood Flow Metab14:574–580
37.
ZhangZGChoppMBaileyFMalinskiT (1994) Nitric oxide changes in the rat brain after transient middle cerebral artery occlusion. J Neurol Sci128:22–27