
Abstract
The multiple ligand concentration assays (MLCRA) method provides researchers with the ability to measure in vivo receptor characteristics in a stable condition. Measurements of the density and affinity of the dopamine D2 receptors with [11C]raclopride, using a sequential method (three scans throughout 1 day) or a nonsequential method (three scans spread over several weeks but at the same time of the day), yield similar values. However, after an acute challenge with drugs that affect dopamine neurotransmission, the concentration of endogenous ligand may vary over the course of the in vivo sequential MLCRA. Combined PET-microdialysis studies after acute amphetamine showed that during the imaging time frame the concentrations of extracellular dopamine vary widely, but that nonetheless the decrease in raclopride binding potential is sustained and nearly constant over time. These observations apparently contradict the simple competitive displacement model if the changes in extracellular concentration are taken to reflect necessarily comparable changes at the binding sites. To understand the effect of the delay between drug administration and start-to-end of data acquisition on the MLCRA results, we compared the outcomes of the sequential and nonsequential methods after methamphetamine. Comparison of the binding potential, density, and affinity of D2 receptors in both experimental conditions revealed good concordance between the data sets, suggesting that methamphetamine produces sustained and stable increases in synaptic dopamine.
Most positron emission tomography (PET) studies of living neuroreceptors yield a binding potential (BP) (Mintun et al., 1984) or an equivalent. The BP, however, reflects both the density of the receptors in the tissue of interest and the apparent affinity between these receptors and the tracer (Bmax/Kd). The multiple ligand concentration assays (MLCRA) method introduced by Farde et al. (1986) and adapted by others (Delforge et al., 1995; Holden et al., 2002; Ito et al., 1998) provides researchers with the ability to measure in vivo receptor characteristics, but the peculiarity of the acquisition protocols and conditions precludes strict comparison with similar data obtained in in vitro Scatchard studies (Holden et al., 2002; Riffee et al., 1982; Scatchard, 1949; Seeman et al., 2002). Most MLCRA studies are based upon multiple (minimum of two) administrations of the tracer with increasing occupancy of the receptors by various means, usually throughout the course of one day, and thus last over several hours, which for a living organism, tissue or cell, may lead to significant alterations in environment.
We reported recently (Doudet et al., 2003) that in stable conditions, measurements of the density and affinity of the dopamine (DA) D2 receptors with [11C]raclopride, using a sequential method (three scans throughout 1 day) or a nonsequential method (three scans spread over several weeks but at the same time of the day), yield similar values. Thus the circadian changes in endogenous ligand and the ordinary daily alterations in environment may not represent a large source of variability in the measurements of receptor density and affinity, or, more plausibly, the changes are sufficiently small that we are unable to detect them with the available current technologies. In contrast, after an acute challenge with drugs that affect DA neurotransmission, the concentration of endogenous ligand DA may vary over the course of the in vivo sequential MLCRA.
Recently, Carson et al. (2002) with amphetamine and the present authors (Doudet and Holden, 2003) with methamphetamine, using two different acquisition protocols and data analyses, found that the routinely reported decrease in raclopride BP after an acute dose of amphetamine or methamphetamine can be accounted for by a decrease in the affinity of D2 receptors for the benzamide tracers (increased Kdapp). However, in both studies, the data were acquired over several hours after acute drug challenge. Carson et al. (2000) demonstrated that within that time period, the raclopride BP remains constant. In contrast, combined PET-microdialysis studies by Breier et al. (1997) and Laruelle et al. (1997) showed that during the time frame of imaging studies, the concentrations of extracellular DA peaked shortly (within 15 to 30 minutes) after amphetamine and returned towards baseline levels within a couple of hours of administration. This short-lived increase in extracellular DA after amphetamine is well documented (Butcher et al., 1988; Kuczenski and Segal, 1989; Tsukada et al., 1999). The stable and sustained decrease in benzamide BP during significant variations in the concentrations of the endogenous competitor in extracellular fluid is thus difficult to explain in the context of a simple competitive inhibition model, if these extracellular concentrations are taken to reflect necessarily comparable changes at the raclopride binding sites. The decrease in both BP and affinity found by both Carson et al. (2002) and the present authors could be the result of a variety of factors throughout the hours after amphetamine/methamphetamine administration, including progressive changes in affinity and/or receptor internalization/externalization processes. Because one of the most striking and obvious issues with the in vivo sequential method is the delay between pharmacologic challenge and the start to end of data acquisition, we compared the outcomes of our sequential and nonsequential MLCRA acquisition methods after methamphetamine administration.
We used [11C]raclopride and our previously published method of acquiring and analyzing MLCRA data (Holden et al., 2002) to measure the density and affinity of the DA D2 receptors after methamphetamine administration. The measurements were obtained after a single administration before the three scans in the sequential method and after administration of a dose of methamphetamine before each scan in the nonsequential method. Although repeated administrations of this psychotropic substance could eventually lead to sensitization of the DA system, the effect of which upon our measurements are unknown, comparison of the density and affinity of D2 receptors in both conditions revealed good concordance between the two sets of data.
MATERIALS AND METHODS
Four male rhesus monkeys that had not participated in other drug studies or challenges at the time of data acquisition were used. All animal procedures were approved by the Committee on Animal Care of the University of British Columbia. The animal procedures, anesthesia regimens, PET scans acquisition, and analysis are described in detail elsewhere (Doudet et al., 2000; Holden et al., 2002). Briefly, the animal was positioned prone in a stereotactic headholder. A Siemens ECAT 953-31B allowed the simultaneous acquisition of 31 coronal slices through the head and brain of the monkey (in plane resolution, 6 mm FWHM; axial resolution, 5 mm). Scan data were acquired in 2D mode over 1 hour. For all scans, raclopride was administered as a bolus (2.5 mCi in 1 minute in 10 mL saline) followed by constant infusion (2.5 mCi in 59 minute in 30 mL saline) to create a true equilibrium condition (Carson et al., 1997). The sequential versus nonsequential acquisition methods are described in detail in Doudet et al. (2003). Briefly, for the sequential acquisition method, three bolus/infusion injections of raclopride (specific activity [SA]: SA1 > 1,000 Ci/mmol; 40 < SA2 < 20 Ci/mmol and 10 < SA3 <4 Ci/mmol) were performed throughout the day, 2 to 3 hours apart. For the nonsequential studies, the SA for each corresponding scan were similar, but each raclopride scan was performed on a separate day, at least 2 to 3 weeks apart. The order of scan acquisition was random.
Methamphetamine (2 mg/kg i.p.) (mAMPH) was administered 15 to 20 minutes before the first high SA scan for the sequential studies and 15 to 20 minutes before each raclopride administration in the nonsequential studies. Thus the anesthetized monkey received up to four methamphetamine injections over a 6-month period. As previously described (Doudet et al., 2000), regions of interest were positioned over the striatum and cerebellum, and the kinetic data were analyzed using the graphical analysis of Logan et al. (1996) using the time activity course in the cerebellum as the input function, between 30 and 60 minutes after injection, by which time equilibrium was reached at all SA used. The Logan analysis yielded a distribution volume ratio (DVR), which, minus one (DVR-1), was interpreted as an estimate of the equilibrium ratio of bound and free ligand (B/F) in striatal regions (the y-axis). This was then fitted against the bound ligand concentration in striatum using the routine Scatchard straight line method (Scatchard, 1949) used in most PET studies. The method yielded a value for the density, Bmax (the x-axis intercept), the apparent ligand-receptor dissociation constant Kdapp (inverse of the slope), and the BP (y-axis intercept). For the sequential method, the data from the three sequential scans were combined to yield BP-s, Bmax-s, and Kdapp-s; for the nonsequential method, the data from the three scans obtained on 3 separate days were combined to yield BP-n, Bmax-n, and -n. As we already had data showing that in the baseline condition there is no difference between the sequential and nonsequential data, to spare the monkeys further radioactivity exposure and anesthesia, we only acquired sequential data in the control baseline condition (BP, Bmax, and Kdapp).
The densities, affinities, and BP yielded by the sequential method were compared with those yielded by the nonsequential method using paired t-tests. The two sets of data were compared with the control data obtained in the sequential condition using paired t-tests.
Finally, to demonstrate the sensitivity of our approach to the time-varying affinity between raclopride and receptor that would result from a transient pulse of elevated DA levels at the binding sites, we calculated the locus of B/F versus B under these transient conditions, based upon the assumption of competitive displacement. The same linearization (Scatchard plot) used to fit the measured data was used:
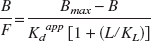
In this expression, the dependence of the equilibrium relationship on the concentration L of competing ligand DA is made explicit rather than absorbed into the value as in our previous report (Holden et al., 2002). As in that reference, KL is the dissociation constant for DA. In the scenario modeled, each value of the independent variable B was associated linearly with an increasing elapsed time after methamphetamine administration. The rationale for this association is that in our MLCRA method, the SA values are intentionally chosen to distribute the resulting data points evenly over the B axis, and the studies performed to yield these progressively increasing values of B are evenly spaced over time. The concentration L was assumed to decline exponentially over this elapsed time, with a mean life of 120 minutes, such that the denominator in Equation 1 would fall from approximately 20 pmol/mL at the beginning of the series to approximately 12 pmol/mL at the time of the final scan, between 5 and 6 hours later. Sequential MLCRA studies performed under these circumstances according to the design just described, such that progressively decreasing SA values would yield B values proportional to the elapsed time after intervention, would thus yield data points drawn from the calculated curve. The sensitivity of our data to this putative time variation in the competing DA concentration was demonstrated by comparing the resulting hypothetical curve with our actual data using the sequential approach under both the baseline and drug intervention conditions.
RESULTS
The data are summarized in Table 1 and Fig. 1. There were no significant differences between the mAMPH data obtained with the two acquisition methods for any of the parameters of interest. There was however, a significant increase in (80% for both sequential and nonsequential methods, P < 0.03) and a significant decrease in BP (41% and 43% respectively, P < 0.002) after methamphetamine compared with the baseline condition. There was no significant change in Bmax after methamphetamine challenge. Figure 1 shows the straight line plots in two representative animals. The bottom panel in particular gives support to the hypothesis of a single population of receptors, which is consistent with the known single affinity state for raclopride binding (Seeman, 1993). All points appear to fall on the same straight line, despite the very different values for the lowest SA, and the consequent very different values for the bound mass B.
Density (Bmax) affinity (Kdapp), and BP obtained in four monkeys using the sequential paradigm in the baseline condition and using either the sequential, 1-day acquisition paradigm, or the nonsequential 3-day acquisition paradigm after administration of mAMPH
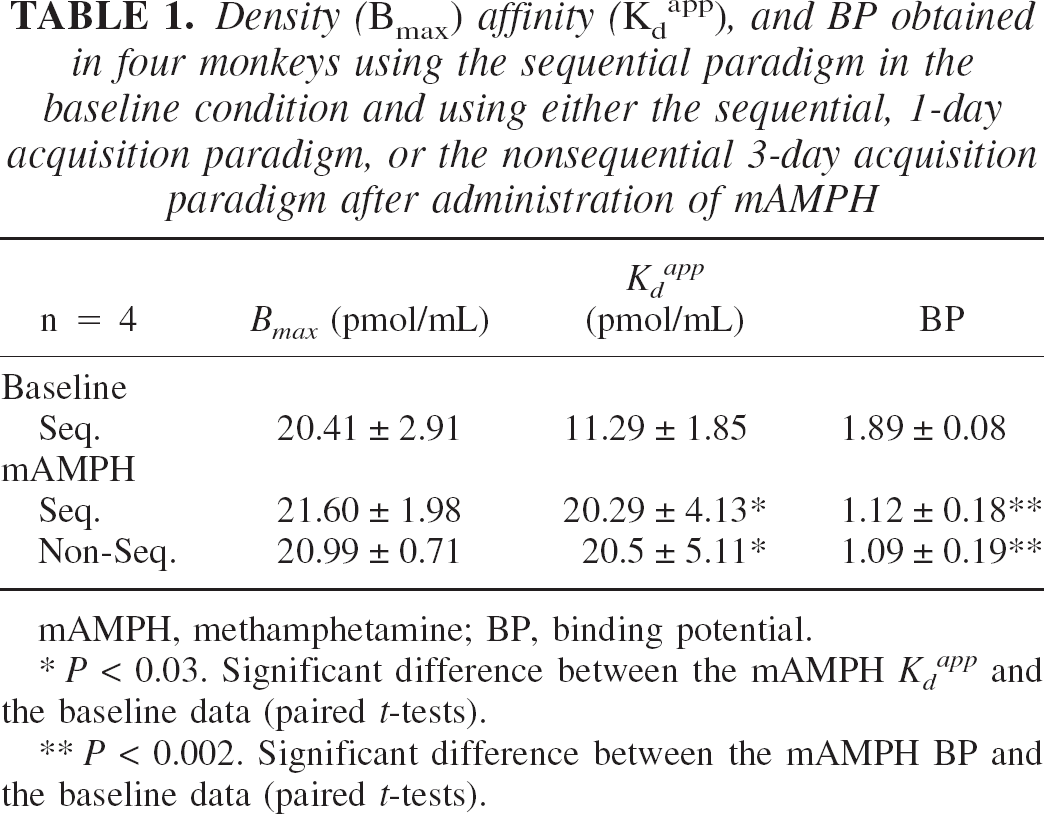
mAMPH, methamphetamine; BP, binding potential.
P < 0.03. Significant difference between the mAMPH Kdapp and the baseline data (paired t-tests).
P < 0.002. Significant difference between the mAMPH BP and the baseline data (paired t-tests).
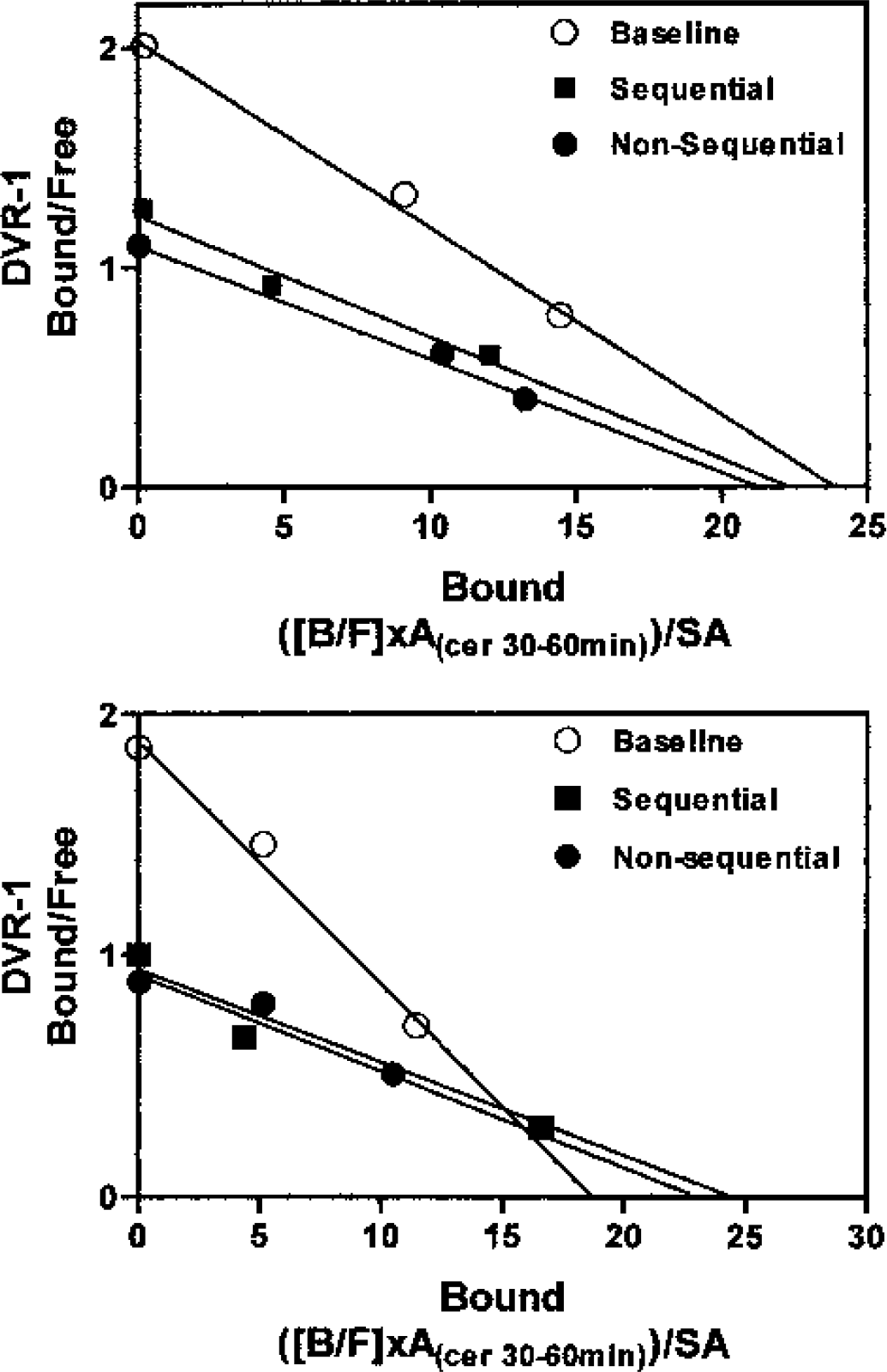
Straight line Scatchard plots of the data in two monkeys at baseline (open circle) and after methamphetamine (filled symbols) using a sequential (filled square) and a nonsequential (filled circle) method of acquisition. The bottom panel in particular gives support to the hypothesis of a single population of receptors, consistent with the known single affinity state for raclopride binding. After methamphetamine, there is an increase in Kdapp and a decrease in BP in the striatum without significant changes in Bmax.
The results of the simulation of a time-variant are presented in Fig. 2. The curve showing the dependence of the B/F values upon B under these conditions is plotted together with those corresponding to the baseline and methamphetamine conditions. The curves confirm that our method can clearly distinguish between the two possible scenarios with high sensitivity. At low values of B, the behavior of the hypothetical curve is dominated by the decline of and actually takes on a small positive slope. As B approaches Bmax, it becomes dominated by the numerator of Equation 1 and declines rapidly towards zero. A sequential study with bound masses B of approximately 0, 6, and 12 pmol/mL, typical of our 3-point studies with raclopride, would yield a degree of non-straightness never observed in this or any other of our MLCRA studies. A fit of these data points would predict values of both Kdapp and Bmax in excess of 40 pmol/mL.
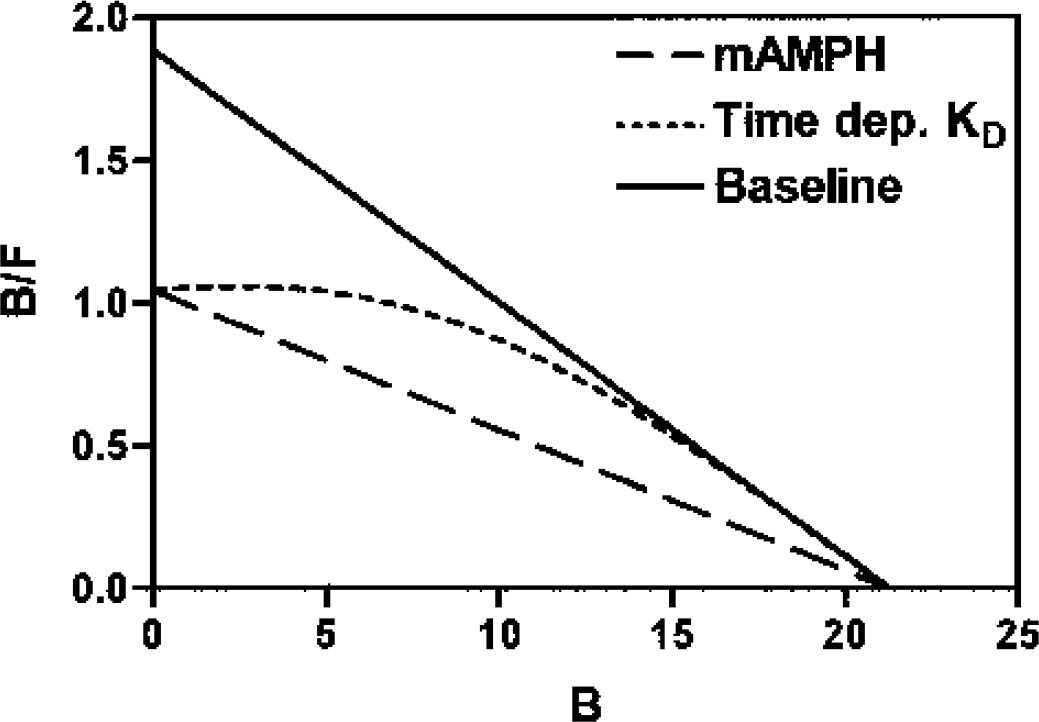
Hypothetical Scatchard plot in the situation of a time-dependent, together with the straight line plots reflecting our baseline and mAMPH results. In the simulation, each B value on the horizontal axis was assumed to be acquired at a proportionately increasing time after mAMPH administration, consistent with our MLCRA experimental design. The apparent dissociation constant was assumed to decline from approximately 20 pmol/mL early in the series to approximately 10 pmol/mL for the largest B values, to reflect a hypothetical exponential decrease in the concentration of competing DA. The simulation demonstrates the sensitivity with which the measured data exclude this possible scenario.
DISCUSSION
Previous work by the present authors and others (Carson et al., 2002; Doudet and Holden, 2003) established that the reduction of raclopride binding potential after acute administration of amphetamine or methamphetamine in nonhuman primates is predominantly caused by a decrease in affinity between raclopride and its target receptors, with only minimal changes in receptor density. In the present study we showed that this reduction of affinity is the same, within experimental uncertainty, whether the raclopride equilibrium period used to determine the binding parameters begins 50, 200, or 350 minutes after a single intraperitoneal dose of methamphetamine.
In our development of an equilibrium approach to the distinction of density from affinity effects in receptor binding assay using multiple ligand concentrations, attention was paid to the question of the multiple administrations of cold ligand interfering with each other. For multiple studies performed sequentially on the same day, we chose the obvious expedient of performing the studies in order of decreasing specific activity. Interference between studies in the absence of other interventions was assessed both by searching for trends in apparent affinity as the cold ligand mass increased (Holden et al., 2002) and by direct comparison of sequential studies performed on the same day with otherwise similar studies performed on different days several weeks apart (Doudet et al., 2003). These assessments confirmed the independence of the multiple studies from each other and validated the use of the more convenient sequential method in the absence of acute interventions.
In the event of a drug intervention, however, this necessary ordering of the specific activity levels means that each specific activity is studied at a different point in the time course of the drug effect, and if this is not stable over time, the receptor assay results will be distorted. The present study serves as an example of our routine use of sequential versus nonsequential comparisons to determine the influence of the drug effect time course on the receptor assay results. This comparison must be performed for each new intervention studied, as the concordance between the two approaches after any particular drug intervention implies nothing about any other.
The review of in vivo binding competition techniques by Laruelle (2000) cites evidence in the literature that the phenomenon of displacement of test ligands by endogenous neurotransmitter primarily involves synaptic rather than extrasynaptic receptors. As much as 50% of D2 receptors are not associated with a synapse (Yung et al., 1995), but the effects of raclopride/DA competition can be measured as readily after increase in intrasynaptic DA, which are accompanied with only negligible increase in extrasynaptic DA (Kim et al., 1998) as after a dose of amphetamine, which increases dramatically both intra and extrasynaptic concentrations, and both conditions lead to similar decrease in binding potential. This suggests that maybe the extrasynaptic receptors are mainly in the agonist low affinity state and thus, although they can be bound by raclopride, are not sensitive to competition. In normal conditions, DA is largely prevented to spill over into the extracellular space through action of the DA transporter (for review, see Grace, 1991). For example, low electrical stimulation of the DA neurons, while increasing synaptic DA, is unable to produce significant increases in extracellular DA (Kuhr and Wightman, 1986; May, 1988). Presynaptic fibers are thus the most important source of the displacing DA. Additional evidence in support of this idea is described in our recent report (Doudet and Holden, 2003) on the correlation between the degree of displacement of raclopride by methamphetamine and quantitative PET markers of presynaptic integrity. In one extreme case, raclopride binding in the lesioned striatum of a rhesus monkey unilaterally lesioned with MPTP was completely unaffected by amphetamine, despite the presence a 50% upregulation of receptor density.
There was no similarity between our measured data and the results predicted by the competitive displacement model when the synaptic DA concentration is declining over time in proportion to that in extracellular fluid (Fig. 2). Our results therefore strongly support the existence of an altered but stable synaptic milieu after methamphetamine administration that persists for many hours. We conclude with brief discussions of the possible character of these synaptic changes.
Increase in the time-averaged concentration of synaptic dopamine
In their analysis of simultaneously measured PET image and microdialysis data by compartmental modeling, Endres et al. (1997) hypothesized proportionality between synaptic and extracellular concentrations of DA following amphetamine administration. This was, of course, the most plausible assumption in the absence of more information, but there are many reasons why the two concentrations would not necessarily be linked. Each of the compounds known to mobilize DA has a unique profile of pharmacologic actions, and each has its own distinct effect on the many different pools of intraneuronal DA available for mobilization. Based upon a number of their own combined PET-microdialyis studies, Tsukada et al. (1999) have questioned the relationship between changes in DA levels and changes in raclopride binding. In a comprehensive review, Laruelle (2000) cites two examples. The binding potential changes induced by cocaine apparently follow the expected transient time course of mobilized DA (Volkow et al., 1999), although these results were preliminary. Nicotine was observed to cause a reduction of the raclopride BP in rodent that is comparable with that by amphetamine, while exciting a pulse of extracellular DA with a magnitude of only a few percent of that of amphetamine (Kim et al., 1998). Similarly, the behavioral effects of amphetamine outlast the increase in extracellular DA (Kuczenski and Segal, 1989). Thus amphetamine and methamphetamine, with their many actions and effects throughout every part of the dopaminergic system, could most certainly have their own unique uncoupling between the synaptic and extrasynaptic time courses of elevated DA concentrations. The pulse of extracellular concentration reflects the uncontrollable passage of the combined DA mobilized from all pools and released by all mechanisms. On the contrary, the concentration in synapse after amphetamine would be the complex result of the many different effects that amphetamine may have on DA synthesis, release, reuptake, and catabolism. The possibility that amphetamine/methamphetamine induce an acute, abrupt, and instantaneous but long-term stable elevation of synaptic DA levels, resulting in the observed decrease in the affinity between raclopride and receptors consistent with the competitive displacement model, cannot be excluded.
Receptor internalization
Access of free test ligand to free receptors is one of the most important determinants of the apparent dissociation constant observed in in vivo studies (Holden et al., 2002). This access would be reduced for raclopride if postsynaptic receptors were to be internalized after amphetamine/methamphetamine administration. Agonist-mediated receptor internalization has been reported for D2 receptors (Chugani et al., 1998) and appears to be a slow process from 15 minutes to more than 2 hours and primarily involves receptors in the low affinity state (Itokawa et al., 1996; Ko et al., 2002). Raclopride can cross the membrane and access these internalized D2 receptors, thus preserving Bmax. However, its moderate lipophilicity, the lower intracellular sodium concentrations, and the higher intracellular pH, would all result in a decreased affinity of raclopride for these receptors (D'Souza and Strange, 1995; Lepiku et al., 1996). Thus internalized receptors could in principle continue to be counted as part of Bmax in appropriately designed experiments, and the reduced on-rate would appear as a change in apparent affinity rather than a change in the total number of receptors, as with the present data.
Features of these data argue against this interpretation, however. Most importantly, these data are consistent with the presence of a single population of receptors and with the purely competitive binding of raclopride with these receptors. However, particularly if the fraction of postsynaptic receptors that are internalized is small, the possibility of a combination of DA-based and internalization effects, in which noninternalized receptors have normal access but increased Kdapp caused by elevated DA concentrations and internalized receptors have normal competition with DA but elevated Kdapp because of the factors listed above, cannot be absolutely excluded.
In conclusion, using our equilibrium approach to receptor assay by multiple tracer studies at several different ligand concentrations, we showed that the reduction of affinity between receptor and raclopride after acute methamphetamine is the same, within experimental uncertainty, whether the raclopride equilibrium period used to determine the binding parameters begins 50, 200, or 350 minutes after the amphetamine dose. This observation is fully consistent with the simple competitive displacement picture and supports the induction by amphetamine or methamphetamine of changes in the synaptic milieu that are stable over many hours and that do not track with the passage of mobilized DA through the extrasynaptic fluid. A tightly regulated long-term elevation of the time-averaged levels of synaptic DA would appear to be the simplest of the many possible explanations of our results.
Footnotes
Acknowledgment:
The authors thank Astra Research Center for the gift of the raclopride precursor; the staff of the UBC/TRIUMF PET program for assistance and contribution to this work; Dr. T.J. Ruth (Head, PET program), Dr. J.A. Stoessl (Director, Pacific Parkinson Research Center), Ms. S. Jivan and M. Pronk (chemists), C. English and C. Williams (technologists), and J. Grant (AHT), as well as Dr. J. Love and Mr. M. Boyd and the personnel of the UBC Animal Care Facilities for their outstanding care of the animals. TRIUMF is funded by a contribution by the National Research Council of Canada.