
Abstract
Transient brain ischemia induces an inhibition of translational rates and causes delayed neuronal death in selective regions and cognitive deficits, whereas these effects do not occur in resistant areas. The translational repressor eukaryotic initiation factor (elF) 4E-binding protein-2 (4E-BP2) specifically binds to eIF4E and is critical in the control of protein synthesis. To link neuronal death to translation inhibition, we study the eIF4E association with 4E-BP2 under ischemia reperfusion in a rat model of transient forebrain ischemia. Upon reperfusion, a selective neuronal apoptosis in the hippocampal
INTRODUCTION
Transient ischemia induces an inhibition of translational rates and causes selective neuronal death and cognitive deficits. Ischemia is a stressful situation involving energy metabolism inhibition due to lack of oxygen. Faced with this adverse situation, cells respond with an inhibition of protein synthesis. Ischemia induces a period of hypoxia and energy depletion that is restored in the subsequent reperfusion period upon reoxygenation.
1
Moreover, the initial period of reperfusion increases reactive oxygen species production and causes additional stress.
2
The stress that results from ischemia and ischemia reperfusion (IR) affects different tissues. The brain, because of its high metabolic rate, limited energy stores, and critical dependence on aerobic metabolism, is known to be particularly sensitive to these stresses, where the restoration of translation inhibition is delayed compared with that of energy metabolism or ion homeostasis.2,3 Nonetheless, if the state of abnormally low protein synthesis is maintained over time, it can lead to cell death.4,5 The neuronal degeneration induced by IR stress is followed in time by secondary cell death in adjacent and/or connected areas, which is known as delayed neuronal death.
6
This delayed neuronal death occurs in the selectively vulnerable regions of the penumbra in focal ischemia,7–9 and mainly in the hippocampal
An important control point in the translation process in eukaryotic organisms is the recruitment of the 40S ribosomal subunit to the 5’ end of mRNA. A key step in this process is the assembly of eukaryotic initiation factor (eIF) 4F complex, which contains the initiation factor eukaryotic initiation factor 4E (eIF4E). eIF4E recruits eIF4G and eIF4A to assemble the eIF4F complex and bind to the 5’ cap. 13 The availability of eIF4E is a limiting step in translation initiation and is a primary factor in the control of gene expression. The family of translational repressors named eIF4E-binding proteins (4E-BPs), which in mammals comprise three members (4E-BP1, 4E-BP2, and 4E-BP3), share with eIF4G a common binding motif to eIF4E that is mutually exclusive.13,14 Active forms of 4E-BPs bind to eIF4E, compete with eIF4G, and inhibit eIF4G binding to eIF4E, which prevents eIF4F complex formation and inhibits cap-dependent translation.5,13
To link neuronal death to translation inhibition, we study the eIF4E association with 4E-BP2, the predominant 4E-BP expressed in brain, 15 under IR stress in a rat model of transient forebrain ischemia. In the present report, we investigate the critical participation of 4E-BP2 in delayed neuronal death after ischemic reperfusion. Interestingly, we discovered specific changes in the association of 4E-BP2 to eIF4E, demonstrating that the association of 4E-BP2 to eIF4E correlates with translation inhibition and delayed neuronal death induced by IR stress in brain.
MATERIALS AND METHODS
Materials
Rabbit polyclonal anti-4E-BP2 antibodies were from Sigma (Madrid, Spain) and Cell Signaling (Beverly, MA, USA). Mouse monoclonal anti-eIF4E antibody was from BD Transduction Labs (Erembodegem, Belgium). Goat polyclonal anti-eIF4G1 antibody was from Santa Cruz Biotech (Santa Cruz, CA, USA). Rabbit polyclonal anti-4E-BP1 antibody was from Cell Signaling. Rabbit polyclonal anti-β-tubulin antibody was from Covance (Emeryville, CA, USA). Mouse monoclonal anti-α-spectrin antibody was from Chemicon (Temecula, CA, USA). The chemicals used in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad (Madrid, Spain) and GE Healthcare (Barcelona, Spain). Peptides RIIYDRKFLLDR and RIIADRKFAADR linked to penetratin and amidate were from Mimotopes (Melbourne, VI, Australia). siRNA targeted rat 4E-BP2 was purchased from Life Tech (Madrid, Spain) and TransMessenger reagent was from Qiagen (Hilden, Germany). The 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent was from Roche (Barcelona, Spain). All general chemicals were purchased from Sigma unless stated otherwise.
Animal Model of Ischemia
Transient forebrain ischemia was induced in adult Wistar rats (10-12 weeks, from Charles River, L'Arbresle, France) by the standard four-vessel occlusion model described previously.16–18 Briefly, both vertebral arteries were irreversibly occluded by electrocoagulation under anesthesia with a mixture of atropine, ketamine, and diazepam (0.25, 62.5, and 5 mg/kg, respectively) delivered by intraperitoneal injection. After 24 hours, ischemia was induced by carotid occlusion with small atraumatic clips for 15 minutes and then clips were removed from the carotid arteries for reperfusion. After 3 or 7 days of reperfusion (R3d and R7d, respectively), the animals were killed. Sham control (SHC3d) animals were prepared in the same way as the R3d animals but without carotid occlusion. In some experiments, animals were treated with 1.0 mg/kg cycloheximide (CHX), diluted in saline vehicle, by intraperitoneal injection 45 minutes before ischemia induction. Ninety-six animals were used in the investigation. All procedures associated with animal experiments were approved by The Ethics Committee of the Hospital Ramon y Cajal, Madrid, Spain, and in accordance with the ARRIVE (Animal Research: Reporting
Hippocampal Slices
Hippocampal slices were prepared as described in Supplementary Methods. Briefly, brain coronal sections were prepared in KHH solution and the hippocampus dissected. Sections containing the hippocampus were kept in KHH for 3 hours at 37 °C in a 6.5% CO2 atmosphere, before incubation with or without additives.
Sample Preparation
Cerebral cortex and the hippocampal CA1 region from control and ischemic animals or from slices were rapidly dissected. The samples were homogenized 1:5 (w/v) with buffer A (20 mM Tris-HCl, pH 7.5; 140 mM potassium chloride; 5 mM magnesium acetate; 1 mM dithiothreitol; 2 mM benzamidine; 1 mM EDTA; 2mM EGTA; 10 μg/mL pepstatin A, leupeptin, and antipain; 20 mM sodium β-glycerophosphate; 20 mM sodium molybdate; 0.2 mM sodium orthovanadate), and centrifuged at 12,000 × g for 15 minutes to obtain a postmitochondrial supernatant (PMS). All procedures were performed at 4 °C. The sample corresponding to each animal was separately kept at − 80 °C until used and protein concentrations were determined for each sample.
Binding Assay of 4E-BP2 to eIF4E
To study eIF4F complex formation and the binding of 4E-BP2 and eIF4G to eIF4E, a cap-containing matrix was used 19 as described previously. 18 Postmitochondrial supernatant samples (300 μg) for each experimental condition were added to 7-methyl-GTP (m7GTP)-Sepharose 4B (GE Healthcare; 30 μl of 50/50 w/v slurry), incubated for 30 minutes at 4 °C and the beads were centrifuged at 2,500 × g for 5 minutes and washed, and the bounded proteins subjected to SDS-PAGE and western blotting.
Western Blot Analysis
Samples of PMS (35 μg) or m7GTP-Sepharose of each different experimental condition were analyzed by SDS-PAGE, transferred onto PVDF membranes (GE Healthcare), developed with the antibody against the specific protein, and after the blots were quantified. More detailed protocol and procedure is included in Supplementary Methods.
Immunohistochemistry and Confocal Fluorescence Microscopy
Brain sections containing the hippocampus were prepared as described in Supplementary Methods. Sections were incubated with primary antibodies followed by specific fluorochrome-conjugated secondary antibodies, mounted in antifade solution, and examined using an MRC-1024 confocal laser scanning microscope (Bio-Rad) at 488 and 568 nm wavelength excitation controlled by Bio-Rad LaserSharp software. The software provides quantitative analysis of the degree of colocalization evaluating the percentage of green objects co-localizing with red objects in the area of interest. More detailed protocol and procedure is included in Supplementary Methods.
Polysome Profile Analysis
Postmitochondrial supernatant fractions from fresh cerebral cortex and hippocampal CA1 region were homogeneized 1:2 (w/v) with buffer A containing 100 units/mL RNasin (Promega, Madison, WI, USA) and 0.2 mg/mL heparin in diethyl pyro-carbonate-treated-water, and after were layered (45 μl, ∼50 μg of RNA) onto 4 mL of 15%-55% linear sucrose gradient containing 20 mM Tris-HCl, pH 7.6, 3 mM magnesium acetate and 100 mM KCl as described previously. 16 Ultracentrifugation was performed in an SW60 rotor (Beckman, Madrid, Spain) at 164,000 × g for 180 minutes. Gradient profiles were eluted from the top of the gradient using a density gradient fractionator and monitored at 254 nm with an on-line UV detector. The profiles were registered, peaks were quantified by valley-to-valley integration method shaping the baseline, and the ratio of polysomes/80S species calculated. The entire procedure was performed at 4 °C.
TUNEL Assay and Hoechst Nuclear Staining
Apoptotic cells within brain sections or hippocampal slices were detected using the terminal deoxynucleotidyl transferase-mediated dUTP Nick-End Labeling (TUNEL) assay (Promega). Detailed protocol and procedure is included in Supplementary Methods. The number of TUNEL-positive apoptotic cells was blindly counted by two independent observers as described in Supplementary Methods.
Protein Synthesis Labeling
Hippocampal slices (200 μm thick) were prepared from control and ischemic animals as described above and 1.25 mCi/mL [35S]Met was added for 2.5 hours by modification of the procedure described in. 20 The CA1 regions from slices were homogeneized 1:2 (w/v) with buffer A containing 1% Nonidet-P40, and lysates were centrifuged at 12,000 × g for 15 minutes. Total extracts (12 μl) were analyzed by SDS-PAGE, transferred onto nitrocellulose membranes, and then exposed on phosphoimager Typhon-9200 (GE Healthcare). Total protein was analyzed by SDS-PAGE in twin separate gels stained with Coomassie Blue (Sigma).
4E-BP2 siRNA Assay
Eukaryotic initiation factor 4E-binding protein-2 siRNA transfections were performed in primary neuronal cultures (7 days
Statistical Analysis
The different animals from each experimental condition or group were independently analyzed in duplicate and their averaged values were used for statistical analysis. Data were from three to six different animals run in duplicate, represented in arbitrary units and expressed as mean ± s.d. Statistical analysis was performed using analysis of variance following after Newman-Keuls’ post test, when analysis of variance was significant, to compare the data between experimental groups. Comparisons between two groups were done by Student's
RESULTS
Apoptosis Induction in Cornu Ammonis 1 Neurons upon Reperfusion
In the rat four-vessel occlusion model of cerebral ischemia, a brief period of ischemia induces delayed neurodegeneration in the hippocampal CA1 region.6,10,22 In our experimental model, 3 days of reperfusion after ischemia (R3d) induced significant CA1 neuronal death, and 7 days of reperfusion (R7d) resulted in even more neurodegeneration (Supplementary Figure S1A). The selective neurodegeneration of CA1 neurons was evident in comparison with the cerebral cortex where that did not occur (Supplementary Figure S1B). Induction of apoptosis was demonstrated specifically in the CA1 region in the R3d group by TUNEL assay (Figure 1A, CA1 R3d). Control and R3d cryosections from the cerebral cortex showed very few TUNEL-positive cells (Figure 1A, C SHC3d and C R3d). Nucleated cells within the brain sections were co-stained with the fluorescent DNA-binding dye Hoechst that detects condensed chromatin, a feature of apoptotic nuclei, 23 thereby confirming apoptotic nuclear characteristics. All TUNEL-labeled nuclei matched with condensed Hoechst-dye nuclei (not shown). Quantitative analysis data of TUNEL-positive cells showed a significant apoptosis induction in the CA1 region during R3d with a further increase after R7d (Figure 1A, bar graph). To characterize further the cell death induced by IR with R3d, we studied the activation of caspase-3. Caspase-3 activation was assessed by the presence of a specific 120-kDa cleavage fragment from α-spectrin. 24 Levels of the caspase-3-dependent 120-kDa fragment were significantly increased in CA1 R3d samples when compared with those of their control or cerebral cortex samples, while there were no changes between the cerebral cortex and their control (Figure 1B). These results demonstrated a specific neuronal apoptosis induction in the CA1 region after reperfusion.
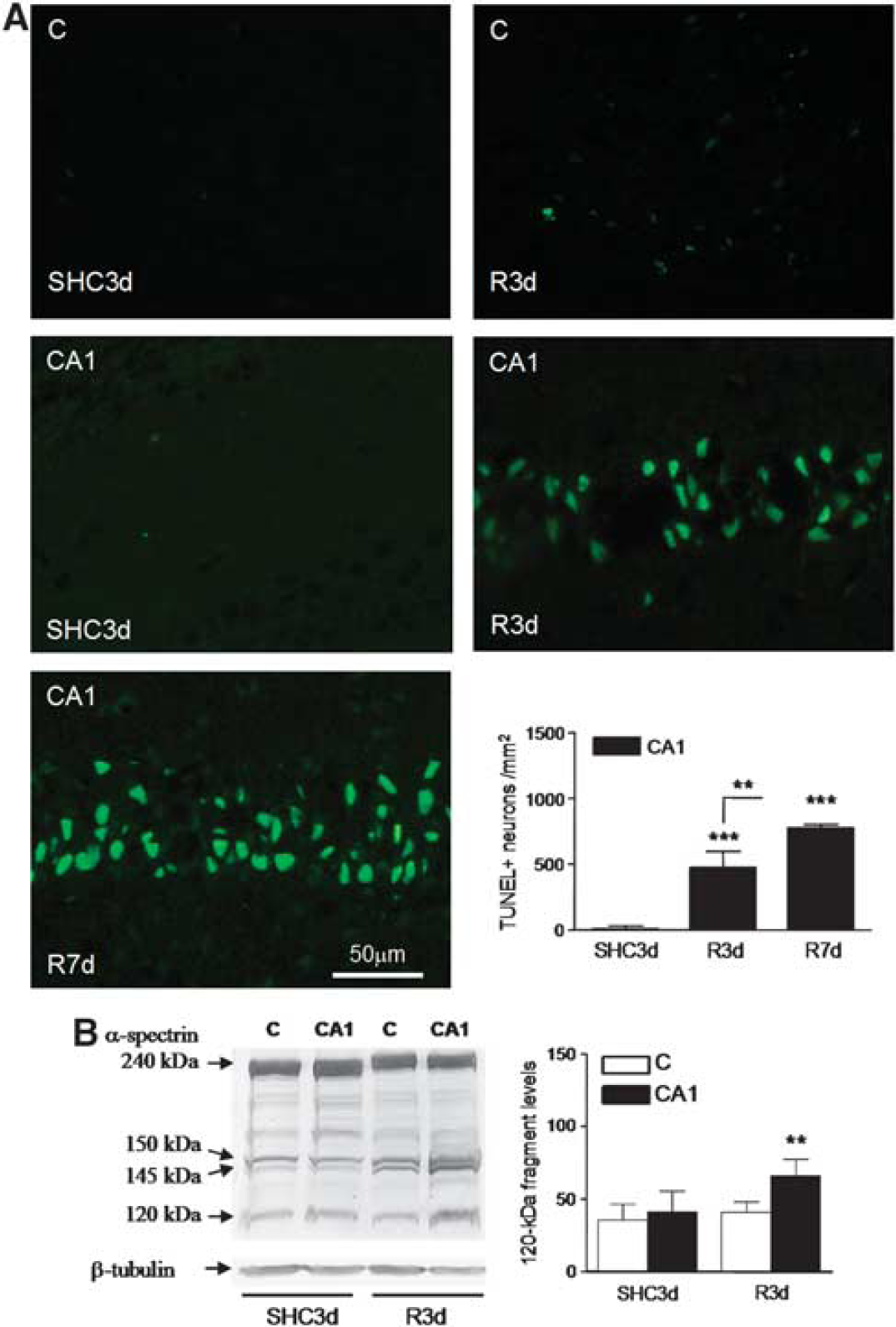
Nuclear labeling for apoptosis detection in the hippocampal
Colocalization of 4E-BP2 and eIF4E Induced by Ischemia-Reperfusion Stress During Reperfusion in the Hippocampal Cornu Ammonis 1 region
We previously observed the specific binding of 4E-BP2 to eIF4E in the CA1 region compared with cerebral cortex at R3d in a binding assay (Ayuso and Alcazar, unpublished results). Because the binding of 4E-BP2 to eIF4E is essential to prevent eIF4F complex formation and inhibit protein synthesis, a colocalization study of 4E-BP2 and eIF4E was performed in brain sections. Control and ischemic brains with 3-day reperfusion were prepared for cryosectioning and the coronal sections obtained were used for analysis of 4E-BP2 and eIF4E colocalization by confocal fluorescence microscopy. In the SHC3d control, a diffuse signal for 4E-BP2 was localized to the cytoplasm and nucleus; eIF4E was localized primarily to the cytoplasm of both cortical and CA1 regions (Figure 2A, SHC3d panels). The overlay image shows cell nuclei labeling with 4E-BP2 (Figure 2A, SHC3d, merged panels; in green) and colocalization of 4E-BP2/eIF4E in the cytoplasm (in yellow). The degree of colocalization for these control cortical and CA1 regions was determined to be 11.9% and 18.3%, respectively (
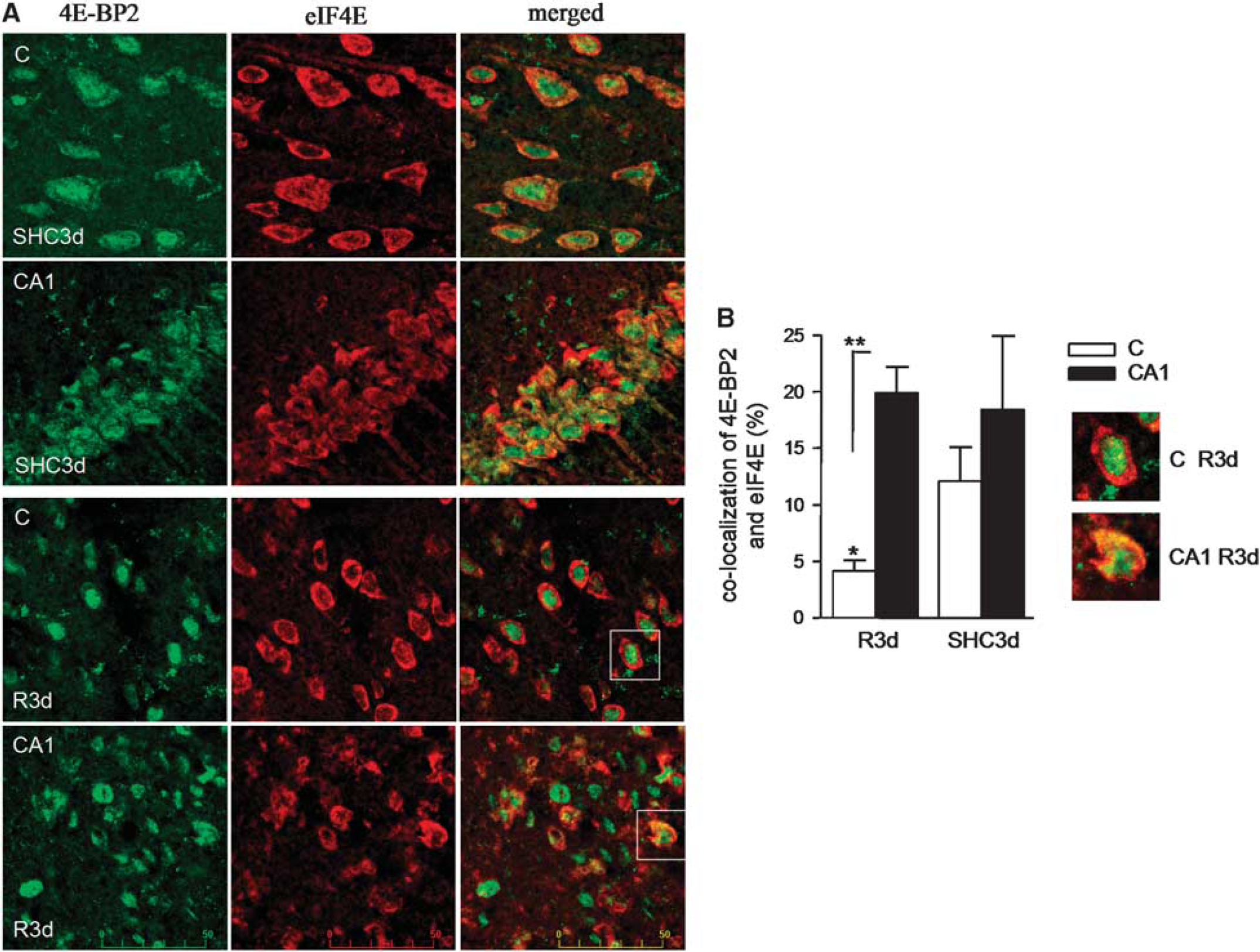
Colocalization analysis of 4E-BP2 and eIF4E in the cerebral cortex and hippocampal CA1 regions. (
Polysome Dissociation Induced by Ischemia Reperfusion During Reperfusion in the Hippocampal Cornu Ammonis 1 Region
The inhibition of eIF4F complex formation inhibits translation at the level of initiation and causes dissociation of polysomes to 80S monosomes.
25
Because the binding of 4E-BP2 to eIF4E inhibits eIF4F complex formation and R3d resulted in higher levels of 4E-BP2 bound to eIF4E in CA1, whereas low levels of 4E-BP2 associated to eIF4E were found in the cerebral cortex (see above), we analyzed polysome profiles in the R3d condition in both brain regions. The polysome profiles obtained were quantified and the polysomes/80S species ratio was calculated (Figure 3). The ratio in the CA1 region was significantly lower when compared with that of the cerebral cortex (0.51 ± 0.04 for CA1 R3d, compared with 0.73 ± 0.01 for C R3d;
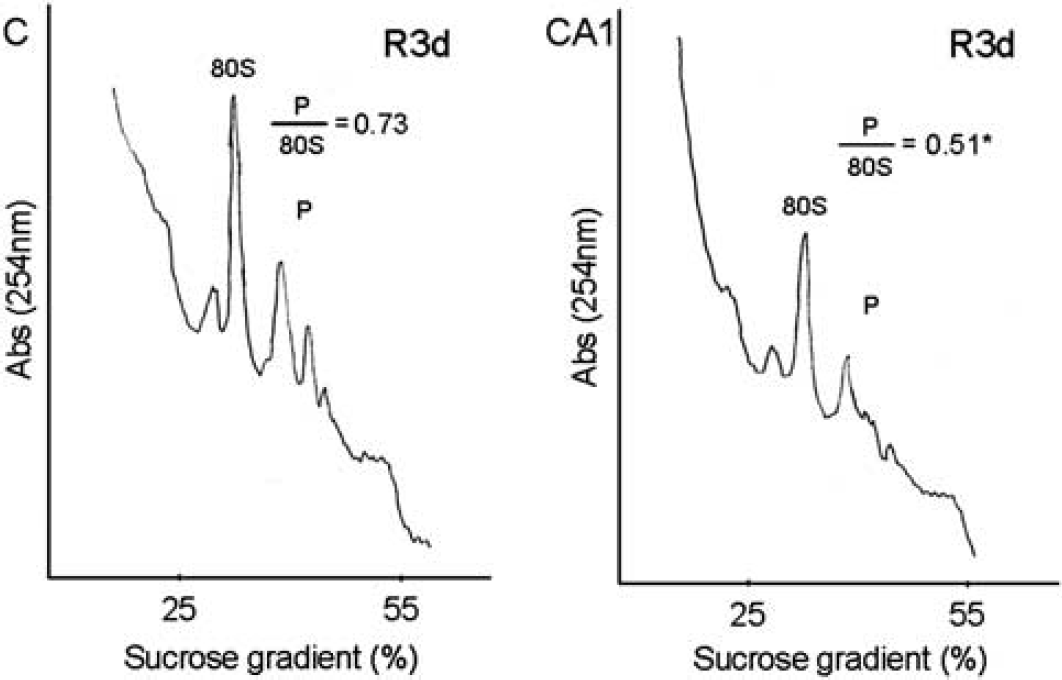
Polysome dissociation upon reperfusion in the hippocampal
Induction of Apoptosis by 4E-BP2 Peptide in Cornu Ammonis 1 Neurons
We next studied whether the association of 4E-BP2 to eIF4E in CA1 neurons can directly cause the death of these cells by incubating hippocampal CA1 slices with a peptide based on the eIF4E-binding site of 4E-BP2. We used the RIIYDRKFLLDR peptide (BP2 peptide) that is derived from the conserved eIF4E-binding motif within 4E-BP2 and which has been demonstrated to be able to bind eIF4E
Hippocampal slices were treated with 10 μM BP2 or YLL-AAA peptides for 1 hour and apoptosis detection by the TUNEL assay was performed in cryosections obtained from the slices. Induction of apoptosis by the BP2 peptide, but not by YLL-AAA peptide, was detected in the hippocampal CA1 region (Figure 4A). All TUNEL-labeled nuclei matched with the condensed Hoechst-dye-stained nuclei that are characteristics of apoptosis (Figure 4A, inserts). Quantitative analysis data of TUNEL-positive cells with characteristics of apoptotic nuclei present in the CA1 region are shown (Figure 4A, bar graph). Thus, these results demonstrated that neuronal apoptosis in the CA1 region can be significantly induced by the binding of 4E-BP2 to eIF4E. Caspase-3 activation was assessed to further underpin this result. Levels of the caspase-3-dependent 120-kDa cleavage fragment from α-spectrin were significantly increased by the BP2 peptide when compared with those untreated (not shown) or treated with YLL-AAA peptide (Figure 4B).
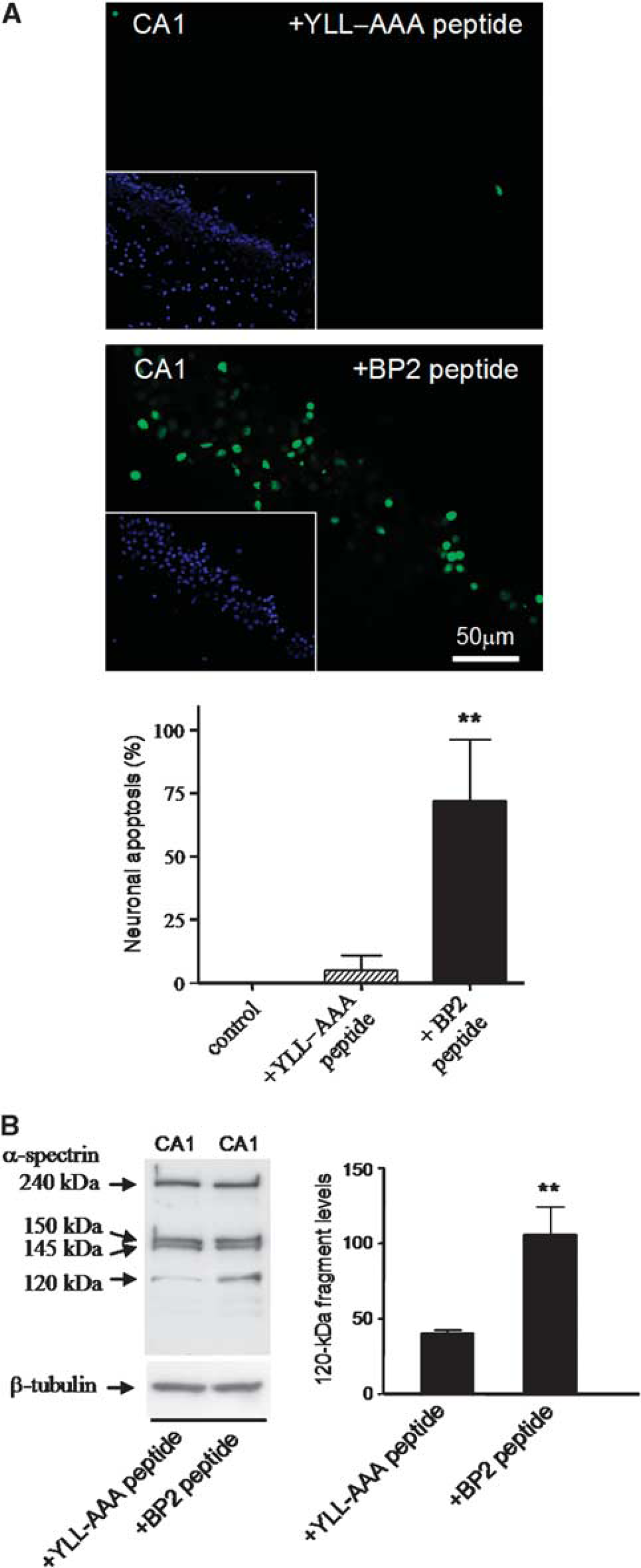
Induction of apoptosis by 4E-BP2 peptide in hippocampal slices. (
Cycloheximide Treatment Decreases 4E-BP2/eIF4E Association as well as the Apoptotic Neuronal Death Induced in the CA1 Region After Reperfusion
As CHX has been described as a neuroprotective agent in ischemia models when given before ischemia,27,28 and it is also known to be an anti-apoptotic agent,
29
we treated animals with a sublethal dose of 1 mg/kg CHX before ischemia induction, and we studied both neuronal death and 4E-BP2/eIF4E association induced by R3d. The results showed that animals treated with CHX displayed significantly decreased neuronal apoptosis, as detected by the TUNEL assay, in the CA1 region at R3d (Figures 5A and 5B). The protective effect of CHX against the IR stress-induced neuronal death was also detected by Fluoro-Jade B staining and visualized by fluorescence microscopy, which also showed that CHX treatment significantly decreased neuronal death in the hippocampal CA1 region (Supplementary Figure S3). Interestingly, when the colocalization study of 4E-BP2 and eIF4E was performed in brain sections from CHX-treated animals, a re-localization of 4E-BP2 to the nucleus was evident in the CA1 region, resulting in decreased colocalization of 4E-BP2/eIF4E (Figure 5C, CA1 R3d±CHX; no yellow mark). Quantification of the colocalization of 4E-BP2 and eIF4E in the CHX-treated group showed a significant difference with respect to that found in the CA1 region of untreated animals (Figure 5D). It is of interest to note the similarity of the images in the cellular localization of both 4E-BP2 and eIF4E between the cerebral cortex in R3d (Figure 2A, C R3d, merged panel) and the CA1 region with CHX treatment (Figure 5C, CA1 R3d±CHX, merged panel). In addition, we studied the association of 4E-BP2 with eIF4E in samples from the CA1 region in CHX-treated animals in the R3d group in a binding assay. Cycloheximide induced a significant decrease in the levels of 4E-BP2 bound to eIF4E when compared with CA1 R3d from untreated animals or with the SHC3d control group (Figure 6A), mirroring the results of the colocalization studies. This functional study demonstrated that the CHX-induced decrease in 4E-BP2/eIF4E association was correlated with neuronal apoptosis inhibition and protecting the CA1 region against ischemic injury. Finally, we evaluated the effect of CHX treatment on protein synthesis. As expected, CHX treatment increased translational rates in the CA1 region of ischemic animals as demonstrated in the protein labeling visualized by autoradiography (Figure 6B, CA1 R3d + CHX). This result showed that CHX treatment recovered shut-off of protein synthesis induced by IR stress in the CA1 region. Polysome profiles were performed to assess further this result. The polysomes/80S species ratio was calculated (Figure 6B), and the ratio in the CA1 region of CHX-treated ischemic animals was significantly higher when compared with that of the untreated animals (0.77 ± 0.08 for CA1 R3d + CHX, compared with 0.52 ± 0.09 for CA1 R3d + vehicle;
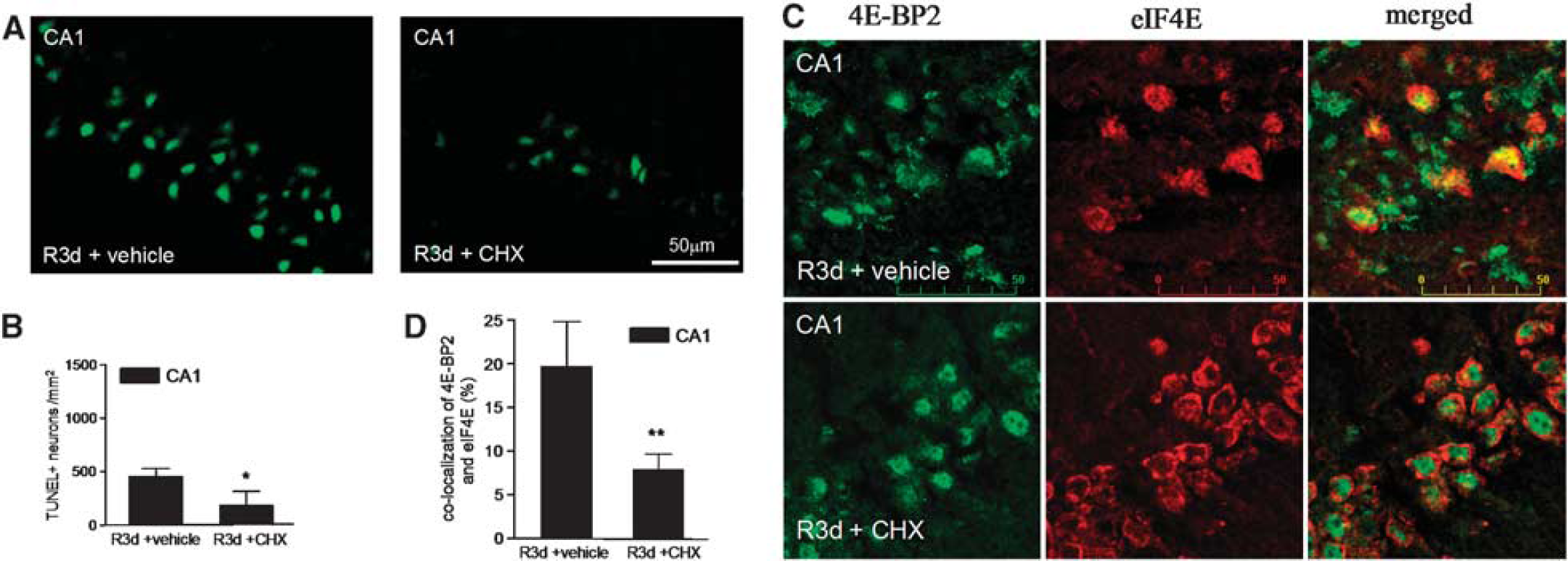
Cycloheximide (CHX) prevents the ischemia reperfusion (IR)-induced neuronal apoptosis and 4E-BP2 association with eIF4E in the
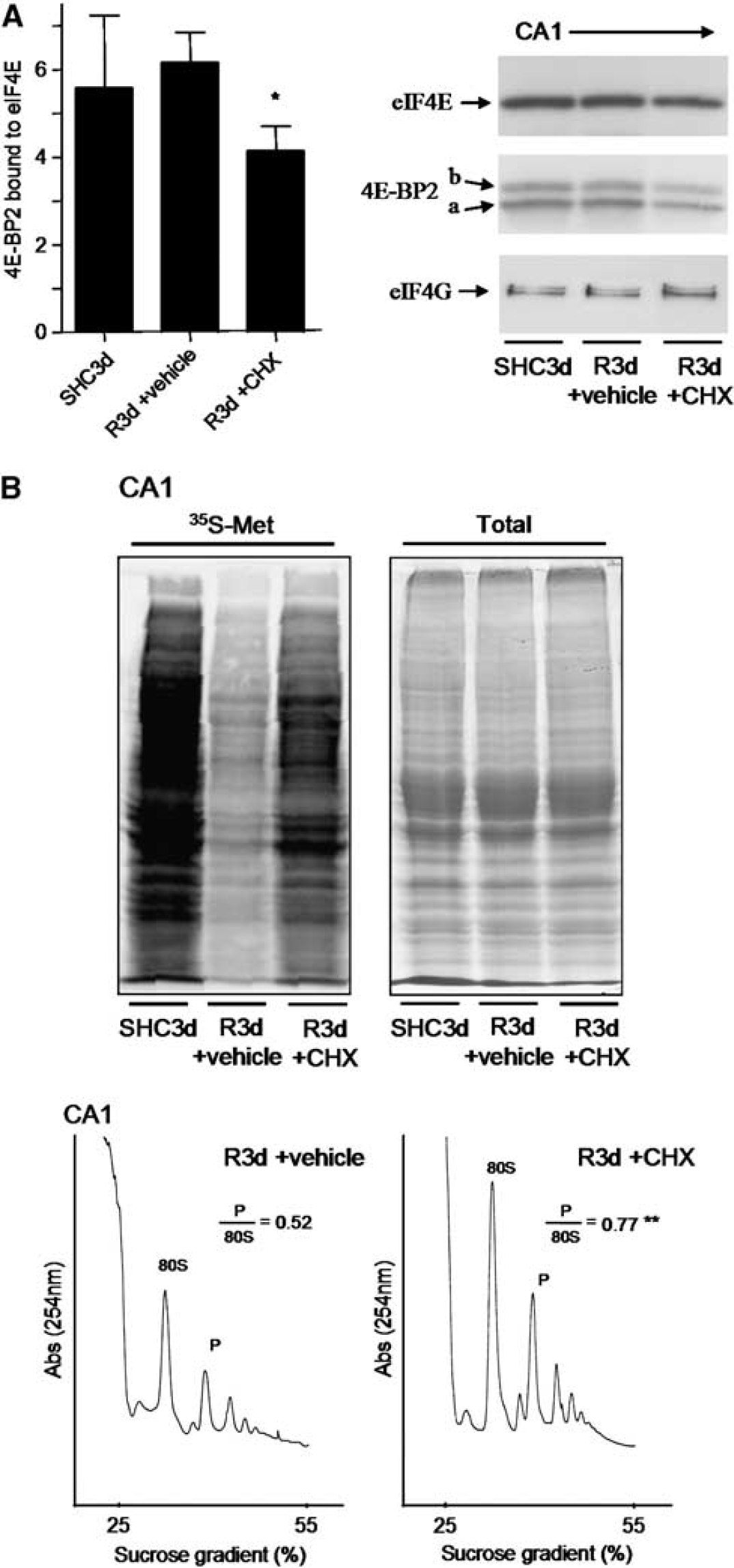
Cycloheximide (CHX) prevents the binding of 4E-BP2 to eIF4E and restores protein synthesis in the hippocampal
RNA Interference Targeting 4E-BP2 Protects Neuronal Cells in Culture from Ischemia Reperfusion In Vitro
Primary neuronal cells in culture were transfected with siRNA targeting 4E-BP2 and the cell lysates were after analyzed to verify the effectiveness of RNA interference. 4E-BP2 siRNA transfection decreased significantly 4E-BP2 expression by 35% (65 ± 12.0 for transfected cells, compared with 100 ± 1.3 for mock transfected,
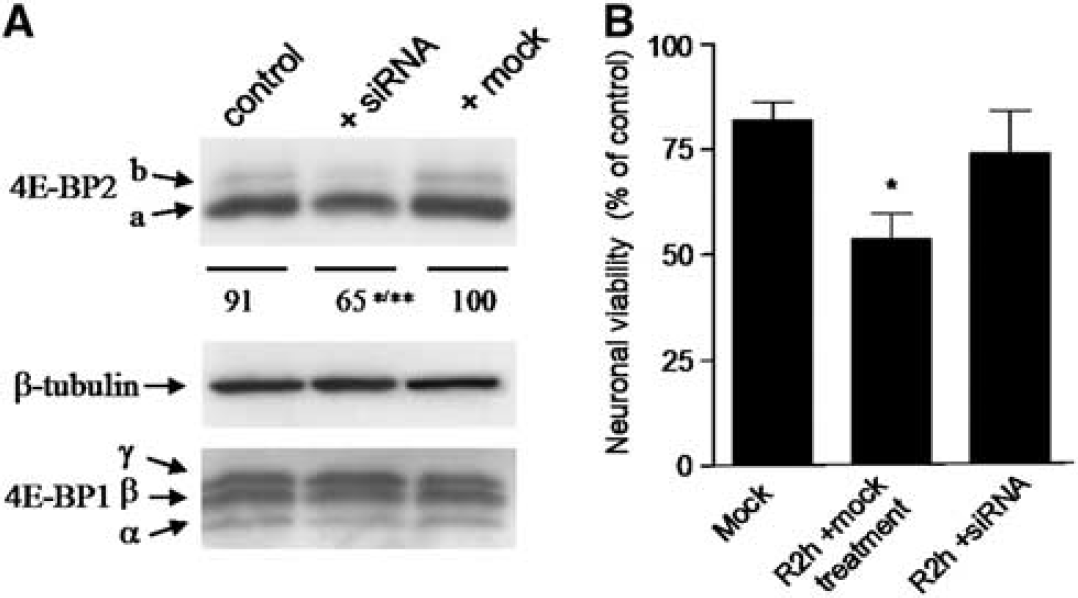
4E-BP2 siRNA prevents the ischemia reperfusion-induced cell damage in primary neuronal cells in culture. (
DISCUSSION
These results demonstrated the specific association of 4E-BP2 to eIF4E in the vulnerable CA1 region upon reperfusion, while the resistant cortical region had significantly lower levels of 4E-BP2/eIF4E association. 4E-BP2 is a translational repressor through their binding to eIF4E, and these results agreed with the polysome profile results, which showed inhibition of protein synthesis in the CA1 region compared with the cerebral cortex.
The selective vulnerability of the hippocampal CA1 region to ischemic injuries is manifested in delayed neuronal death.6,10,22 The delayed neuronal death induced by IR stress appears to result in a mechanism of cell death with apparent apoptotic characteristics, although this feature is still under discussion.3,9 Here, we have demonstrated apoptotic features in the delayed neuronal death induced by IR. Induction of apoptosis was demonstrated in the CA1 region by detecting (i) DNA fragmentation by TUNEL labeling, (ii) nuclear condensation by Hoechst staining, and (iii) caspase-3 activation, an executing caspase in the apoptotic pathway. All these results were specific for the CA1 region with very few signs of apoptosis detected in the cerebral cortex. From 3 to 7 days of reperfusion, the number of TUNEL-positive neurons in the CA1 region increased in a manner that corresponds to the reduction in viable neurons observed by hematoxylineosin staining (Figure 1 and Supplementary Figure S1A).
In addition to caspase-3-dependent 120-kDa α-spectrin fragment, a calpain-dependent 145-kDa fragment was also detected in the R3d group. The activation of both proteases has been described in global and focal ischemia; both may be a substrate of the other, and there is no information about which is the priming event for a subsequent trigger of apoptosis because they coexist in ischemic stress. 9 Other authors have reported calpain activation parallel to eIF4G degradation after 4 days of reperfusion after global ischemia in rat brain. 30 Despite the fact that we found calpain activation, we did not detect degradation of eIF4G (Ayuso and Alcazar, unpublished results), a known substrate of calpain,30,31 which may indicate that in our experimental model the calpain activation detected is a later effect, leading to eIF4G degradation. Those results and our results described here are compatible because 4E-BP2/eIF4E association and eIF4G degradation would contribute to the inhibition of eIF4F complex and protein synthesis inhibition. Additionally, we cannot discard the possibility that calpain activation may be altered depending on the severity of the induced ischemia or animal model conditions.
It is known that persistent protein synthesis inhibition is related to delayed neuronal death in both global and focal ischemia, however, both mechanisms have not yet been connected directly.7,8,12 Thus, the desire to elucidate the mechanism implicated in IR stress-induced translation inhibition and delayed neuronal death led us to study the potential effects of the binding of 4E-BP2 to eIF4E. Interestingly, we demonstrate that micromolar concentrations of a peptide based on the eIF4E-binding site of 4E-BP2 triggers apoptosis in hippocampal CA1 neurons. It has also been reported that binding the initiation factor eIF4E by peptides derived from eIF4E-binding motif can rapidly induce apoptosis in cultured cell lines. 26 In addition, the induction of apoptosis in cultured cells by inhibition of 4E-BP1 phosphorylation, with consequent binding to eIF4E and inhibition of cap-dependent translation, has been reported. 4 This supports the notion that repression of cap-dependent translation is concomitant with proapoptotic events, while apoptotic suppression is cap-dependent. 4 Our results are in agreement with these facts: upon reperfusion after ischemic stress, selective neurons, e.g. those of the CA1 region, have 4E-BP2 bound to eIF4E, translation inhibition, and apoptosis activation.
Finally, we studied the association of 4E-BP2 to eIF4E in CHX-treated animals that underwent transient ischemia. Pharmacological treatment of ischemic animals with CHX at a concentration that was demonstrated to be neuroprotective in the CA1 region, decreased neuronal apoptosis induction in this region and specifically induced a significant decrease in 4E-BP2/eIF4E colocalization and 4E-BP2 bound to eIF4E during reperfusion in CA1 neurons. Cycloheximide was chosen, because it is known to be an anti-apoptotic agent
29
and, at the dose range used, it has been shown to be neuroprotective in ischemia models.27,28 In addition, although CHX is a well-known translation elongation inhibitor, it has also been described to inhibit mRNA granule assembly, a stress-induced ribonucleoprotein complex containing translation initiation factors,
32
interfering in the translation initiation machinery. The fact that CHX had a specific effect on both 4E-BP2/eIF4E colocalization and binding of 4E-BP2 to eIF4E indicates that both effects are not an epiphenomenon and that CHX impairs 4E-BP2/eIF4E association. Because CHX decreased 4E-BP2/eIF4E association, they expect that CHX treatment results in an increase of translational rates. Justly, radioactive amino-acid incorporation and the polysome profile experiments demonstrated how the inhibition of proteins synthesis in the CA1 region of ischemic animals was recovered after CHX treatment. Besides, although this paper had no intention to assess the putative role of CHX, we observed that CHX may interfere in the eIF4E association with 4E-BP2, as shown in additional
We observed that the colocalization of 4E-BP2 to eIF4E was not different between SHC3d and CA1 R3d groups. However, both conditions are not comparable because R3d is a stressed condition where the cells need a trigger mechanism to cell survival (e.g., recovery of protein synthesis) that they cannot be expected in a normal cell function. Thus, the association of 4E-BP2 to eIF4E would be a critical event for the neurons because it inhibits recovery of protein synthesis, which is essential for cell survival under IR stress, but may not be in non-stressed situations. Furthermore, neuronal cells from cerebral cortex responds to IR stress with the dissociation of 4E-BP2 and eIF4E, and they are resistant to transient ischemia.
In this paper, we have demonstrated three critical and specific events that occur in the vulnerable CA1 region upon reperfusion after ischemic stress: first, 4E-BP2 remained bound to eIF4E in neuronal cells; second, translation inhibition; and third, neuronal apoptosis induction. Moreover, converse events occur in the cerebral cortex, such as low levels of 4E-BP2 associated to eIF4E, higher translational rates, as well as neuronal survival after IR stress. The following are the proposed sequence of events during ischemic reperfusion, at the time when delayed neuronal death occurs:
Vulnerable hippocampal neurons: 4E-BP2/eIF4E association → cap-dependent translation inhibition, apoptosis induction → neuronal death
Resistant cortical neurons: 4E-BP2 dissociation from eIF4E → cap-dependent translation, apoptosis inhibition → neuronal survival
The results show in an
Ischemia reperfusion induces delayed neuronal death and it causes loss of neurologic function. Since these cells are not initially affected by the ischemic episode, but can later be fatally affected as a result of it, protection of damaged neurons in these regions are the main objective of drug therapy. 9 The great social relevance of cerebral ischemia requests to develop additional treatment interventions for ischemic stroke, other than thrombolytic agents, in order to directly protect brain cells and/or improve brain recovery from post-ischemic injury. 33 To this end, it is necessary to elucidate the molecular targets that may be critical to rescue neuronal cells directed to die. In this direction, these findings identify 4E-BP2 as a novel target for neuroprotection against ischemic injury. Given the importance of translation regulation in neuronal viability and repair during ischemic stress, strategies that modify the association between 4E-BP2 and eIF4E during reperfusion could become powerful tools to prevent ischemic injury.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.