
Abstract
During focal cerebral ischemia, matrix metalloproteinase-2 (MMP-2) can contribute to the loss of microvessel integrity within ischemic regions by degrading the basal lamina. MMP-2 is secreted in latent form (pro-MMP-2), but the activation of pro-MMP-2 in the ischemic territory has not been shown. Immunohistochemical and in situ hybridization studies of the expression of the direct activators of MMP-2, MT1-MMP and MT3-MMP, and the indirect activation system tissue plasminogen activator, urokinase (u-PA), its receptor (u-PAR), and its inhibitor PAI-1 after middle cerebral artery occlusion/reperfusion were undertaken in basal ganglia samples from 26 adolescent male baboons. The expressions of all three MMPs, u-PA, u-PAR, and PA1-1, but not tissue plasminogen activator, were increased from 1 hour after middle cerebral artery occlusion in the ischemic core. mRNA transcripts confirmed the increases in latent MMP-2, u-PA, u-PAR, and PAI-1 antigen very early after middle cerebral artery occlusion. The expression patterns are consistent with secretion of pro-MMP-2 and its activators in the ischemic core, perhaps from separate cell compartments. The rapid and coordinate appearance of pro-MMP-2 and its activation apparatus suggest that in the primate striatum this protease may participate in matrix injury during focal cerebral ischemia.
Degradation of the extracellular matrix (ECM, or basal lamina) within cerebral microvessels contributes to the loss of microvessel integrity and the appearance of hemorrhagic transformation during focal cerebral ischemia (Hamann et al., 1995; Rosenberg et al., 1996). These alterations in vascular matrix-integrin receptor interactions accompany changes in the vascular microenvironment that are associated with cell injury (Frisch and Francis 1994; Re et al., 1994). Matrix metalloproteinases (MMPs) and plasminogen activators are protease families with members capable of degrading many ECM components. Among them, pro-MMP-2, which in active form can degrade type IV collagen and laminins within the microvascular basal lamina, is significantly upregulated in the ischemic striatum within 2 hours after middle cerebral artery occlusion (MCAO) in the nonhuman primate (Hamann et al., 1995; Heo et al., 1999; Hosomi et al., 2001). Pro-MMP-2 expression is highly significantly correlated with neuron injury and the region of detectable cellular injury (Heo et al., 1999; Tagaya et al., 1997). Several studies have raised the question whether active MMP-2 is generated and contributes significantly to microvascular ECM degradation and neuron injury.
There is scant evidence of the generation of the active MMP-2 after MCAO in the several experimental systems in which it has been examined (Clark et al., 1997; Gasche et al., 1999; Heo et al., 1999; Rosenberg et al., 1996). Rosenberg et al first reported that in rats, pro-MMP-9 expression was associated with significantly increased blood brain barrier permeability at 3 and 48 hours after MCAO (Rosenberg et al., 1998). pro-MMP-2 increased 5 days or 3 hours after MCAO in separate studies with spontaneously hypertensive rats (Rosenberg et al., 1996; Rosenberg et al., 2001). However, the demonstration of active MMPs and their location in ischemic brain tissue has remained elusive. Active MMP-9 and MMP-2 were extracted from the ischemic tissues in one experiment (Gasche et al., 1999), but have not been detected with any significance in the majority of studies (Ahn et al., 1999; Asahi et al., 2000; Heo et al., 1999; Rosenberg et al., 1996; Rosenberg et al., 1998).
Pro-MMP-2 requires cleavage of the propeptide domain for activation by membrane-type MMPs (MT-MMPs) (Sato et al., 1994) or plasmin (Mazzieri et al., 1997). MT-MMPs are involved in the cell surface activation of extracellular pro-MMP-2, and are present in the human brain (Shofuda et al., 1997). The precise location of the MMP-2 and the MT1- and MT3-MMP antigens has not been described, although evidence for rapid pro-MMP-2 synthesis after MCAO supports the search for these sources (Heo et al., 1999; Rosenberg et al., 2001).
Urokinase (u-PA) and tissue plasminogen activator (t-PA) activate plasminogen to plasmin, which can degrade laminin, collagen IV, fibronectin, and myelin basic protein directly or indirectly, activate pro-MMP-2 and pro-MMP-9 (Keski-Oja et al., 1992; Mazzieri et al., 1997). u-PA and plasminogen activator inhibitor-1 (PAI-1), but not t-PA activities, are rapidly generated after MCAO in ischemic primate brain tissue in the same time-frame as pro-MMP-2 (Hosomi et al., 2001). The specific cellular receptor for u-PA, u-PAR, serves to localize and concentrate u-PA in other cell systems (Andreasen et al., 1997; Blasi 1999). However, it is not known whether and how u-PA and u-PAR are presented in the CNS, most particularly during focal cerebral ischemia.
Because pro-MMP-9 was associated with hemorrhagic transformation in the primate (Heo at al. 1999), we focused on the potential role of pro-MMP-2 in the maturation of ischemic injury in these studies. We hypothesize that both direct and indirect activators of pro-MMP-2 are upregulated rapidly on discrete vascular structures and cells within the ischemic region, and that their expressions persist during focal ischemia. Here, we demonstrate for the first time the very early regional coexpression of MMP-2 with its direct MT-MMP activators, the indirect activator u-PA, its receptor u-PAR, and PAI-1 together after MCAO. These findings suggest the coordinate appearance of the pro-MMP-2 activation apparatus, which may contribute to the degradation of microvessel matrix and neuron injury.
MATERIALS AND METHODS
The experimental procedures used here were approved by the institutional Animal Research Committee and were performed according to standards published by the National Research Council in The Guide for the Care and Use of Laboratory Animals and the United States Department of Agriculture Animal Welfare Act. Every effort was made to ensure that the subjects were free of pain and discomfort. The veterinarians, primate handling staff, research personnel, and the principal investigator attended all procedures. All animals were neurologically normal before MCAO and apparently free of infection or inflammation before and during the experiments.
Experimental stroke model and tissue processing
Cerebral tissues from 26 adolescent male baboons (Papio anubis/cynocephalus) were used for these studies. The detailed surgical and experimental approaches to the awake nonhuman primate stroke model have been reported previously (del Zoppo et al., 1986; Hamann et al., 1995). Before entry into the experiments, all animals were allowed a 7-day procedure-free interval after surgical implantation of the proximal MCA balloon device. The experimental procedures involved extrinsic compression of the proximal MCA by inflation of the device for 1 hour (n = 4), 2 hours (n = 4), or 3 hours followed by reperfusion for 1 hour (n = 4), 4 hours (n = 3), or 24 hours (n = 3). Additional tissues were provided by a group of subjects that did not undergo any preparation procedure (normal) (n = 4), and a group of subjects that suffered MCAO at surgical implantation that were harvested 7 days later (n = 4).
Tissues were removed under thiopental Na+ anesthesia after left ventricular transcardiac perfusion of the cranial structures at 180 to 220 mm Hg with chilled isosmotic anticoagulated perfusate containing heparin (200 IU/L), nitroprusside (1 mg/L), and bovine serum albumin (50 g/L) (Sigma, St. Louis, MO, U.S.A.). Tissue blocks (1.0 × 1.0 × 0.5 cm) were excised from symmetrically located sites of both left and right basal ganglia. Alternate blocks were either embedded in Tissue-Tek OCT compound (Miles, Inc., Elkhart, IN, U.S.A.), immediately frozen in 2-methylbutane/dry ice, and stored at −80°C until use, or immediately immersed in 2% paraformaldehyde for 24 hours before paraffin embedding for in situ hybridization.
Reagents
Well-characterized antibodies against the human protein haptens were used throughout. The murine monoclonal antibody raised against purified human recombinant MMP-2 (1A10, British Biotech Pharmaceuticals, Inc., Oxford, UK), and a purified murine anti–human MMP-2 monoclonal antibody (IM33L, Calbiochem, Cambridge, MA, U.S.A.) both detected MMP-2 and pro-MMP-2. MT1-MMP immunoreactivity was signaled with a protein A Sepharose-purified rabbit polyclonal antibody (RP63, British Biotech Pharmaceuticals) and confirmed with a murine monoclonal antibody (MAB 3319, Chemicon, Temecula, CA, U.S.A.). MT3-MMP was detected with a rabbit polyclonal antibody (British Biotech Pharmaceuticals). Cross-competition studies demonstrated no cross-reactivity between 1A10 or IM33L and among RP-63, MAB3319, or the anti–MT3-MMP antibody by Western blotting (data not shown).
The t-PA antigen was detected with a goat polyclonal antibody (387, American Diagnostica Inc., Greenwich, CT, U.S.A.), whereas PAI-1 antigen was detected with a rabbit polyclonal antibody (the kind gift of Dr. David J. Loskutoff, TSRI, La Jolla, CA, U.S.A.). The u-PA antigen was detected with a rabbit polyclonal antibody, raised against purified u-PA from human embryonic kidney cells. The u-PAR antigen was detected with a rabbit polyclonal antibody against the purified protein (Mazar et al., 2001). In separate studies, we demonstrated that none of these antibodies detected baboon immunoglobulin G or albumin by Western blot at concentrations used for immunohistochemistry (data not shown).
To prepare biotinylated u-PA, recombinant human single-chain u-PA (scu-PA) was expressed in Drosophila S2 cells by transfection with a cDNA encoding for scu-PA (amino acids 1–411) (DES System, Invitrogen). A clone expressing high levels of scu-PA was selected for scale-up using an enzyme-linked immunosorbent assay for u-PA (American Diagnostica). scu-PA in the culture supernatant was purified on an SP-Sepharose column (5 × 100 cm) equilibrated with 20-mmol/L BICINE (pH 8.8) and eluted with a NaCl gradient (0–0.5 mol/L) in the same buffer. scu-PA was detected by SDS-PAGE to be more than 90% pure, and was purified to homogeneity using RP-HPLC on a C8 column followed by lyophilization. Its identity was confirmed by sequence analysis and mass spectroscopy.
Lyophilized scu-PA was reconstituted to 1 mg/mL in PBS (20 μmol/L) and combined with 1-mmol/L sulfo-NHS-LC-biotin (Pierce) in PBS to a final molar ratio 5:1, then incubated at neutral pH at room temperature for 3 hours. Unreacted sulfo-NHS-LC-biotin was removed using a PD10 (Bio-Rad) column. Mass spectroscopy analysis revealed a ratio of 1.5 to 2.0 biotin per molecule scu-PA. Biotinylation was confirmed on a blot probed with avidin-HRPO and the retention of binding activity was confirmed in a whole-cell binding assay using HeLa cells. A small peptide inhibitor of u-PA binding to u-PAR was used to demonstrate the specificity of biotin-scu-PA (Ploug et al., 2001). The peptide [Asp-cyclohexylalanine-Phe-(D-Ser)-(d-Arg) Tyr Leu Trp Ser] was prepared using standard solid phase methodology (Star Biochemical).
Riboprobes for the in situ hybridization studies were subcloned from cDNAs (Table 1). A 443-bp fragment of human MMP-2 cDNA (GenBank J03210) was subcloned from pBSGEL (a kind gift of Dr. Lynn Matrisian, Vanderbilt University, Nashville, TN, U.S.A.) (Collier et al., 1988; Rodgers et al., 1994). The 310-bp t-PA probe was prepared from a full-length cDNA (a kind gift of Dr. Gene Levin, TSRI). The PAI-1 probe was a kind gift of Dr. David Loskutoff (TSRI). Others probes were prepared from known cDNA sequences (e.g., GenBank).
Riboprobe sequences for in situ hybridization

Immunohistochemistry
Immunoperoxidase studies were performed on serial 10-μm frozen sections using published methods (Abumiya et al., 1999). Avidin-biotin complexes were generated with streptavidin-horseradish peroxidase (Vector Laboratories, Burlingame, CA, U.S.A.), and detected with 3-amino-9-ethyl carbazole (AEC kit, Biomeda Corp., Foster City, CA, U.S.A.). Studies with the IM33L antibody required tyramine signal amplification (NEN Life Science Products, Inc., Boston, MA, U.S.A.) and another avidin-biotin complex reaction before chromogen development. All sections were counterstained with Mayer's hematoxylin (Biomeda) or a Nissl stain (Polysciences, Inc., Warrington, PA, U.S.A.).
Regions of cellular injury
Evidence of nuclear DNA scission/repair was taken as an indication of significant cellular injury as described by Tagaya et al., (1997) and Abumiya et al., (1999). The DNA polymerase I-based procedure for incorporation of digoxigenin-dUTP (DIG-dUTP) was used for 10-μm cryosections or 3-μm paraffin sections adjacent to those used for the immunohistochemical or in situ hybridization studies, respectively.
In situ hybridization
In situ hybridization was performed on paraffin-embedded sections using 35S-UTP cRNA probes as described previously (Abumiya et al., 1999; Seiffert et al., 1991). Deparaffinized PFA-fixed sections were incubated in 0.2-mol/L HC1 for 10 minutes and digested with 5-μg/mL proteinase K for 10 minutes. After incubation with prehybridization buffer [50% formamide (vol/vol), 0.3-mol/L NaC1, 20-mmol/L Tris-HC1 (pH 8.0), 5-mmol/L EDTA, 1% Denhardt's solution, 10% dextran sulfate (weight/volume), and 10-mmol/L DTT] at 42°C for 3 hours, the sections were hybridized with 1 × 106 cpm of 35S-labeled riboprobes at 55°C for 18 hours. The sections were then washed twice in 2 × SSC/2-mmol/L EDTA, treated with RNase A (20 μg/mL) for 30 minutes, and washed twice again in 2 × SSC/2-mmol/L EDTA. A high-stringency wash in 0.1 × SCC/-mercaptoethanol/EDTA was performed at 55°C for 2 hours followed by washing in 0.5 × SSC. The sections were coated with NTB emulsion (Kodak, New Haven, CT, U.S.A.) and exposed in a sealed box at 4°C for 4 to 6 weeks. After development, the sections were counterstained with Richardson solution.
Western blot preparations
Western blot studies confirmed the specificity of the rabbit anti-human primary antibodies against u-PA, u-PAR, and PAI-1 in ischemic tissue. Samples were separated under nonreducing or reducing conditions on 8% SDS polyacrylamide gels, and electrophoretically transferred to nitrocellulose (Schleicher & Schuell Inc., Keene, NH, U.S.A.) by the semidry blotting method. Nitrocellulose membranes were blocked with 5% nonfat milk in PBS containing 0.1% Tween-20 (PBST), then incubated with the primary antibody for 1 hour at 37°C. The membranes were then incubated with the horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, U.S.A., or Vector Laboratories) at 2°C for 1 hour. After each incubation, the blots were washed in PBST to remove any unbound antibody. Bound antibody was detected by enhanced chemiluminescence (NEN Life Science Products Inc., Boston, MA, U.S.A.) according to the manufacturer's instructions.
For these studies, recombinant u-PA, u-PAR, and PAI-1 served as target antigens. Rabbit nonimmune immunoglobulin G was used as a primary antibody for the control. Absorption studies were performed by using a primary antibody incubated with recombinant u-PA, u-PAR, or PAI-1 for 1 hour at 37°C. Multiple correlation studies were consistent with the unique nature of primary antibodies for the immunohistochemical studies.
Enzyme-linked immunosorbent assay for u-PAR
An enzyme-linked immunosorbent assay was used to detect the u-PAR antigen content of homogenized brain tissue samples, according to the manufacturer's directions (American Diagnostica). Antigen concentrations were calculated in nanograms per milligram of extracted protein (by the Bradford method).
Quantitative analysis
In ischemic tissue, the ischemic peripheral (Ip) region lay outside the ischemic core (Ic) region. Regions of cell dUTP incorporation defined the Ic region. The Ic regions were demarcated and scanned under the mode of 12 bits per pixel digital resolution and 50-μm pixel size. This mode provided satisfactory images of sections with 4,096 levels of digital resolution and 4,000 data points/cm2. The total section and target surface areas were measured using the NIH Image 1.61 Program staged on a Macintosh platform (Tagaya et al., 1997). The absolute number and the minimum transverse diameter of immunoreactive microvessels were quantified with the aid of computerized video imaging microscopy. A matrix of 25 or 100 nonoverlapping microscopic fields at 400× magnification comprised 1.5 or 6.0 mm2 regions of interest (ROI), respectively. Two identical ROI each were chosen within the Ic and Ip regions, and one ROI was chosen within the contralateral nonischemic region and in the matched basal ganglia of control subjects. Coregistration of these ROI on consecutive sections allowed assessment of epitope coexpression.
Statistical analysis
All data are presented as the mean ± SD. For each antigen, differences in time courses between ischemic and matched nonischemic samples were assessed by two-way analysis of variance, or Student's t-test with Bonferroni correction for multiple comparisons, where appropriate. Significance was set at P < 0.05.
RESULTS
Expression of immunoreactive MMP-2 after MCAO
Within the gray matter of normal and nonischemic basal ganglia, immunoreactive MMP-2 was found only on select individual microvessels (Fig. 1A). Beginning 2 hours after MCAO, clear and detectably increased MMP-2 antigen expression was strictly confined to and contained within the Ic parenchyma (Fig. 1B). MMP-2 antigen persisted within the Ic regions (but not the Ip regions) throughout all times after MCAO. However, the detectable association with microvessels was lost by 2 hours MCAO, in part because of the increased parenchymal expression of MMP-2.
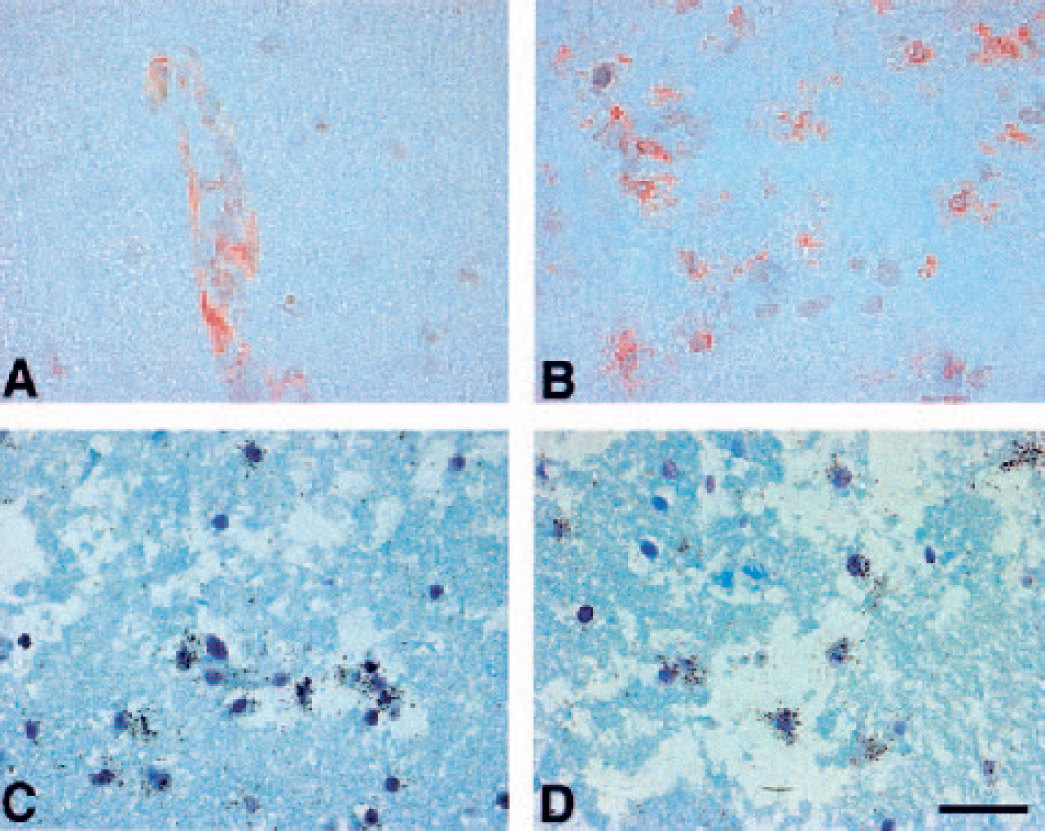
MMP-2 antigen in the media of a 24-μm-diameter microvessel in the nonischemic basal ganglia (
In the Ic region, the number of microvessels (including capillaries) displaying MMP-2 mRNA transcripts increased significantly by 2 hours after MCAO, compared with the normal basal ganglia (Figs. 1C and 1D). Few microvessels displaying MMP-2 transcripts were detected in the Ip region. After MCAO, MMP-2 transcripts also appeared to be associated with glial cells, as suggested by nuclear morphology and separate immunohistochemical studies.
Expression of the direct pro-MMP-2 activators
The parenchymal expression of both MT1-MMP and MT3-MMP antigens increased significantly within the Ic regions by 1 to 2 hours after MCAO. The regions of increased MMP-2, MT1-MMP, and MT3-MMP immunoreactivity were highly significantly cross-correlated at all time points (Fig. 2), as confirmed by dual immunohistochemistry. Neither MT1- nor MT3-MMP was detected in normal or nonischemic tissues.
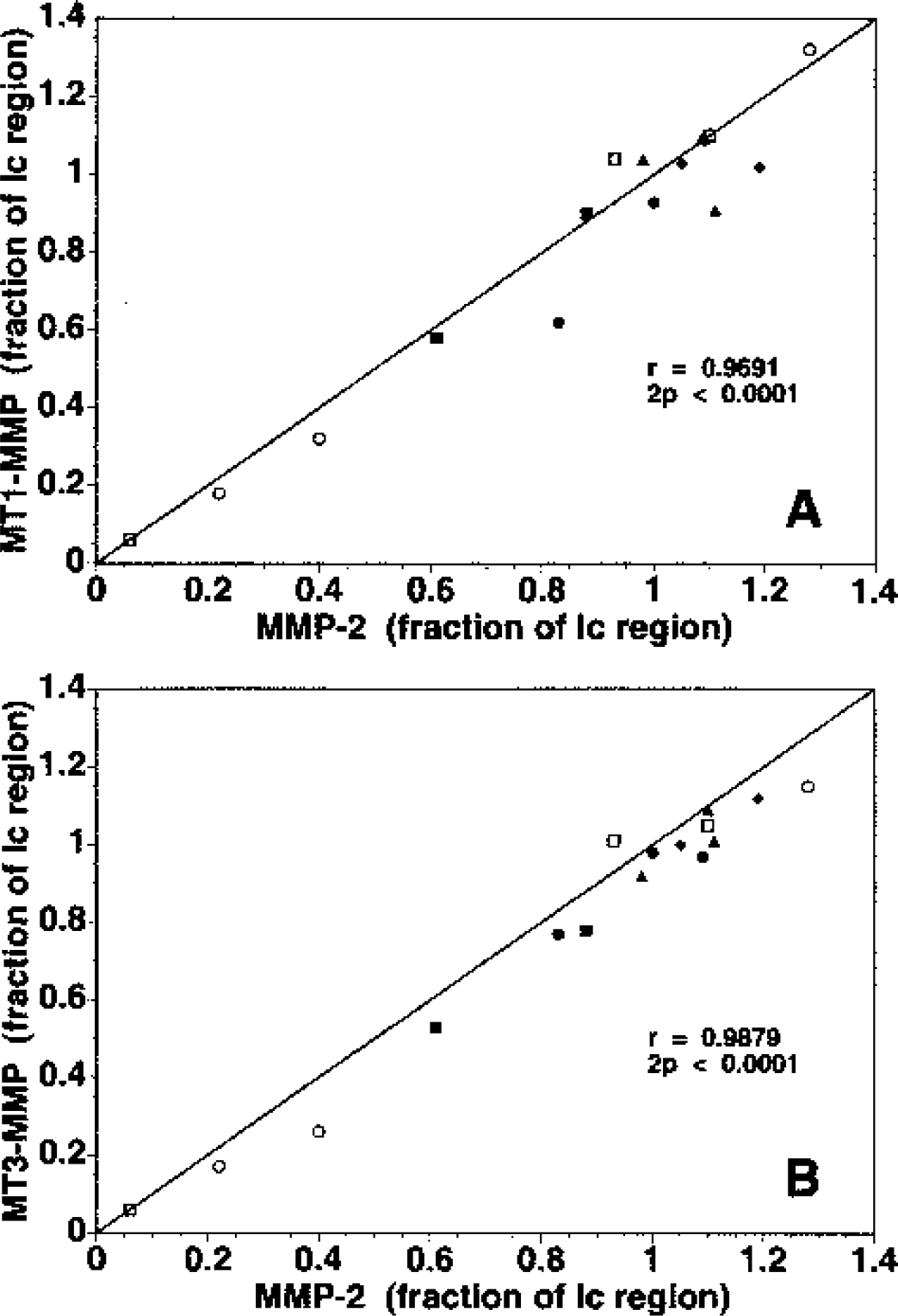
Identity of the regions of expression of the activators MT1-MMP (
Expression of the indirect activator t-PA, and its inhibitor PAI-1
Although t-PA interacts with its inhibitor PAI-1 in plasma, it is not known whether there is a meaningful interaction within brain tissue. Select small arterioles (7.5–30.0 μm diameter) expressed t-PA antigen within the normal and nonischemic basal ganglia, as previously described (Levin and del Zoppo, 1994). There was no detectable change in the number of microvessels expressing t-PA at 2 hours and 7 days after MCAO in either the Ic or Ip regions (Fig. 3), and no detectable t-PA in the Ic parenchyma at any time point. In contrast, t-PA mRNA was noted on select microvessels by 2 hours MCAO.

Microvascular distribution of tissue type plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), urokinase type plasminogen activator (u-PA), urokinase receptor (u-PAR), and laminin by transverse diameter (in micrometers) in the ischemic core (Ic) and peripheral (Ip) regions of ischemic subjects at various periods after MCAO (normal [n = 4], 2 hours [n = 4], and 7 days [n = 4]. , 4.0–7.5 μm;  , 7.5–30 μm; □, 30–50 μm;
, 7.5–30 μm; □, 30–50 μm;  , 50–100 μm.
, 50–100 μm.
In the normal basal ganglia, PAI-1 antigen was associated with arterioles and small arteries (3.8 ± 0.9 microvessels/mm2). In contrast to t-PA, PAI-1 immunoreactivity increased significantly and exclusively within the Ic region (Figs. 3–5). By 2 hours after MCAO, PAI-1 immunoreactivity appeared on capillaries and larger microvessels in the Ic region. The density of capillaries displaying PAI-1 increased modestly from 0 to 2 hours after MCAO from 0 to 2.2 ± 1.1 (Ic) and 2.0 ± 0.6 (Ip) capillaries/mm2 (P = 0.0006 and P < 0.0001, respectively). Parenchymal PAI-1 expression was homogenous and sustained within the Ic region from 2 hours to 7 days. In comparison to nonischemic tissue, increased PAI-1 mRNA was seen on microvessels within the Ic region by 2 hours MCAO, and on larger microvessels up to 7 days.
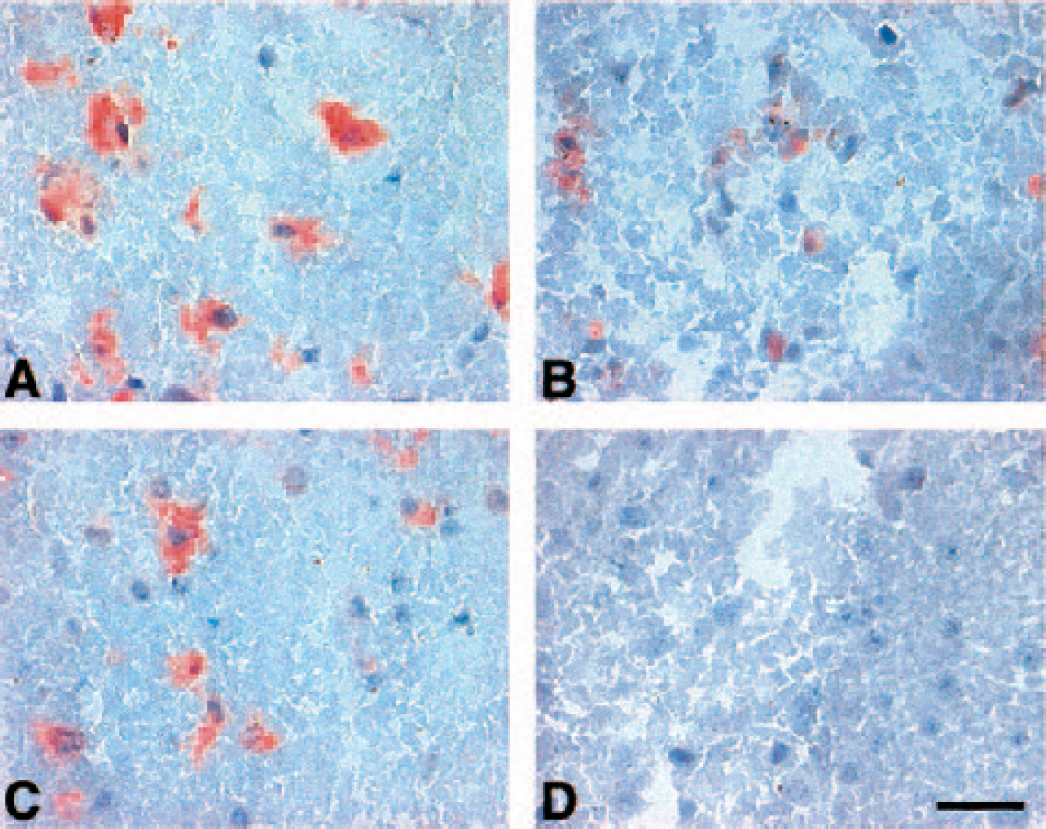
Representative photomicrographs of immunoreactive u-PA (
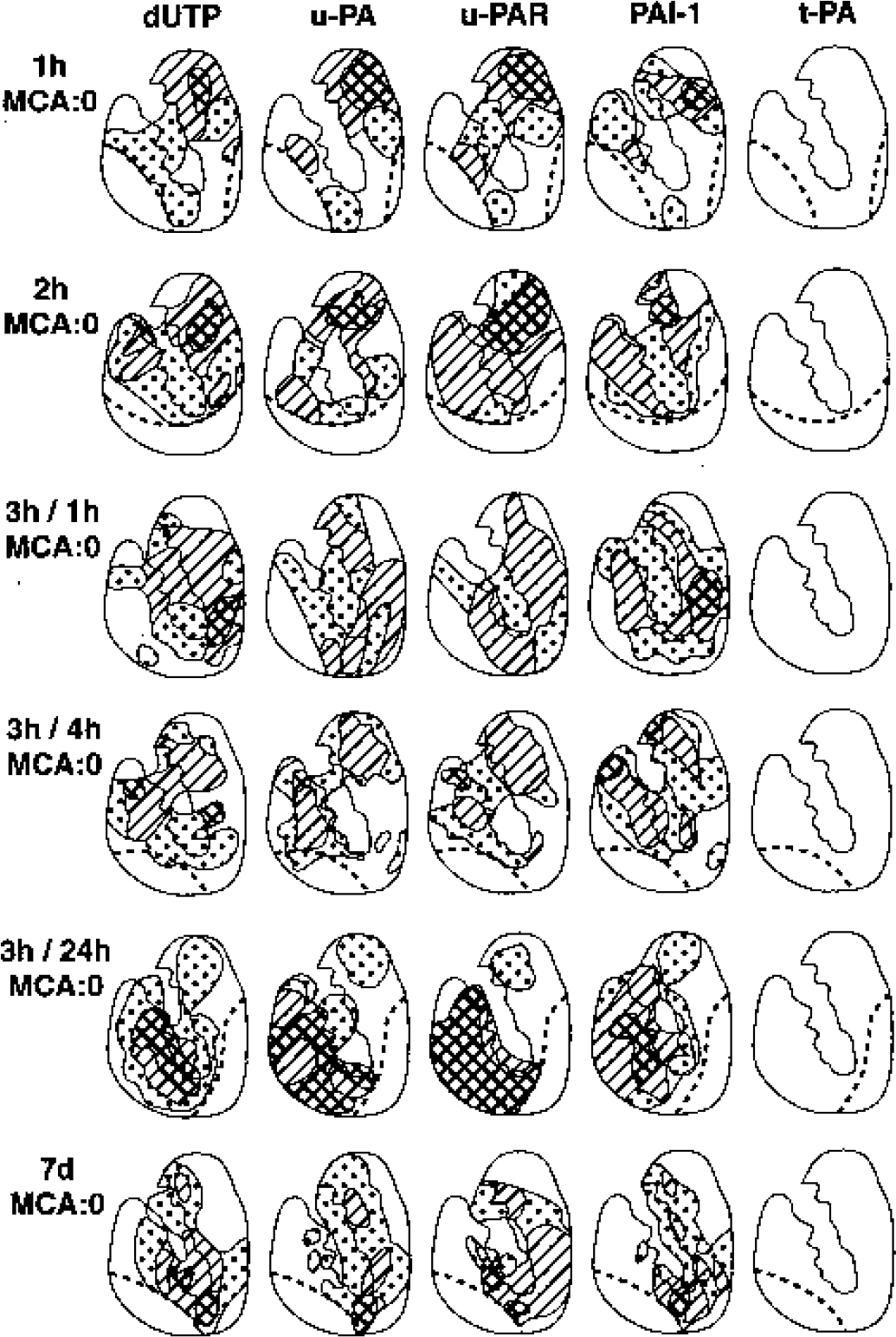
Maps of cellular dUTP incorporation and cell-associated immunoreactive u-PA, u-PAR, PAI-1, and t-PA in the ischemic basal ganglia of each subject after MCAO and MCAO/reperfusion (1 hour [n=4], 2 hours [n=4], 3 hours/4 hours [n=3], 3 hours/24 hours [n = 3], and 7 days [n = 4]. The distribution u-PA, u-PAR, and PAI-1 expression were comparable to the dUTP+ regions. Clear zone = 0%, speckled zone = < 50%, hatched zone = > 50%, cross-hatched zone = 100% of subjects displaying respective antigen expression.
Expression of the indirect activator u-PA, and its receptor u-PAR
u-PA and u-PAR were confined to the Ic regions, increasing significantly as early as 1 to 2 hours after MCAO, and were sustained up to 7 days (Fig. 6). The increased parenchymal u-PAR expression coincided with increased u-PA activity and antigen and PAI-1 antigen previously observed under the same conditions (Hosomi et al., 2001). Highly significant pair-wise correlations were found among the Ic regions of injury for u-PA, u-PAR, and PAI-1 expression: dUTP+ vs u-PA (r = 0.893, P < 0.0001), dUTP+ vs u-PAR (r = 0.860, P < 0.0001), dUTP+ vs PAI-1 (r = 0.838, P < 0.0001), u-PAR vs PAI-1 (r = 0.716, P = 0.0003), u-PA vs PAI-1 (r = 0.705, P < 0.0004), and u-PA vs u-PAR (r = 0.793, P < 0.0001) (Fig. 5).
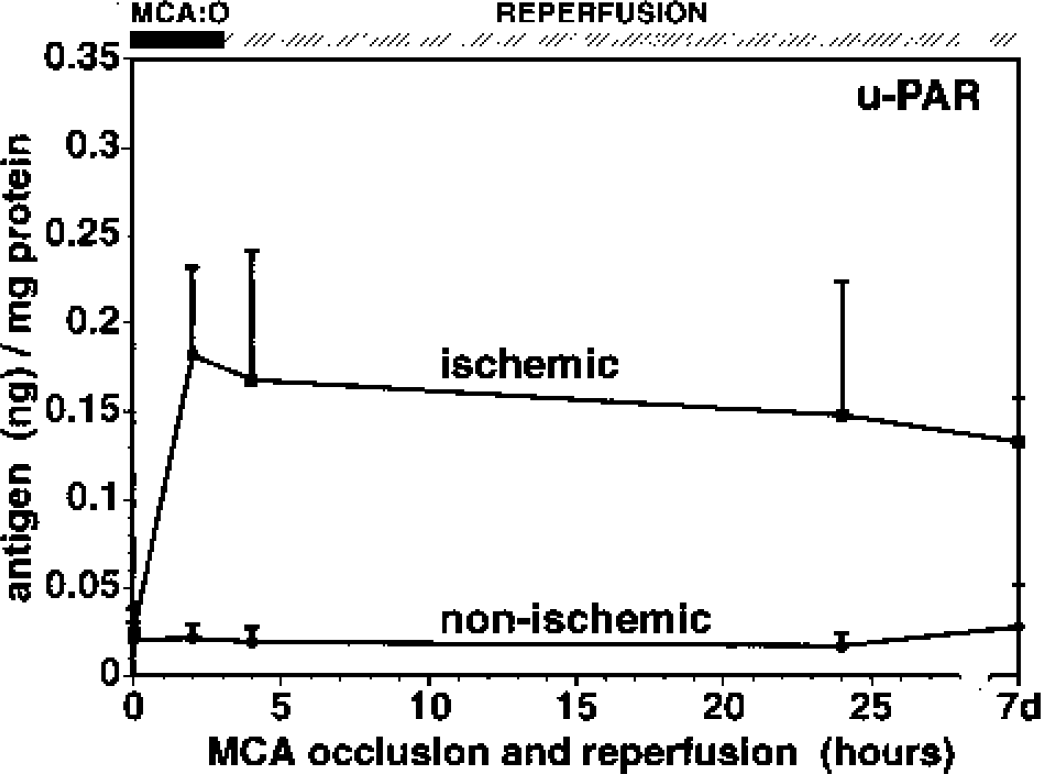
Time course of u-PAR antigen appearance after MCAO in the ischemic basal ganglia. n = 3 or 4 for each time point.
In the normal and nonischemic basal ganglia, weak but detectable u-PA immunoreactivity was observed on select small arterioles (7.5–50 μm and 50–100 μm diameter), but not capillaries. In contrast, u-PAR immunoreactivity was seen on capillaries, small arterioles, and small arteries (Figs. 3). Those vascular signals were completely blocked by recombinant u-PA and u-PAR, respectively. After MCAO, the densities of all u-PA and u-PAR immunoreactive microvessels (microvessels/mm2) remained substantially unchanged up to 7 days, compared with the decrease in laminin expression by 2 hours MCAO (Fig. 3). The density of capillaries expressing u-PA and u-PAR increased significantly in this interval, and the density of u-PAR+ microvessels exceeded those expressing u-PA. Capillary-associated u-PA expression increased from 0 to 2 hours after MCAO within the Ic regions (0 to 2.0 ± 0.9 capillaries/mm2, P = 0.0004) and in the Ip regions (2.2 ± 1.2 capillaries/mm2, P < 0.001) (Fig. 3). Although there was no significant change in u-PAR+ microvessel density in the Ic region by 2 hours MCAO, a substantial increase in capillary density in both the Ic and Ip regions was observed (0.3 ± 0.7 capillaries/mm2 to 3.5 ± 2.4 (Ic) and to 9.2 ± 5.5 (Ip) capillaries/mm2, P = 0.0013 and P = 0.005, respectively).
Serial dual immunofluorescence experiments showed that microvessels that express u-PA could also express u-PAR or PAI-1 (Fig. 7).
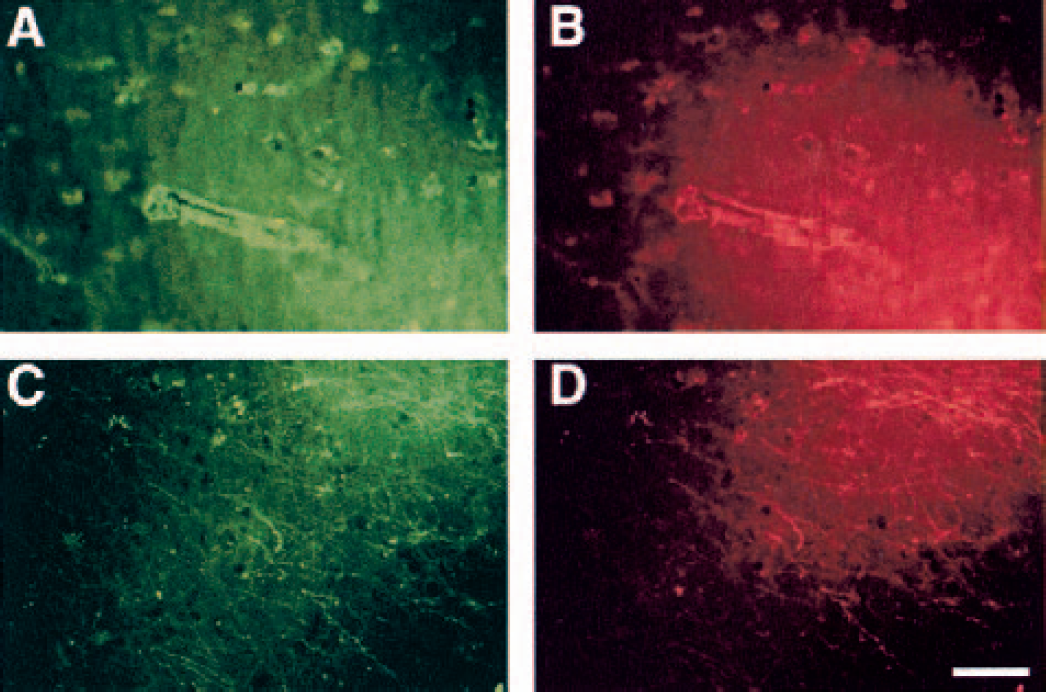
Colocalization of u-PA, u-PAR, and PAI-1 antigens in the same microvessels 2 hours after MCAO, by dual immunofluorescence. Panels A and B, u-PAR (FITC) and u-PA (TRITC). Panels C and D, PAI-1 (FITC) and u-PA (TRITC). Magnification bar = 50 μm.
Cellular expression of u-PA and u-PAR
To further determine whether u-PA and u-PAR associate with one another in the ischemic regions, serial sections were incubated with biotinylated recombinant scu-PA (scu-PA*). Control studies on cultured HeLa cells confirmed the specific interaction of scu-PA* with the constitutively produced u-PAR, and set conditions for the tissue studies (Figs. 8A–8D). No binding of scu-PA* within the Ic region after MCAO was seen on tissue sections (data not shown). However, dissociation of unlabeled u-PA from its receptor under acidic conditions allowed binding of scu-PA* to neurons and microvessels within the Ic regions at both 2 hours and 7 days (Figs. 8E–8H). Compared with the normal and nonischemic basal ganglia, scu-PA* bound significantly to neurons both at 2 hours and 7 days after MCAO, accounting for 88.8 ± 10.2% of neurons overall (compared to 0.32 ± 0.35% in nonischemic tissue, n = 6 each). Neurons accounted for 60.9 ± 7.4% of all cells (n = 6) in the ischemic tissue at both time points.
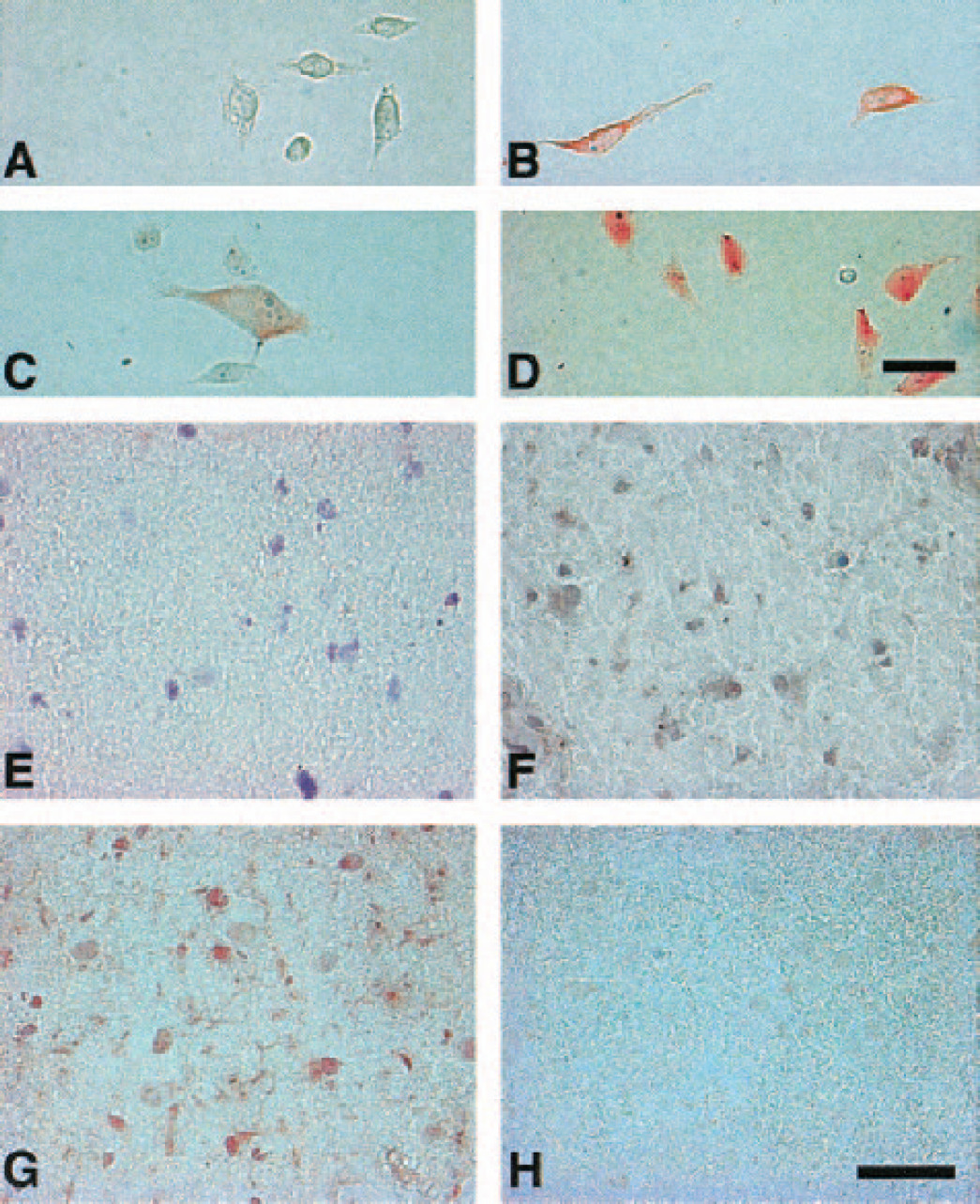
Representative photomicrographs of the localization of biotinylated single chain urokinase (scu-PA*) on HeLa cells (
DISCUSSION
The immediate and coincident loss of vascular integrin antigens (Tagaya et al., 2001), vascular matrix integrity (Hamann et al., 1995), and normal neuron morphometry (Eke et al., 1990; Garcia and Kamijyo, 1974; Garcia et al., 1983) very early after MCAO suggests that active proteolytic ECM degradation may cause or accentuate neuronal injury (Heo et al, 1999). The potentially matrix-degrading protease “activities” of pro-MMP-2 and u-PA, determined by zymography, increase immediately after MCAO within the ischemic basal ganglia of the nonhuman primate (Heo et al., 1999; Hosomi et al., 2001). Striatal neuron injury is directly related to pro-MMP-2 generation in this primate model (Heo et al., 1999). MMP and PA expressions differ from those in rodent MCAO models, however (Ahn et al., 1999; Heo et al., 1999; Rosenberg et al., 1996; Rosenberg et al., 1998). The distribution and compartmentalization of pro-MMP-2, its activation systems, and the relevant receptors are likely to be important in cerebral vascular and neuronal injury, although nothing is known about these relationships. Pro-MMP-2 expression in the normal brain has been described variously as absent (Lim et al., 1996; Nakagawa et al., 1994) or confined to microvessels, leukocytes, microglia, or astrocytes (Anthony et al., 1997; Cuzner et al., 1996; Maeda and Sobel, 1996). However, the sources of the increased pro-MMP-2 expression 1 to 2 hours after MCAO and the nature of its significant association with neuron injury have not been further defined (Heo et al., 1999; Hosomi et al., 2001). For this, activation of pro-MMP-2 must be shown. Here, we demonstrate the coordinate expression of the direct pro-MMP-2 activators, MT1-MMP and MT3-MMP, and the indirect activator u-PA, its receptor u-PAR, and its principal inhibitor PAI-1 (but not t-PA) immediately after MCAO. Furthermore, the rapid appearance of MMP-2 antigen and mRNA in the Ic region confirms that pro-MMP-2/MMP-2 is rapidly synthesized and secreted by select structures (Heo et al., 1999). These observations are the first to suggest that activation of increased pro-MMP-2 is possible within the Ic region during focal ischemia.
Activation of pro-MMP-2 occurs directly by MT-MMPs, which are bound to the membranes of select cells, in contrast to most MMPs, which are secreted and activated by the serine proteases plasmin, trypsin, chymotrypsin, or cathepsin G (Nagase et al., 1990; Okada et al., 1988). Since Sato et al. first identified a novel MT-MMP (designated MT1-MMP), MT2-MMP, MT3-MMP, and MT4-MMP have been identified (Puente et al., 1996; Sato et al., 1994; Takino et al., 1995; Will and Hinzmann 1995). After MCAO, MT1- and MT3-MMP increase together with MMP-2 antigen throughout the Ic regions, indicating coexpression of the direct activators for pro-MMP-2 (Corcoran et al., 1996; Strongin et al., 1995). Although both MT-MMPs are expressed by microglia in normal white matter (Yamada et al., 1995; Yoshiyama et al., 1998), no clear cell association was identified here. This is consistent with their membrane expression on cells distributed throughout the Ic region, and the loss of cell compartments later after MCAO.
MT-MMPs can also degrade a number of ECM proteins including fibronectin, collagen IV, and laminin (Koshikawa et al., 2000). Coexpression of MMP-2 and MT-MMPs has also been reported in malignant brain tumors, liver fibrosis, and vascular neointima (Jenkins et al., 1998; Takahara et al., 1997; Wang and Keiser, 1998; Yamamoto et al., 1996). Recently, it was demonstrated in vitro that plasmin can activate pro-MMP-2 via an MT1-MMP–dependent mechanism, which leads to increased degradation of type IV collagen (Mazzieri et al., 1997; Monea et al., 2002). Hence, pro-MMP-2 could be activated indirectly by plasminogen activators via their generation of plasmin from plasminogen (Mazzieri et al., 1997), and could thereby degrade microvascular ECM collagen.
During ischemia, it is possible that of the plasminogen activators, u-PA is involved in pro-MMP-2 activation (Hosomi et al., 2001). This is because parenchymal t-PA antigen is unaltered after MCAO in the primate, despite a transient decrease in activity due to its binding with PAI-1 in tissue (Hosomi et al., 2001). Although t-PA transcripts appeared on more microvessels in the Ic region than in nonischemic tissue, detectable t-PA production did not increase during ischemia. In contrast, PAI-1 increased in the Ic region. PAI-1 also appears to derive from microvascular endothelium, because both the plasma and parenchymal PAI-1 content increased together (Hosomi et al., 2001). By blocking t-PA or u-PA in plasma, PAI-1 could alter vascular fibrinolytic activity, but its roles in modulating ischemic tissue injury in the parenchymal compartment are not yet defined.
Urokinase plasminogen activator is secreted as the inactive single-chain u-PA (scu-PA), whose activation by plasmin to the two-chain form is accelerated when u-PA is bound to its specific, high-affinity cell surface receptor u-PAR (Conese and Biasi, 1995). u-PAR has been detected on endothelial cells, neurons, and microglia in vitro, where its expression can be stimulated by hypoxia (Graham et al., 1999; Hayden and Seeds, 1996; Washington et al., 1996). Here, u-PAR was identified on neighboring microvessels and neurons only after MCAO. Active u-PA is stably associated via its growth factor domain with u-PAR at the cell plasma membrane (Corti et al., 1989). PAI-1 specifically binds to and inhibits both free and receptor-bound u-PA in plasma (Cubellis et al., 1989), but the binding of u-PA to PAI-1 on u-PAR also allows the u-PA–PAI-1 complex to be internalized and degraded while u-PAR is recycled back to the cell surface (Nykjaer et al., 1997). By inhibiting u-PAR bound u-PA, PAI-1 can potentially modulate the propagation of u-PA-mediated vascular matrix injury.
The density of capillaries displaying immunoreactive u-PA, u-PAR, and PAI-1 increased shortly after MCAO, and coincided with increased parenchymal pro-MMP-2 expression (Heo et al., 1999). This suggests a potential link between u-PA synthesis and pro-MMP-2 activation (He et al., 1989; Kazes et al., 1998; Mackay et al., 1990; Mazzieri et al., 1997). Other investigators have described increased u-PA activity significantly later (at 12–24 hours, and up to 5 days), decreased t-PA activity, or increased u-PA and t-PA in the rat brain after MCAO (Ito et al., 2000). In human postmortem materials, immunoreactive u-PA, u-PAR, and PAI-1 were expressed by endothelial cells, reactive astrocytes, and microglia around, but not within, the infarct (Dietzmann et al., 2000). These inconsistencies probably reflect differences in tissues, resolution of the detection techniques, and other factors. Here, immunoreactive u-PA, u-PAR, and PAI-1 increased together after MCAO in the same Ic subregions as pro-MMP-2, MT1-MMP, and MT3-MMP (Figs. 2 and 3).
The rapid appearance of u-PAR is consistent with the cell-surface concentration of u-PA for matrix degradation and vascular cell migration. These studies also raise the interesting question whether u-PA bound to neurons that express u-PAR generates pericellular proteolytic activity for the degradation of ECM around neurons, or serves other purposes. Nearly all nonvascular cells derived from the CNS have been shown to express u-PA in vitro (Dent et al., 1993; Kalderon et al., 1990; Masos and Miskin, 1996; Nakajima et al., 1992; Presta et al., 1999; Tranque et al., 1992; Washington et al., 1996). Except for u-PAR, however, the topographical distributions of u-PA, PAI-1, and pro-MMP-2/MMP-2 antigens after MCAO in the primate do not suggest consistent and exclusive cell associations, in contrast to those reported in rodents (Mun-Bryce et al., 2002; Rosenberg et al., 2001). Instead the findings here are compatible with their secretion into the Ic region.
In vitro studies also support the interpretation that the cellular expressions of u-PA, u-PAR, and PAI-1, but not t-PA, are stimulated by the cytokines tumor necrosis factor-α, interleukin-1β, and/or transforming growth factor-β (Ahn et al., 1999; Docagne et al., 1999; Ninomia et al., 2000; Schleef et al., 1988; Van Hinsbergh et al., 1990). Transforming growth factor-β can also stimulate u-PA and u-PAR expression (Mori et al., 2000). Tumor necrosis factor-α and interleukin-1β are generated early in the ischemic territory after MCAO, as demonstrated in rodents (Wang et al., 1994). Hence, the very early differential responses of u-PA, u-PAR, t-PA, and PAI-1 to focal ischemia in the primate may be specific responses to cytokine release.
Finally, the proposed cell-surface activation of pro-MMP-2 by MT-MMPs and u-PA, and their vascular expression, suggests that both MMP-2 and u-PA may also play roles in angiogenesis and vascular permeability. Abumiya et al. (1999) demonstrated that vascular endothelial growth factor and integrin αVβ3 are rapidly and coordinately upregulated within precapillary arterioles in the ischemic basal ganglia, in the same site and time frame as the changes noted here. Vascular and endothelial growth factor also increases MMP-2, u-PA, and u-PAR expression in endothelial cells (Koolwijk et al., 1996; Lamoreaux et al., 1998; Zucker et al., 1998), which accompany neovascularization (Kroon et al., 1999; Korff et al., 2001). u-PAR and a variety of vascular integrins including αVβ3 and α3β1 are associated (Conforti et al., 1994; Tagaya M., Chang D-I, del Zoppo, unpublished data, 2000). Because u-PAR greatly potentiates cell-surface proteolytic activity by u-PA (Montuori et al., 2000), the results here also imply that u-PA/u-PAR complexes could disrupt microvascular matrix integrity after MCAO. A unique relationship between increased microvessel permeability and active MMP-2 is not certain, however, because other causes of vascular permeability (including u-PA, vascular endothelial growth factor, and thrombin) also coincide with protein extravasation in these models (Abumiya et al., 1999; del Zoppo et al., 1986; Okada et al., 1994).
The changes observed here were confined to the ischemic core as defined by neuron injury. However, particularly the expression of u-PAR on microvessels was increased in the regions peripheral to the core. Recent findings of microvessel integrin responses to ischemia (Tagaya et al., 2001) support the possibility that, early after MCAO, multiple “penumbrae” around injury subregions exist within the ischemic core. The appearance of the (pro-)MMP-2 and u-PA antigens here are most compatible with their secretion in the core.
In summary, focal cerebral ischemia acutely increases the secretion of pro-MMP-2 in parallel with its direct cell membrane activators, MT1-MMP and MT3-MMP, throughout the ischemic core. The rapid increase in u-PA and u-PAR expression after ischemia suggests that u-PA expressed by surviving cells throughout the Ic region, whether microvascular endothelial cells, neurons, microglia, or astrocytes, could also indirectly activate pro-MMP-2 via plasmin and MT1-MMP in response to the ischemic insult. The broad expression of these activators implies that the small amounts of active MMP-2 detected (Heo et al., 1999; Hosomi et al., 2001) may represent high local concentrations. It is plausible that locally active MMP-2 contributes to early matrix degradation, loss of vascular integrity, neuron injury, and maturation of the ischemic lesion. Although the data here indicate the very rapid appearance of pro-MMP-2 and its activators in the Ic region, further studies are needed to determine whether and how activation occurs. If true, these findings imply that inhibitors of direct or indirect activation of pro-MMP-2 could have a beneficial effect on vascular integrity, injury maturation, and neuron demise.