
Abstract
Cerebral microvessels have a unique ultrastructure form, which allows for the close relationship of the endothelium and blood elements to the neurons they serve, via intervening astrocytes. To focal ischemia, the cerebral microvasculature rapidly displays multiple dynamic responses. Immediate events include breakdown of the primary endothelial cell permeability barrier, with transudation of plasma, expression of endothelial cell-leukocyte adhesion receptors, loss of endothelial cell and astrocyte integrin receptors, loss of their matrix ligands, expression of members of several matrix-degrading protease families, and the appearance of receptors associated with angiogenesis and neovascularization. These events occur pari passu with neuron injury. Alterations in the microvessel matrix after the onset of ischemia also suggest links to changes in nonvascular cell viability. Microvascular obstruction within the ischemic territory occurs after occlusion and reperfusion of the feeding arteries (“focal no-reflow” phenomenon). This can result from extrinsic compression and intravascular events, including leukocyte(-platelet) adhesion, platelet-fibrin interactions, and activation of coagulation. All of these events occur in microvessels heterogeneously distributed within the ischemic core. The panorama of acute microvessel responses to focal cerebral ischemia provide opportunities to understand interrelationships between neurons and their microvascular supply and changes that underlie a number of central nervous system neurodegenerative disorders.
From a distance, the microvascular bed of the central nervous system (CNS) appears functionally adynamic and apparently inactive compared with neurons and their supportive cells (del Zoppo, 1994). The microvasculature (defined as vascular elements less than 100 μm diameter, which include the capillaries and their afferent–efferent connections) maintains and, with states of arousal, increases flow and cellular nutrition through local regulatory mechanisms (Hoge et al., 1999; Lovick et al., 1999; Gotoh et al., 2001). These local changes in flow appear more obvious in the cerebral cortex where the regional blood flow is greater than that of the striatum (Horton et al., 1980; Fenstermacher et al., 1991; Santizo et al., 2000). The effects also appear to be hierarchical. Local mechanical (Madden and Christman, 1999; Bryan et al., 2001), neuronal (Krimer et al., 1998; Lovick et al., 1999; Barbelivien et al., 1999; Norup and Lauritzen, 2001), and autocrine (Iadecola et al., 1993; Malonek et al., 1997; Medhora et al., 2001; Niwa et al., 2001; Horiuchi et al., 2002) mechanisms appear to control flow. These mechanisms involve cross-talk between the components of the vascular wall (e.g., endothelial cells and smooth muscle cells), and with cellular components of the blood (e.g., platelets and leukocytes) (Peters et al., 1991). Microvessels are targeted in cerebral disease states, including focal ischemia, hyperglycemia, amyloid deposition and amyloid angiopathy disorders, hypertensive angiopathy (as in lacunar syndromes), autoimmune vasculitides, and perhaps others that may contribute to Alzheimer's type dementia, vascular dementia, and inflammatory disorders. These observations support the notion that cerebral microvessels are the target of numerous insults.
Experimental preparations demonstrate that within the core regions the microvasculature displays multiple rapid responses to focal ischemia (del Zoppo et al., 1991; Mori et al., 1992; Tagaya et al., 1997; Wagner et al., 1997; Abumiya et al., 1999; Heo et al., 1999; Tagaya et al., 2001; Hosomi et al., 2001). Following focal ischemia, with recovery of flow in the occluded feeding arteries, the downstream microvascular bed bears the brunt (del Zoppo, 1994). Significant alterations in microvessel ultrastructure accompany changes in vascular cell receptor expression. Local breakdown in the permeability barrier allows transudation of fluid and solutes from the plasma compartment. Furthermore, the microvasculature provides the scaffold and entry point for inflammatory cells into the neuropil. Alterations in integrin–matrix relationships during focal ischemia are also consistent with early events in vascular remodeling and angiogenesis. These events imply possible vascular contributions to the plasticity and functional recovery of brain tissue after the ischemic insult. Moreover, an attractive notion derives from the rapid microvascular responses to focal ischemia, which suggests that cerebral microvessels and their neighboring neurons may participate together in a “neurovascular unit.” Hence, early views of the cerebral microvasculature as inert conduits for blood transport have been largely dispelled.
MICROVESSEL ULTRASTRUCTURE
Microvessel ultrastructure in the CNS is remarkably preserved across species. Pial and cortical penetrating arteries consist of an endothelial cell layer, the basal lamina derived from the extracellular matrix (ECM), myointimal layers of smooth muscle cells encased in ECM, and an adventitia derived from the leptomeninges (Peters et al., 1991). In the cortex an extension of the subarachnoid space forms the Virchow-Robins space, which surrounds cortical penetrating arterioles until it disappears into the glia limitans, the abluminal boundary formed by the astrocyte end-feet. With further arborization of the microvasculature, the glia limitans at the capillary level fuses with the thin basal lamina. The postcapillary venule network bears close ultrastructural resemblance to the capillaries (Fig. 1).
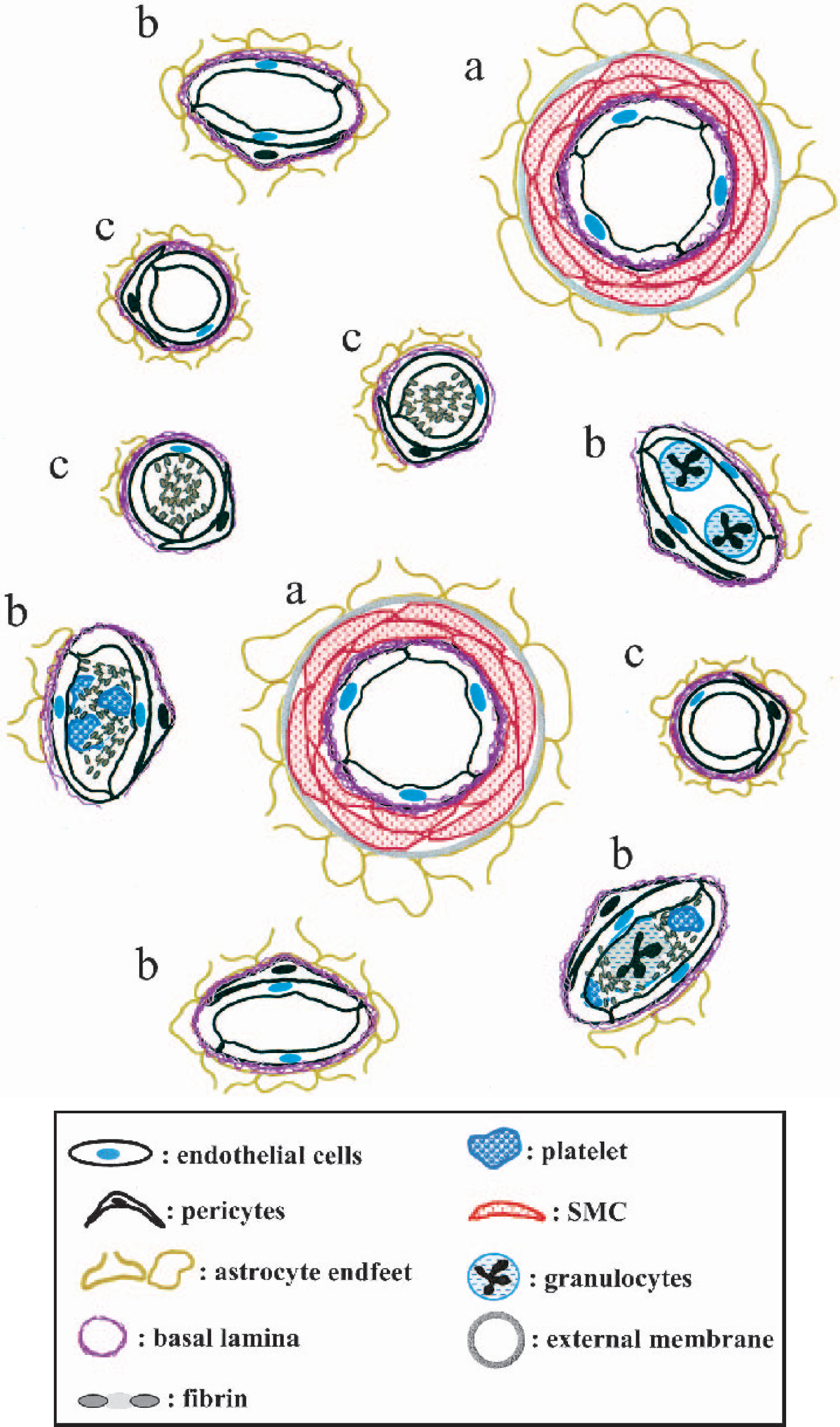
The focal “no-reflow” phenomenon following MCA occlusion and reperfusion in the CNS is in part dependent on occlusion of the different portions of the microvascular bed. Focal “no-reflow” can occur with only partial obstruction of the ischemic bed.
The permeability barrier characteristics of the microvasculature are represented by (1) interendothelial cell tight junctions (blood brain barrier) and (2) an intact subtending basal lamina. Both the permeability barrier and the basal lamina derive from the concerted interaction of the endothelial cells and astrocytes (Bernstein et al., 1985; Kusaka et al., 1985; Janzer and Raff, 1987; Tagami et al., 1992; Webersinke et al., 1992; Nagano et al., 1993). The proximity of the cellular components of intact cerebral capillaries implies ready communication between endothelial cells and astrocytes, while flow alterations with increased (neuronal) metabolic demand during arousal imply coupling of neuronal requirements to microvessel function (Peters et al., 1991). Focal ischemia can abruptly alter the stable relationships among endothelial cells, astrocytes, resident histiocytes or pericytes, and the intervening ECM and myointimal cells within the vascular wall.
Individual cerebral microvascular endothelial cells display regional functional specialization along the microvascular axis (Spatz et al., 1978, 1989). Glucose and amino acid transporter density vary with the microvessel diameter (Spatz et al., 1978, 1989; McCarron et al., 1991; Micic et al., 1993; Honkanen et al., 1995). Adhesion receptors for leukocyte trafficking are expressed predominantly by the postcapillary venule endothelium (del Zoppo et al., 1991). Whether similar specialization of astrocytes occurs is uncertain, but the presence or absence of microvessels displaying integrin α6β4 on astrocyte end-feet suggests at least a binary expression state (Wagner et al., 1997). Given regional and individual specialization of neurons, local subspecialization of the microvascular bed seems possible, although it is as yet unproven. In vitro coculture studies indicate that astrocytes can modulate endothelial cell function (e.g., expression of hemostatic elements) (Minakawa et al., 1991; Wang et al., 1997; Tran et al., 1998). Tissue plasminogen activator (t-PA) expression is limited to a select population of microvessels within normal brain tissue (Levin and del Zoppo, 1994). Those observations support the notion that microvessel function may be locally or regionally specialized and dependent on the interaction of its cellular components.
THE CEREBRAL MICROVASCULAR NETWORK
The architecture of the microvascular array of the brain reflects its specific location.
Microvessel architecture
With ascending phylogeny, branching of the leptomingeal anastomoses shows increasing complexity (Edvinsson et al., 1993). Penetrating arteries from the pial network dive orthogonal from the surface to serve columns of cortical gray matter. Bär (1978; 1983) described hexagonal arrays of vessels descending from pial arteries into the cortex, interconnected to vertical and horizontal microvessels, in the rat. At the gray matter–white matter junction, the density of microvessels significantly decreases. In contrast, the microvascular networks of the striatum, thalamus, and subthalamic structures are not well-described. In humans, these structures are served by the lenticulostriate arterial branches of the middle cerebral artery (MCA), the internal carotid artery, or the posterior cerebral artery, but the arterial supply varies with the species (Edvinsson et al., 1993). In the primate basal ganglia where the lenticulostriate arterial branches derive predominantly from the MCA, the three-dimensional capillary array branches at approximately 30 μm intervals (del Zoppo et al., 1986; del Zoppo et al., 1991; Hamann et al., 1995). Although the collateral vascular protection of the cortex, from leptomeningeal anastamoses across the brain surface are well-defined, the origins of a separate vascular protection of the striatum are unknown.
Local protection derives in part from retrograde flow behind obstructions within the low pressure-high flow microvessel bed (Zulch, 1985). However, despite local protection, proximal (M1) MCA occlusion (MCA:O) produces selective injury to the striatum (often sparing the thalamus). The relative homogeneity of the distribution and density of striatal microvessels in the nonhuman primate make it a suitable setting for examining microvessel responses. It should be mentioned that these vascular relationships, and therefore their responses to ischemia, may also be influenced by genetic factors (Edvinsson et al., 1993; Irikura et al., 1995; Niwa et al., 2000; Cavaglia et al., 2001; Bale et al., 2002; Demchenko et al., 2002).
Microvascular reserve in the central nervous system
During normal blood flow, not all capillaries are perfused (Keyeux et al., 1995). Hypercapnia and anesthesia elicit substantial increases in local blood flow, which may involve the recruitment of closed capillaries or vasodilatation of extraparenchymal arterioles (Weeks et al., 1990; Keyeux et al., 1995). Those observations suggest that a cerebrovascular reserve exists, which can be recruited on short notice, but does not involve neovascularization. However, direct measurements indicate that all capillaries of the pial and cortical circulation have plasma flow, and there is no capillary recruitment during forebrain ischemia (Seylaz et al., 1999; Pinard et al., 2000). It is uncertain whether in subcortical regions with lower baseline flows such restitution of flow is possible (Fenstermacher et al., 1991). Observations in humans and in nonhuman primates suggest that the low pressure-high flow character of the basal ganglia does not perfuse all microvascular channels and that microvascular obstruction is “managed” by reversal of flow through adjacent channels (Zulch, 1985). This may afford a type of protection. However, the presence of a nascent microvascular bed that can be recruited in the early moments of ischemia remains uncertain.
INTEGRIN-MATRIX INTERACTIONS AND MICROVESSEL INTEGRITY
Microvessel integrity in the CNS requires maintenance of the endothelial permeability barrier and the ECM-containing basal lamina. The primary barrier, formed by endothelial cell tight junctions and limited endothelial cell pinocytosis, may also involve adhesion of endothelial cells and astrocytes to the ECM (Risau et al., 1986; Risau and Wolburg, 1990; Risau et al., 1998). Exclusively in the brain, microvascular endothelial cells and astrocytes contribute to the primary permeability barrier and the vascular ECM, which protect the brain parenchyma from edema and hemorrhage (Risau and Wolburg, 1990). The secondary barrier involves integrin receptor cell-associated matrix adhesion (Hamann et al., 1995; Haring et al., 1996a; Wagner et al., 1997; Tagaya et al., 2001; Hamann et al., 2002).
The ECM-containing basal lamina separates the specialized endothelium from the astrocytes (the glia limitans) and their connections to neurons. Components of the vascular ECM include the laminins, collagen type IV, fibronectin, and other components including heparan sulfate proteoglycans, heparan sulfates, entactin, and nidogen. Generated by endothelial cells and astrocytes in concert during development, the basal lamina forms a biologically active connection between the two cell components, which requires their juxtaposition (Bernstein et al., 1985; Kusaka et al., 1985). In culture, astrocytes secrete laminin, fibronectin, and chondroitin sulfate proteoglycan, while collagens stimulate astrocyte-induced endothelial cell maturation (Ard and Faissner, 1991; Tagami et al., 1992; Webersinke et al., 1992). Conversely, endothelial cell-derived ECM components stimulate astrocyte growth and function (e.g., glutamine synthetase activity) (Nagano et al., 1993; Kozlova et al., 1993). These developmental interrelationships highlight the close functional association of endothelial cells and astrocytes.
The blood–brain barrier also relies on the interactions of endothelial cells and astrocytes, as demonstrated by elegant xenograft experiments (Hurwitz et al., 1993). Astrocytes promote many microvascular blood–brain barrier properties, including endothelial cell tight junctions, Evan's blue exclusion, and HT7/neurothelin generation (Risau et al., 1986; Schlosshauer and Herzog, 1990; Lobrinus et al., 1992). It has been postulated that soluble factors generated by astrocytes maintain the endothelial blood–brain barrier characteristics, including the tight-junctions, transendothelial resistance, and glucose/amino acid transport polarity (Estrada et al., 1990; Minakawa et al., 1991; Hurwitz et al., 1993).
Proteoglycans within the ECM have been detected around neurons and glial cells and are thought to support their viability (Carlson and Hockfield, 1996). Glycosaminoglycans, heparan sulfate proteoglycans, and chondroitin sulfates are found in the microvascular compartment of the adult brain (Margolis et al., 1975; Margolis and Margolis, 1993). Cat-301 proteoglycan is an ECM component that is expressed by subsets of neurons and that coats the perikaryon and the proximal portions of dendrites and axonal segments (Carlson and Hockfield, 1996). It is possible that processes that disrupt the vascular matrix may also affect the ECM, which separates cells within the neuropil. Little is known about the interplay of the vascular and nonvascular matrix compartments under normal conditions and during ischemia.
Adhesion of vascular cells to the subtending basal lamina requires the interaction of cellular integrin receptors to their matrix ligands. Although nonvascular cells of the CNS are known to express integrin receptors (Milner and Campbell, 2002), the appearance of integrins in vivo is most readily evident in the microvasculature (Hamann et al., 1995; Haring et al., 1996a). Haring et al. (1996a) demonstrated patterns of integrin subunit expression in normal CNS microvessels and their expression by specific microvessel subclasses. Integrin α1β1 on endothelial cells appears on all cerebral microvessels, including cerebral capillaries (Gehlsen et al., 1989; Kramer et al., 1990; Korhonen et al., 1990; Paulus et al., 1993; Haring et al., 1996a; Tagaya et al., 2001). Integrin α6β4 is expressed on astrocyte end-feet around select microvessels, whereas integrin α1β1 is found on their fibers (Wagner et al., 1997; Tagaya et al., 2001).
Integrins subserve cellular adhesion, sense environmental change, and signal both extra- and intracellular events (e.g., platelet adhesion and activation) (Shattil and Ginsberg, 1997; Hynes and Bader, 1997; Shattil, 1999). In the CNS, these properties appear to maintain endothelial cell-astrocyte proximity across the thin matrix barrier within capillaries. We have postulated that these relationships may be perturbed by loss of integrin–matrix attachments following MCA:O. However, so far, little is known about the signaling functions of the integrins expressed in cerebral microvessels. Based on the properties of specific integrins, it would be expected that microvascular endothelial cells and astrocytes could also sense the environment and respond to changing conditions (e.g., flow or changes in matrix composition).
EFFECTS OF MIDDLE CEREBRAL ARTERY OCCLUSION ON THE MICROVASCULATURE
Cerebral microvessels have been regarded as less sensitive to ischemia than the neurons they serve, in part because of the relative resistance of endothelial cells to hypoxia (Tagaya et al., 1997), but in fact microvascular responses to ischemia are very rapid and highly coordinated, although they vary topographically with the degree of ischemic injury. Within 1 to 2 hours following MCA:O, initial loss of the primary microvessel permeability barrier occurs, producing transudation of small molecules, fibrinogen, and plasma (Okada et al., 1994a). Loss of selective K+ channels is associated with the swelling of astrocyte end-feet (Fig. 2) (Petito and Babiak, 1982; Maxwell et al., 1989; Chan et al., 1990; Bender and Norenberg, 1994). On subpopulations of microvessels, endothelial cells express (1) leukocyte adhesion receptors, (2) elements of angiogenesis, including vascular endothelial growth factor (VEGF) and integrin αvβ3, and (3) nutrient transporters, including glucose transporter–1 (GLUT-1) (Kalaria et al., 1988; Harik et al., 1988; Pardridge et al., 1990; Honkanen et al., 1995; Abumiya et al., 1999).
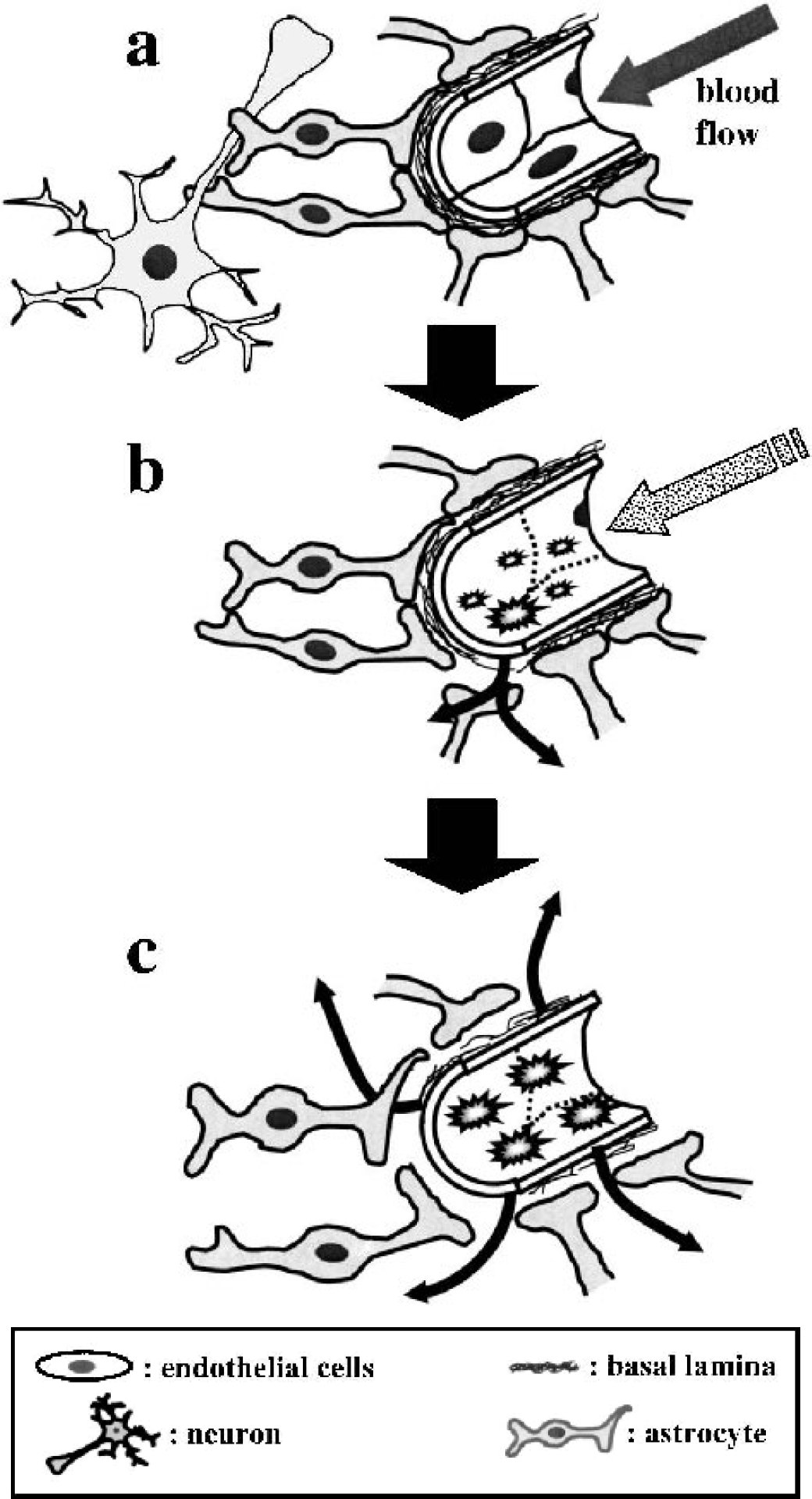
Effects of MCA:O on the microvessel ultrastructure.
In the same time frame, there is coordinate loss (or downregulation) of structural integrins on both endothelial cells and astrocytes. Detectable alterations in the major vascular matrix constituents laminin-1 and −5, collagen type IV, and fibronectin occur more gradually (Hamann et al., 1995; Wagner et al., 1997). The loss of integrin receptors coincides with the expression of proteases known to degrade matrix proteins and the secondary vascular barrier (Heo et al., 1999; Hosomi et al., 2001). The changes in microvessel-associated integrin–matrix interactions are confined to the regions of documented neuron injury (Wagner et al., 1997). Loss of specific integrin immunoreactivity within the ischemic regions appears to coincide with altered cellular function, loss of matrix ligand immunoreactivity, the appearance of signals for vascular remodeling, or a combination of these elements.
In both clinical and experimental settings, edema within the region of injury follows leakage of the vascular permeability barrier shortly after the onset of focal ischemia (Fig. 2) (Gotoh et al., 1985; Olesen, 1986; Dietrich et al., 1993; Okada et al., 1994a; Risau et al., 1998). There is evidence that loss of the blood–brain barrier and the microvascular ECM results from the actions of bradykinin (Kamiya et al., 1993; Aschner et al., 1997), VEGF (Abumiya et al., 1999; Zhang et al., 2000), thrombin (Okada et al., 1994a; Aschner et al., 1997), active matrix metalloproteinases (MMPs) (Heo et al., 1999), proteases released by activated leukocytes (Hasty et al., 1990; Garcia et al., 1994; Armao et al., 1997; Opdenakker et al., 2001), and other protease activities (Hosomi et al., 2001). Blockade of bradykinin receptors has been associated with reduced injury and edema formation (Relton et al., 1997). Thrombin can increase edema formation by direct action on the cerebral microvascular endothelium (Aschner et al., 1997; Lee et al., 1997; Kubo et al., 2000). VEGF disrupts the interendothelial cell tight-junctions, which involve the gap junction complex containing connexin 43 (Suarez and Ballmer-Hofer, 2001). Rosenberg et al. (1994; 2001; Rosenberg and Navratil, 1997) have described a temporal correlation between pro–MMP-9 “activity” and edema formation in rat strains following MCA:O. In contrast, in the nonhuman primate, early extravasation of plasma constituents coincides with pro–MMP-2, urokinase (u-PA), and plasminogen activator inhibitor-1 (PAI-1) expression, but not pro–MMP-9, by the cerebral microvasculature in the ischemic core (Heo et al., 1999; Hosomi et al., 2001). Whereas it has been hypothesized that members of the PA and MMP families can contribute to matrix degradation, proof of pro-MMP activation is not available, and the manner of protease compartmentalization is not yet defined. Furthermore, roles for proteases in endothelial cell permeability cannot be ruled out. It is now apparent that the rapidly increased protease expression coincides with loss of both the primary and secondary permeability barrier.
Endothelial cell leukocyte adhesion receptors
The microvasculature is a staging platform for secondary injury processes ischemia (Fig. 1 and Fig. 3). Endothelial cell leukocyte adhesion receptors respond to focal ischemia in a rapid and orderly way. Transmigration of polymorphonuclear (PMN) leukocytes from microvessels in the ischemic core occurs as early as 1 hour following MCA:O (Abumiya et al., 1999). The appearance of P-selectin, intercellular adhesion molecule–1 (ICAM-1), and E-selectin on microvessel endothelium, together with their counter-receptors (e.g., P–selectin glycoprotein–1 (PSGL-1) and the β2-integrin CD18) on PMN leukocytes accompany the initial movement of inflammatory cells into the ischemic region (Fig. 3) (Mori et al., 1992; Okada et al., 1994b; Haring et al., 1996b). P-selectin appears on the endothelium by 2 hours MCA:O, followed by ICAM-1 by 4 hours and E-selectin between 7 and 24 hours following MCA:O in the ischemic regions of the awake nonhuman primate (Okada et al., 1994b; Haring et al., 1996b). The persistence of endothelial cell P-selectin on 5.5% to 9.5% of microvessels up to 24 hours post-MCA:O could be explained by continued local thrombin generation or persistent local hypoxia (McEver, 1991; McEver et al., 1995). In contrast, ICAM-1 appeared maximally on 10.0 ± 6.8% of noncapillary microvessels (Okada et al., 1994b). E-selectin upregulation occurred in the same territory but also in the contralateral and ipsilateral nonischemic regions by 24 hours after MCA:O (Haring et al., 1996b). Both P- and E-selectin expression occurred only on microvessels with an intact basal lamina (Okada et al., 1994b; Haring et al., 1996b). Observations in rodent models of focal cerebral ischemia have confirmed the appearance of leukocyte adhesion receptors on microvessels, but the details vary (Zhang et al., 1994; Zhang et al., 1995; Zhang et al., 1998).
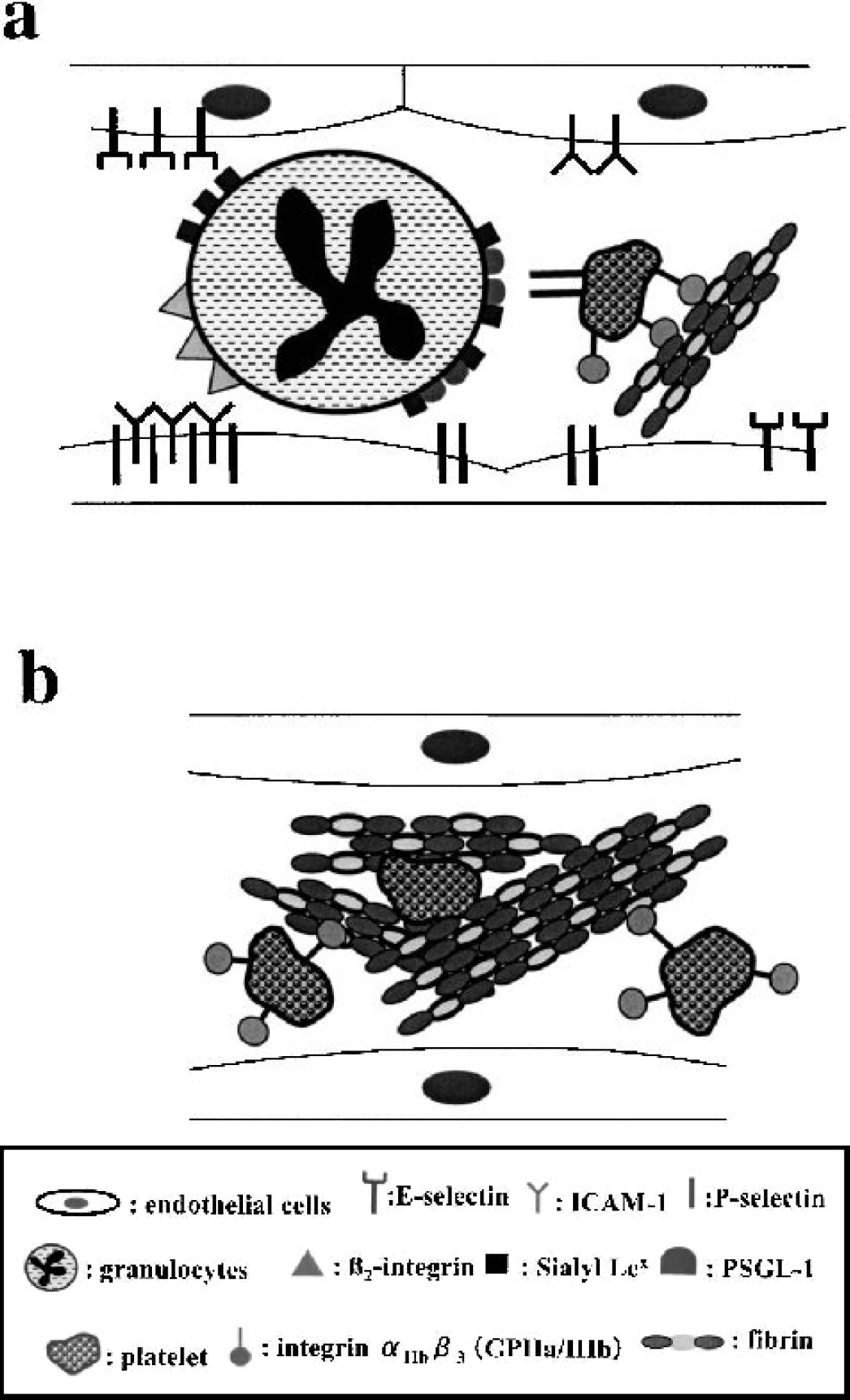
Microvascular responses to MCA:O that may contribute to focal ‘no reflow.’
The focal “no-reflow” phenomenon and vascular wall reactivity
Focal cerebral ischemia is associated with obstruction of the downstream microvascular bed after reperfusion of the occluded supply arteries (Fig. 1) (del Zoppo et al., 1991). This accords with regions of territorial vascular compromise following transient MCA:O, as demonstrated by carbon tracer techniques (Sundt, Jr. et al., 1969; Little et al., 1975; Little et al., 1976; del Zoppo et al., 1991). Although attributed to extrinsic compression from edema, endothelial cell swelling, and endothelial microvillus formation (del Zoppo, 1994), intravascular obstruction also occurs with the local activation of platelets, leukocytes, and coagulation (fibrin) (del Zoppo et al., 1991). Within 4 hours of MCA:O, PMN leukocytes adhere to postcapillary venules through the coordinate appearance of leukocyte adhesion receptors on activated endothelium (del Zoppo et al., 1991) and occlude capillaries in the territory, contributing to focal “no-reflow” (Mori et al., 1992). PMN leukocyte adhesion requires the interaction of leukocyte β2-integrins (CD18) with the endothelial cell receptor ICAM-1 in the CNS as elsewhere (Mori et al., 1992; Okada et al., 1994b). Activated platelets and fibrin(ogen) also contribute to microvascular obstructions in both capillary and noncapillary sectors of the ischemic bed (del Zoppo et al., 1991, 1992; Okada et al., 1994a, 1994b). Platelet-leukocyte interactions are mediated by platelet P-selectin/PMN leukocyte PSGL-1, whereas platelet–fibrin binding requires the integrin αIIbβ3 on activated platelets (Shattil, 1995; Shattil, 1999). The accumulation of activated platelets beginning within 2 hours MCA:O within the ischemic territory was confirmed by an increase in the platelet integrin αIIbβ3 antigen in a subpopulation of microvessels (Okada et al., 1994b). Interference with either interaction by specific interventions against the β2-integrin CD18 (Mori et al., 1992) or integrin αIIbβ3 (Abumiya et al., 2000) before reperfusion significantly increases residual microvessel patency (decreases focal “no-reflow”). Separate work with knockout preparations and specific inhibitors has demonstrated that inhibition of the platelet-fibrin interactions can reduce microvessel obstruction and decrease tissue injury (Walder et al., 1997; Coudhri et al., 1998). In short, the interface between the vascular compartment and the plasma column is significantly and swiftly altered by ischemia, which promotes microvascular obstruction by leukocyte (and platelet) activation and adhesion.
In the ischemic territory, fibrin is also deposited in an increasing proportion of microvessels with time following MCA:O (Thomas et al., 1993; Okada et al., 1994a). The accumulation of fibrin in the vascular lumen implies the intravascular generation of thrombin (Okada et al., 1994a). Tissue factor (TF), which is present preferentially in perivascular gray matter (del Zoppo et al., 1992), catalyzes fibrin formation by generating thrombin from plasma coagulation factors (Mackman et al., 1989; Ruf et al., 1990; Eddleston et al., 1993). TF resides mainly around noncapillary microvessels (del Zoppo et al., 1992). Okada et al. (1994a) demonstrated that blockade of the TF: Factor VIIa complex around microvessels significantly reduced fibrin deposition within the microvessel lumen. Furthermore, luminal fibrin implies locally increased permeability of the ischemic microvasculature soon after MCA:O, which allows filtration of quite large molecules such as fibrinogen in addition to albumin and IgG (Nordborg et al., 1991; Kitagawa et al., 1992; Okada et al., 1994a; Mabuchi et al., 2000). Finally, blockade of TF: Factor VIIa-mediated thrombin generation modestly increases microvessel patency, indicating that fibrin deposition can contribute to focal “no-reflow” (Thomas et al., 1993; Okada et al., 1994a). These findings confirm the relevance of the microvascular wall to the activation of coagulation and cellular inflammation during focal cerebral ischemia. Recently, similar alterations in microvascular patency in the rodent cortex following ischemia have been reported (Wang CX et al., 2001).
Alterations in microvessel-related integrin expression
Endothelial cell and astrocyte integrin α1β1 and astrocyte integrin α6β4 immunoreactivity rapidly decrease in response to MCA:O in the regions of neuron injury. Within the ischemic core, microvessel-related integrin α1β1 expression decreased to 20% of baseline, also implying continued endothelial cell synthesis of integrin α1β1 antigen on select microvessels (Tagaya et al., 2001). The rapid fall and plateau in integrin expression could reflect injury to both endothelial cells and astrocytes initiated by ischemia, alterations of the matrix by processes involving microvascular endothelium or astrocytes with secondary changes in integrin expression, or both. Downregulation of β1 integrin subunit expression in microvessels is in part caused by shut-down of its transcription (Tagaya et al., 2001). In the basal ganglia, these changes are reflected topographically as multiple subregions of increased microvessel-associated β1 integrin mRNA surrounding islands devoid of vascular β1 mRNA within the ischemic core (Tagaya et al., 2001). Increased microvessel-associated integrin β1 transcription also occurred in the boundary between the ischemic core and peripheral regions and was confluent with the subregional boundaries. The β1 subunit transcripts appeared prominently over noncapillary microvessels. It is important to note that the dynamic nature of microvessel β1 integrin responses confirms the presence of viable microvascular endothelial cells within the ischemic core region. These changes suggest that despite widespread loss of integrin immunoreactivity, injury was not yet complete by 2 hours, in contrast to the commonly held view that striatal injury following MCA:O is rapidly complete and irreversible.
Subunit β1 integrins also play an important role in maintaining cell viability. Disruption of interactions between β1 integrins and their matrix ligands can induce apoptosis in specific target cells (Hoyt et al., 1996; Strater et al., 1996; Forsythe et al., 1996). Although apoptotic changes do not appear in the ischemic microvasculature, co-localization experiments suggest that endothelial cell function is compromised in select microvessels soon after MCA:O. By 2 hours MCAO, a reduction in both CD31 antigen and subunit α1β1 expression on endothelial cells was detectable in the ischemic core (Tagaya et al., 2001). These changes were not associated with obvious endothelial cell detachment (del Zoppo et al., 1986; Tagaya et al., 2001). However, preservation of endothelial cell integrin α1β1 expression was associated with the presence of laminin-1 within the microvascular basal lamina. Despite the early loss of integrin α1 β1 immunoreactivity, evidence of endothelial cell DNA scission is sparse up to 24 hours MCA:O, supporting the relative resistance of the endothelium to ischemia (Tagaya et al., 1997).
Moreover, in the regions of evolving tissue destruction the topographic distribution of altered microvessel integrin expression is not homogeneous (Wagner et al., 1997; Abumiya et al., 1999; Tagaya et al., 2001). The early changes in microvessel β1 integrin and integrin α6β4 expression appear heterogeneously interspersed among microvessels displaying apparently normal expression of these integrins (Wagner et al., 1997; Tagaya et al., 2001). Those and separate observations (vide infra) of microvessel integrin αvβ3 expression indicate that the early tissue injury within the ischemic core, where neuron injury is observed, is not homogeneous. This suggests that microvessels with apparently normal features persist long after ischemia has been initiated, even in the most vulnerable territory.
The mechanisms responsible for the reduction in CNS microvessel integrin expression and the dissolution of the microvascular matrix are not worked out. Dynamic changes in endothelial cell–matrix interactions could result from downregulation of integrin receptors of the β1 subfamily by TNF-α and IL-1β (Defilippi et al., 1992). Exposure of endothelial cells to TNF-α for 24 hours resulted in a 60% loss of integrin α1β1 expression from baseline and significantly decreased endothelial cell adhesion to laminin in one study (Defilippi et al., 1992). In addition, IL-1β can participate in early postischemic cerebral edema formation perhaps via alterations in β1 integrin expression (Yamasaki et al., 1992). Functional blockade of the integrin β1 subunit in rat epithelium causes edema formation by inhibiting cell–matrix adhesion and decreasing interstitial fluid pressure (Reed et al., 1992; Rodt et al., 1994). Hence, β1 integrins, through their participation in cell–matrix adherence may play a role in preventing transudation and edema formation in conjunction with other contributors to the intact blood–brain barrier and may be disturbed during ischemia by action of cytokines locally generated by microglia and astrocytes (Liu et al., 1993; Buttini et al., 1996; Loddick and Rothwell, 1996). Similar mechanisms may contribute to the changes in microvessel ECM, which occur in the same territory.
Fate of the vascular extracellular matrix during focal ischemia
The intact basal lamina represents the second functional barrier to cellular exit from the vascular compartment. Significant but gradual local decrease in the microvascular expression or integrity of the major ECM components laminin-1 and −5, collagen IV, and fibronectin (of cellular origin) are initiated by MCA:O (Hamann et al., 1995, 1996). Inasmuch as local degradation of its components may be associated with leakage of blood cells or their active transmigration, Hamann et al. (1996) found a significant association between regional loss of microvascular matrix with the extravascular accumulation of hemoglobin (hemorrhagic transformation) within the regions of ischemic injury. The loss of microvessel ECM is associated also with the loss of endothelial cell reactivity during focal ischemia. Endothelial cell P-selectin and E-selectin, and β1-integrin expression within the ischemic territory, occur only on those microvessels with an intact (laminin and collagen-containing) basal lamina (Haring et al., 1996b; Tagaya et al., 2001).
Microvessel-associated protease generation
Alterations in the expression of microvessel ECM can be explained by local proteolysis, initiated by ischemia. Recent attention has focused on the expression and activity of MMPs and plasminogen activators following experimental MCA:O (Rosenberg et al., 1996; Ahn et al., 1999; Heo et al., 1999; Hosomi et al., 2001; Rosenberg, 2002). Loss of the basal lamina matrix in the primate ischemic striatum follows rapid increase in the regional expression of pro–MMP-2, u-PA, and the inhibitor PAI-1 (Rosenberg et al., 1996; Heo et al., 1999; Hosomi et al., 2001; Rosenberg, 2002). Pro–MMP-2 is generated rapidly by the microvasculature and by nonvascular cells during ischemia (Norton et al., 1978; Saksela and Rifkin, 1988; McGuire and Seeds, 1989; Mackay et al., 1990; Vassalli et al., 1991; Heo et al., 1999; Hosomi et al., 2001; Rosenberg et al., 2001). There is some discrepancy regarding the timing of the increased expression of pro–MMP-2 and pro–MMP-9 following MCA:O (Heo et al., 1999). Recent reports now support the very early expression of pro–MMP-2 seen in primates (Rosenberg et al., 2001). In primates, increased pro–MMP-9 expression within the ischemic tissue is associated with hemorrhagic transformation (Heo et al., 1999). Rosenberg and colleagues (1996; 2001) have recently shown that MMP-2 antigen rests in select astrocytes around microvessels in the ischemic rat brain. Lo and investigators have confirmed that pro-MMP-9, but not pro-MMP-2, expression is associated with focal ischemia in the rodent (Asahi et al., 2000, 2001a,2001b), and that deletion of the MMP-9 gene is associated with a decrease in microvascular permeability, but not in laminin immunoreactivity (Asahi et al., 2001b).
Pro–MMP-2 and pro–MMP-9 are not active in tissue and cannot degrade ECM. Furthermore, experimental work, with one exception, has failed to show substantial active MMP-2 (Gasche et al., 1999). However, focal ischemia stimulates the appearance of both direct and indirect activation systems for pro–MMP-2 in concert in the injured tissue. Membrane type (MT)1- and MT3-MMP, direct activators of pro–MMP-2, are upregulated in the ischemic core region in concert with pro–MMP-2 (D-I Chang and G. J. del Zoppo, unpublished observations, 2003). Collagen, laminin, fibronectin, elastin, and myelin basic protein can be degraded by plasmin, which requires activation of plasminogen. Recently, plasmin has been shown to activate pro–MMP-2 in an MT1-MMP–dependent process (Mazzieri et al., 1997; Monea et al., 2002). Pfefferkorn et al. (2000) described increased PA activity following MCA:O within the caudate putamen of the Wistar rat, although the activator was not defined. Rosenberg et al. (1996; 1998) demonstrated increased u-PA–like, but decreased t-PA–like, activities by 12 to 24 hours after permanent MCA:O in the Wistar-Kyoto and spontaneously hypertensive rats. Ahn et al. (1999) also noted increased u-PA activity without increased t-PA activity in C57-BL/6J mice. The primary t-PA inhibitor in plasma, PAI-1, increased 4 hours following MCA:O in Wistar rats but without a clear relation to u-PA and t-PA activity (Zhang et al., 1999). In nonhuman primates, the very rapid (1 hour) increase in u-PA and PAI-1 and the early regional increase in t-PA·PAI-1 complex with transient decrease in free t-PA are consistent findings matching the rapid changes in other gene products in the microvasculature (van Hinsbergh et al., 1990; Abumiya et al., 1999; Docagne et al., 1999; Hosomi et al., 2001). U-PA secretion has been attributed to a number of CNS cell types, including stimulated endothelial cells, astrocytes, neurons, and microglia in vivo or in vitro (Schleef et al., 1988; van Hinsbergh et al., 1990; Nakajima et al., 1992; Tranque et al., 1994; Masos and Miskin, 1996; Vivien and Buisson, 2000), and PAI-1 can be generated by endothelial cells, astrocytes, and neurons (Patterson, 1985; Schleef et al., 1988; van Hinsbergh et al., 1990; Nakajima et al., 1992; Tranque et al., 1994; Masos and Miskin, 1996; Vincent et al., 1998). The relative absence of t-PA antigen from the ischemic core may seem curious, despite its localization on select noncapillary microvessels in nonischemic brain (Levin and del Zoppo, 1994). However, the differential upregulation of u-PA and PAI-1 in the ischemic parenchyma may be explained by stimulation by TNF-α, TGF-β, or 1L-1β, which do not promote endothelial cell t-PA synthesis or secretion (Schleef et al., 1988; van Hinsbergh et al., 1990; Docagne et al., 1999). Therefore, microvascular endothelial cells and select nonvascular cells may be the sources of the acutely increased u-PA and PAI-1, but not t-PA, following MCA:O (Hosomi et al., 2001). PAI-1 increased early following ischemia in both plasma and tissue, consistent with an endothelial cell source (Hosomi et al., 2001). It has been detected on cerebral microvessels following MCA:O in the Wistar rat (Zhang et al., 1999). Together these observations suggest that proteases that could contribute to vascular and parenchymal matrix degradation are generated shortly after MCA:O in the striatum and that microvascular cells are a likely local source of pro–MMP-2, u-PA, and PAI-1. The appearance of the u-PA activity in the ischemic core (relative to PAI-1) does not clearly allow the conclusion that pro–MMP-2 is activated but suggests that u-PA may be an indirect activator. At this point the evidence is circumstantial. The acute appearance of pro–MMP-2, u-PA, and PAI-1 together with the alterations in vascular ECM coincide with increased microvascular permeability and hemorrhage. They also appear in concert with elements known to be associated with vascular remodeling.
ANGIOGENESIS
The formation of new microvessels from existing vessels (angiogenesis) is a hallmark tissue response to injury (Dvorak et al., 1995; Carmeliet, 2000; Papetti and Herman, 2002; Hynes, 2002). The combination of microvessel matrix changes, protease generation, vascular integrin signals common to angiogenesis, and altered downstream cell function are consistent with vascular remodeling and new vessel formation (neovascularization). Although the existence of a cerebral microvascular reserve has been proposed, plasma flow through extant microvessels following MCA:O indicates that new conduits do not appear acutely (Keyeux et al., 1995; Seylaz et al., 1999; Pinard et al., 2000). Early studies by Tsutsumi et al. (1986; 1986) demonstrated new capillary bud formation within the ischemic bed by 7 days post-MCA:O in a canine model.
One hypothesis states that angiogenesis results from local hypoxia, leading to the sustained expression of hypoxia-inducible factor (HIF)-1, which stimulates VEGF and integrin αvβ3 expression (Namiki et al., 1995). VEGF promotes microvessel proliferation, which requires the expression of the two endothelial cell receptors flt-1 and flk/KDR (Nicosia et al., 1997; Chan et al., 1998; Straume and Akslen, 2001). Their interaction results in endothelial cell integrin αvβ3 expression. Integrin αvβ3 is necessary for angiogenesis, neovascularization in organ development, and tissue remodeling (Brooks et al., 1994; Varner et al., 1995). Fibrin(ogen), osteopontin, von Willebrand factor, vitronectin, fibronectin, and other ligands (e.g., MMP-2 by another active site) interact with this integrin receptor (Savill et al., 1990; Varner et al., 1995). In arterial segments, smooth muscle cells generate VEGF in response to hypoxia, mechanical forces, and growth factors (e.g., basic FGF) (Brogi et al., 1994; Stavri et al., 1995; Couffinhal et al., 1997; Bausero et al., 2000; Quinn et al., 2002). Endothelial cell migration and proliferation are stimulated by VEGF via the receptor flk-1 (Nicosia et al., 1997), but only smooth muscle cell migration responds to VEGF (Grosskreutz et al., 1999; Wang Z et al., 2001). Following MCA:O, all of these elements are set in motion.
Activation of cerebral microvessel cells, indicated by PCNA expression, occurs within 1 to 2 hours of MCA:O in precapillary arterioles within the ischemic territory of the primate basal ganglia (Table 1) (Abumiya et al., 1999). Okada et al. (1996) first demonstrated the differential upregulation of angiogenesis-related elements, integrin αvβ3 but not integrin αvβ5, on microvessels within the ischemic territory immediately following MCA:O. The microvascular expression of integrin αvβ3 was significantly and continuously associated with the intravascular deposition of fibrin following MCA:O (Okada et al., 1996). Abumiya et al. (1999) demonstrated highly significant coexpression of VEGF and integrin αvβ3 in microvessels displaying PCNA immediately following MCA:O. In that study, the statistical model was highly parsimonious. Moreover, the contribution of time to the coexpression was not essential, consistent with the heterogeneous distribution of vascular coexpression of the three elements during the evolution of the ischemic lesion (Abumiya et al., 1999). In addition to VEGF and integrin αvβ3 expression, αv mRNA appeared on endothelial cells and smooth muscle cells of 30 to 50 μm diameter microvessels (consistent with precapillary arterioles).
Expression of angiogenesis-related factors in microvessels after MCA:O
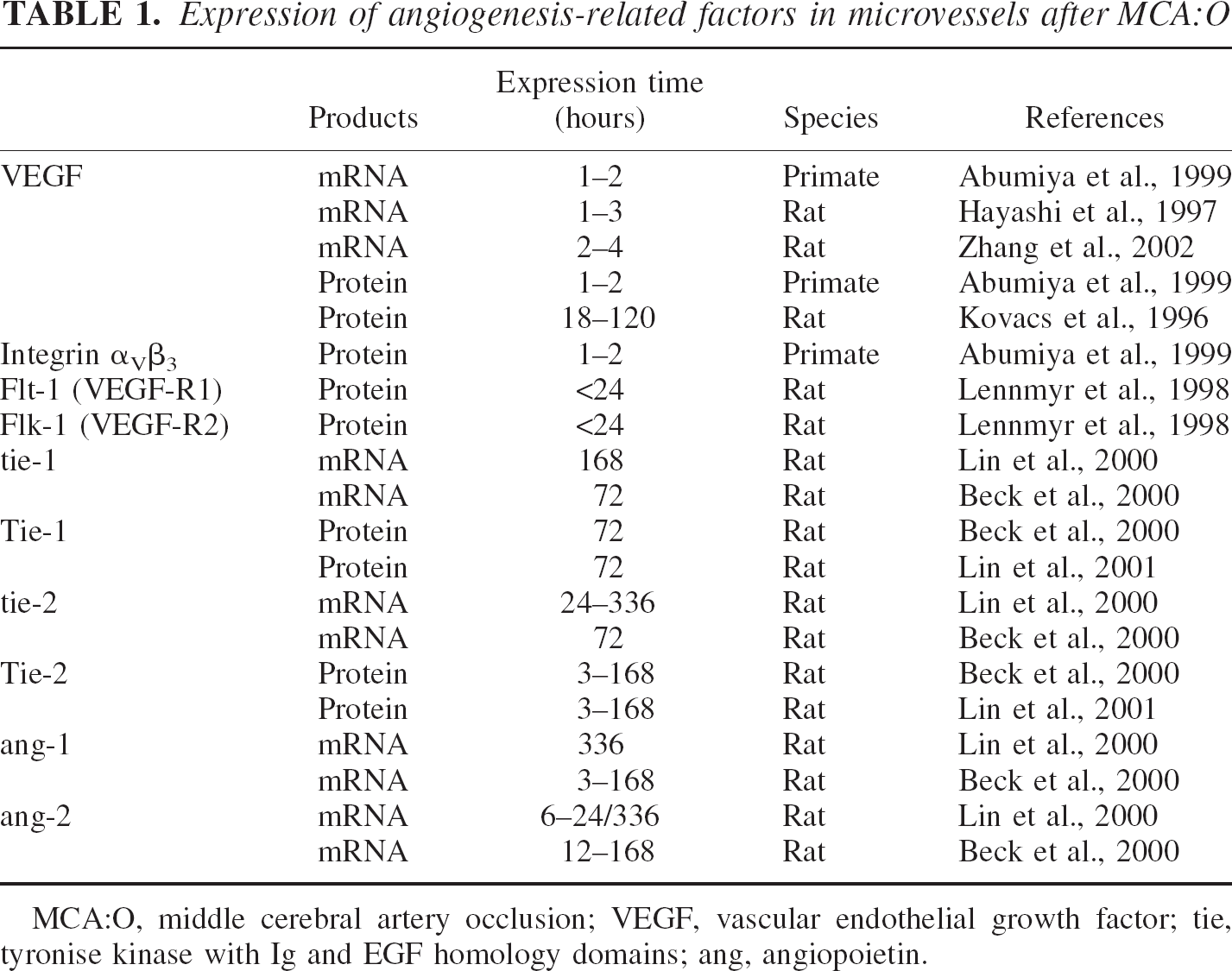
MCA:O, middle cerebral artery occlusion; VEGF, vascular endothelial growth factor; tie, tyronise kinase with Ig and EGF homology domains; ang, angiopoietin.
The group of Risau has demonstrated that hypoxia-induced VEGF expression precedes neovascularization in the border zone of the ischemic neocortex in the rodent (Marti et al., 2000). Focal cerebral ischemia also alters endothelial cell flt-1 and flk/KDR expression. Following transient or permanent MCA:O in the rat, VEGF expression increased in both microvessels and neurons by 24 hours, most prominently in the peri-infarct regions. Under those conditions, flt-1 increased in endothelial cells, neurons, and glial cells, while flk/KDR was most prominent in glial cells with modest expression in endothelial cells (Lennmyr et al., 1998). In separate studies, focal ischemia was associated with an increase in VEGF transcripts and VEGF receptors at the lesion boundary coincident with edema, which was ascribed to leakage of the microvascular permeability barrier (Zhang et al., 2002). Those findings indicate that the specific receptors for VEGF are expressed in concert with the ligand VEGF. A similar relationship of microvessel VEGF and integrin αv to HIF-1α transcription has also been noted (T. Abumiya and G. J. del Zoppo, unpublished observations, 2000). Other nonvascular sources of VEGF transcripts include leukocytes and microglia (Abumiya et al., 1999). Kovacs et al. (1996) and Hayashi et al. (1997) described the appearance of VEGF on endothelial cells, neurons, and glial cells, later following MCA:O in rodent models.
The general observation is that immediately following MCA:O, the cerebral microvasculature provides a scaffold for receptors and their ligands known to promote angiogenesis in other biologic systems. Hypoxia differentially alters tie-2 (tyrosine kinase with Ig and EGF homology domains), angiopoietin (ang)-1, and ang-2 levels, in an organ-specific manner (Abdulmalek et al., 2001). In the rodent, 6 hours after MCA:O, ang-2 transcription increased in endothelial cell-vascular elements within the ischemic core and periphery, which coincided with VEGF and endothelial cell proliferation (Beck et al., 2000). Hsu and colleagues observed an increase in ang-1 transcripts up to 14 days following focal ischemia in the rat, compared with the biphasic appearance of ang-2 transcripts at 24 hours and 14 days, and tie-2 mRNA 1-14 days after reperfusion (Lin et al., 2000). Tie-1 and tie-2 were expressed in the ischemic cortex, with tie-2 appearing on capillarylike structures in the outer cortical layers, and tie-1 detected in layers II to IV (Lin et al., 2001). Based on the appearance of these signals, one view is that angiogenesis/neovascularization is initiated by hypoxic stimulation of early expression of the elements VEGF and integrin αvβ3. However, proof of the entire process and how it affects microvessel integrity, neovascularization, and outcome following focal cerebrial ischemia is still lacking.
SUMMARY
Cerebral microvessels serve not only as conduits of blood but respond dynamically to focal ischemia, initiating mechanisms important for cellular inflammation, secondary injury events, and potentially for vascular and tissue remodeling. The microvasculature is both a target and stage for the interrelated processes of ischemia, thrombosis, and inflammation (del Zoppo, 1994). All three processes stimulate protease secretion and activity. Consequences of their action appear to be (1) loss of the primary blood–brain barrier, (2) alterations and degradation of the microvascular matrix (basal lamina), the second barrier, (3) microvascular hemorrhage, (4) edema formation, and (5) secondary alterations in the neuropil. Immediately after thrombotic occlusion of the arterial supply, integrins important for matrix adhesion of endothelial cells and astrocyte end-feet are lost, leukocyte adhesion receptors on endothelial cells of select vessels appear, leukocytes adhere and emigrate, and fibrin deposits and activated platelets accumulate within select microvessels. Activation of precapillary arterioles is accompanied by the expression of VEGF and integrin αvβ3, which are involved with angiogenesis. In the early moments following MCA:O (in the primate), these microvessel-dependent events occur together in a heterogeneous fashion adjacent to apparently normal microvessels throughout the ischemic core and peripheral regions.
FURTHER CONSIDERATIONS
These and many other observations leave open many interesting questions. For instance, still unanswered is how microvessels relate to their neighboring neurons and vice versa. How the microvascular matrix is related to the intercellular matrix is unclear. However, the emerging importance of integrin–matrix interactions to microvessel and tissue integrity and the dependence of cell viability on these interactions in other systems implies their role in cerebral function and the postischemic injury processes. The known associations of integrins α1β1 and α6β4 with cerebral microvessels suggest the hypotheses that (1) microvascular responses are rapid and temporally linked to neuron injury, and (2) the altered regulation of microvascular integrin expression occurs early in patterns suggesting multiple cores but not a single circumferential “penumbra.”
The acute generation of proteases is of particular interest. The activation of pro–MMP-2 and pro–MMP-9 and their compartmentalization in response to focal ischemia have not been reported. Without this information, the role of pro-MMPs in matrix degradation is uncertain. The reports of u-PA generation imply that other protease families may also be involved in matrix remodeling and brain tissue injury. Certainly the contributions of PMN leukocytes to secondary injury support this notion. The precise cell sources of the MMPs, plasminogen activators, and other proteases are under study. Strategies that could limit hemorrhagic complications of PA treatments and limit neuron injury rely on this information.
Increased integrin αvβ3, VEGF, and angiogenic receptors on select precapillary arterioles during focal cerebral ischemia have not yet been shown to be purposeful. It is uncertain whether their appearance is a stereotypic cellular response to hypoxia or early inflammation. However, the expression of select proteases, together with matrix degradation, increases the possibility that vascular and tissue remodeling are already an early response of the microvasculature to ischemia. The importance of these questions is the light they are likely to shed on the unique vascular biology of the brain, the relationships of neuron to microvascular responses, and the possibilities for new insights into other neurodegenerative disorders of the CNS. These insights are also likely to refine the evolving concept of the “neurovascular unit.”
Footnotes
Acknowledgments
The authors are grateful for the expert assistance of M. Piellucci in the preparation of this manuscript.