
Abstract
During focal cerebral ischemia, the degradation of microvessel basal lamina matrix occurs acutely and is associated with edema formation and microhemorrhage. These events have been attributed to matrix metalloproteinases (MMPs). However, both known protease generation and ligand specificities suggest other participants. Using cerebral tissues from a non-human primate focal ischemia model and primary murine brain endothelial cells, astrocytes, and microglia in culture, the effects of active cathepsin L have been defined. Within 2 hours of ischemia onset cathepsin L, but not cathepsin B, activity appears in the ischemic core, around microvessels, within regions of neuron injury and cathepsin L expression. In in vitro studies, cathepsin L activity is generated during experimental ischemia in microglia, but not astrocytes or endothelial cells. In the acidic ischemic core, cathepsin L release is significantly increased with time. A novel ex vivo assay showed that cathepsin L released from microglia during ischemia degrades microvessel matrix, and interacts with MMP activity. Hence, the loss of microvessel matrix during ischemia is explained by microglial cathepsin L release in the acidic core during injury evolution. The roles of cathepsin L and its interactions with specific MMP activities during ischemia are relevant to strategies to reduce microvessel injury and hemorrhage.
INTRODUCTION
The integrities of cerebral microvessels and the extravascular tissue are rapidly disturbed during ischemic stroke. Extracellular matrix (ECM) proteins of the microvessel basal lamina undergo acute changes within the core of the ischemic lesion.1,2 These events are associated with the appearance of edema and microhemorrhages, which have been attributed to either matrix metalloproteinase (MMP)-2 or MMP-9.1,3 It is still unclear whether the loss of microvessel ECM is due to these or other matrix proteases, and what their precise sources are. Fukuda et al 2 showed that cysteine protease activity coincides with MMP activity and the rapid unrecoverable loss of the microvessel perlecan during focal ischemia.
That observation implies the proteolytic cleavage of matrix proteins. Perlecan core protein, in purified systems, is a substrate for plasmin, stromelysin (MMP-3), and collagenase (MMP-1). 4 Cailhier et al 5 showed that cathepsin L cleaves the biologically active 80 kDa domain V fragment from perlecan, confirming the relevance of cathepsin L to perlecan cleavage in vivo, 2 and the potential biologic importance of the proteolytic fragment. 5 However, the rapid appearance of cathepsin L also coincides with the loss of other microvessel ECM components in non-human primate, and the appearance of (pro-)MMP-2.1,3 This suggests that very early during focal ischemia one or more matrix proteases may degrade perlecan, as well as the major structural components of the basal lamina ECM. In addition, cathepsins L and B are known to degrade the substrates and major ECM components collagen IV, laminin, and fibronectin (FN) in vitro.6–8
Kohda et al 9 and Seyfried et al 10 noted the appearance of cysteine proteases in neurons in the rodent after focal ischemia. In view of the close association of neuron and microvessel function, 11 and the coincident expression of cathepsin L on microvessels and neurons of the neurovascular unit in the early moments after middle cerebral artery occlusion (MCAO), 2 the exact non-neuronal derivation of cysteine protease activity is of interest. Cathepsin B has been implicated in ischemic injury of the rat hippocampus. 12
Cathepsin L is a liposomal/endosomal protease that is converted from its inactive pre-pro-enzyme to the active precursor pro-cathepsin L within the cell, then through cleavage of the 96 residue pro-region (between the N-terminus and signal sequence) in an autocatalytic manner to the active form.13,14 Conversion of cathepsin B into its active form requires removal of its 62 residue pro-region, six residue COOH terminus, and residues 47 and 49 to generate the two chain form. Both single chain and two chain forms are active. Although evidence for rescue of ischemic tissue during focal cerebral ischemia by a cysteine protease inhibitor is sparse, 10 the relation of cerebral tissue injury to neuron injury and to cysteine protease release suggests that the protease activity is not confined to neurons.
These observations raise concerns that cysteine protease activity generated within the ischemic regions of the non-human primate acutely may derive from other sources than neurons alone. Furthermore, the appearance of other families of ECM proteases may necessitate strategies that go beyond the inhibition of gelatinase activity alone if tissue and microvessel degradation during focal ischemia is to be abrogated. Understanding the specific sources of these matrix proteases during ischemia and their potential interactions will be essential for designing such strategies.
Recently, we showed the differential expression and secretion of (pro-)MMP-2 and (pro-)MMP-9 by astrocytes and microglia respectively in response to experimental ischemia. 15 (pro-)MMP-9 release is stimulated by the exposure of resting microglia, but not astrocytes, to the circulating plasma matrix proteins FN and vitronectin (VN) that extravasate with edema. 15 This suggests a precise non-neuronal cellular origin of select matrix proteases during focal cerebral ischemia. Often forgotten in tissue disruption is the role of the pH changes caused by focal cerebral ischemia. Development of the ischemic core involves significant reduction in pH, and in the non-human primate this may reach pH6.2 (AJ Strong, personal observations16,17).
The hypotheses tested here state that cysteine protease activity (1) is generated acutely (within 2 hours) after MCAOs, (2) directly degrades both regulatory (perlecan) and structural (collagen IV) ECM proteins within microvessel basal lamina, and (3) derives from specific non-neuronal sources that affect neurovascular unit integrity. These studies address the concern that microvessel ECM degradation depends on acute gelatinase appearance alone, and refines the specificity of acute matrix protease release observed during focal ischemia.
MATERIALS AND METHODS
All experimental procedures used here were approved by the institutional Animal Care and Use Committees of The Scripps Research Institute (TSRI) and of the University of Washington. They were performed according to the standards published by the National Research Council, and the U.S. Department of Agriculture Animal Welfare Act.
For the preparation of this manuscript, the investigators have adhered to the ARRIVE guidelines 2010.
Model of Focal Cerebral Ischemia
Archived samples of cerebral tissues from 10 adolescent male baboons (Papio anubis/cynocephalus) and from adult mice (C57 Bl/6) were used. The surgical and experimental approaches to middle cerebral artery (MCA) occlusion in the focal ischemia model have been previously described in detail. 3 Here, balloon compression of the M1 segment of the MCA was achieved in the awake animal using an implanted device (via the transorbital approach) 7 days remote from the implantation procedure.3,18 Reperfusion occurred at 3 hours after MCAO. Cerebral tissues were removed after transcardiac perfusion with isosmotic heparinized non-fixative perfusate, then frozen and archived at −80°C. 3
Antibodies and Peptides
The monoclonal (MoAb) and polyclonal antibodies used and their specific uses are detailed in Supplementary Table 1 of the Supplement. For determining cellular and secreted cathepsin L and B activities, the specific cathepsin B substrate Z-arg-arg-MNA and the cathepsin L substrate Z-phe-arg-MNA were used.
dUTP Incorporation
Cells in tissue cryosections with evidence of injury (DNA damage/repair) were detected by in situ incorporation of digoxigenin-dUTP with DNA polymerase I (Promega, Madison, WI, USA). 19 Controls for polymerase-based procedures included exposure of parallel sections to DNase I (Promega) (positive control) and deletion of the respective enzyme in each experiment (negative control). dUTP incorporation defined the ischemic core. 19
Culture Substrates
The purified matrix proteins laminin, collagen IV, perlecan, FN, and vitronectin (VN) (all Sigma, St Louis, MO, USA) were used as growth substrates for endothelial cells, astrocytes, and microglial cells where appropriate. Six-well plates (Nunc, Roskilde, Denmark) were prepared by coating the wells with a solution containing 10 μg/mL of each ECM protein (except perlecan (1 μg/mL) or poly-D-lysine (PDL, 5 μg/mL)) for 2 hours at 37°C, followed by washing each well with sterile phosphate-buffered saline (PBS).
Cell Cultures
Murine primary cerebral astrocytes, microglia, and endothelial cells were prepared according to methods in this laboratory.15,20
Endothelium. Primary cultures of murine brain endothelial cells were prepared from the brains of 2–3-month-old male C57Bl/6 mice (Jackson Laboratories, Sacramento, CA, USA). These were removed, cleaned of meninges and external blood vessels, then finely chopped and dissociated for 1 hour in a solution containing 20 U/mL papain and 250 U/mL DNAse I type IV (Worthington, Lakewood, NJ, USA) in 2 mL MEM-HEPES. 20 The homogenate was added to a 15 mL polystyrene conical tube containing 22% bovine serum albumin, and centrifuged at 1,000 g for 20 minutes to separate the myelin from vascular structures and other cells. The collected vascular tubes and cells were cultured in endothelial cell growth media at 37°C under 5% CO2. After 24 hours, the media was changed and puromycin (3 μg/mL; Sigma) was added for the next 3 days. Endothelial cell growth media was replaced every 3 days until cell confluence. The endothelial cells were used in P1 and P2 passage. The purity (>99%) of each cell preparation was verified by immunostaining for CD31 (PECAM-1) and von Willebrand factor antigens.
Astrocytes and microglia. Murine astrocytes and microglial cells were obtained from 1–3-day-old post-natal C57Bl/6 mouse pups (Charles River Laboratories, Hollister, CA, USA) by the procedures previously described.15,20 Cortices were cleaned of meninges and external blood vessels, chopped finely, then dissociated in a solution containing 30 U/mL papain (Worthington) and 40 μg/mL DNase I (Sigma) in 1 mL MEM-HEPES. After trituration, the cell suspensions, in DMEM containing 10% fetal bovine serum, 4 mM L-glutamine, penicillin, and streptomycin (all Sigma), were plated onto PDL-coated T75 flasks (Corning, Corning, NY, USA). Flasks were incubated at 37°C in 5% CO2 overnight for attachment of cells and then the medium (with unattached debris/cells) was aspirated before adding fresh DMEM containing 10% fetal bovine serum. The established glial cultures were further incubated at 37°C with changes of the DMEM (with 10% fetal bovine serum) at 3-day intervals until the cells were confluent (~10 days).
To obtain cultures enriched for astrocytes or microglia, flasks were shaken 15 minutes at 130 g to separate astrocytes and microglia. The non-adherent microglia were removed and collected. Ten milliliters of fresh DMEM was added onto adherent cells and the flasks were further shaken overnight at 37°C and 300 g. The medium with the remaining microglial cells and oligodendroglia was removed. The resulting basal layer of astrocytes was then washed with PBS, lifted with 0.25% trypsin-EDTA, added DMEM, and harvested by centrifugation at 300 g for 5 minutes.
Microglia (~4.0 × 10 5 /well) and astrocytes (~2.5 × 10 5 /well) were seeded in wells coated with the chosen matrix substrate (PDL, laminin, collagen, FN, VN). The cultures routinely contained >95% of the enriched cell population, as determined by immunostaining with antibodies to GFAP (astrocyte) and CD11b (microglia).
Oxygen-Glucose Deprivation (OGD)
Standardized conditions for normoxia and for OGD for endothelial cells, astrocytes, or microglia have been described recently.15,20 Before induction of OGD, serum was completely removed from the cell cultures. 15 The cells were then cultured in strictly serum-free high glucose medium (4.5 g/L, DMEM containing 4 mM L-glutamine, penicillin, and streptomycin, supplemented with N1 medium) for normoxia or in low-glucose medium (1.0 g/L, supplemented DMEM) for OGD. Cultures containing low-glucose medium were placed in a hypoxia chamber (Billups-Rothenburg, Del Mar, CA, USA), which was flushed through with a mixture of 95% N2/5% CO2 for 1 hour, and then closed for the duration of the experiment. O2 levels decreased to 0.2–0.4% by 4 hours post induction, whereat they were maintained for the rest of the experiment. Cells were processed immediately after removal from the chambers.
Cell Lysates and Supernatants
For cathepsin B and L activities, (conditioned) media from endothelium, astrocytes, or microglia were collected and centrifuged at 17,000 g to remove cells and debris. Cell lysates were prepared by scraping cells with 150 μL PBS, repeating freeze–thaw cycles three times, and centrifugation at 17,000 g for 10 minutes. Cell lysates and supernatants were stored at −80° C until assay.
For cathepsin B and L expression by Western immunoblot, cell lysates were prepared using RIPA Lysis and Extraction Buffer (G-Biosciences, St Louis, MO, USA) containing a protease inhibitor cocktail (Sigma). Conditioned medium was concentrated 20-fold using Amicon Ultra-0.5 Centrifugal Filter devices (Millipore, Billerica, MA, USA). Cell lysates and concentrated supernatants were stored at −80°C until immunoblotting. Protein concentrations were determined by the BCA protein assay method (Pierce, Rockford, IL, USA).
For the effects of pericellular pH change on released cathepsin L activity, culture media was thoroughly removed from microglia after normoxia/OGD treatment, and 0.5 mL Ringer's solution buffered to pHs 7.4, 6.2, or 5.6 was added to each well of cells. 21 The plates were incubated at 37°C for 0, 0.5, 1.0, or 2.0 hours on a shaker (80 rpm). At the end of incubation, the supernatants were collected from each time point and cathepsin L activities were determined. The same samples were also assessed for LDH release.
Cathepsin B and L Activity Assays
Cell-generated cathepsin B and L activities were assayed by a modification of the method of Sheahan et al. 22 Cathepsin B activity was assessed with the specific substrate Z-arg-arg-MNA. Although a specific substrate for cathepsin L is not available, Z-phe-arg-MNA is cleaved much more readily by cathepsin L than cathepsin B, especially at < pH5.0. Cell extracts incubated with Z-phe-arg-MNA at pH3.0 for 5 minutes resulted in 98% inactivation of cathepsin B. 23
For cathepsin B-like activity, Z-arg-arg-MNA was pre-incubated with 100 mM 2-(N-morpholino)-ethanesulfonic acid pH6.0, 1 mM DTT, and 1 mM EDTA, while for cathepsin L-like activity 0.5 mM Z-phe-arg-MNA was pre-incubated with 100 mM 2-(N-morpholino)-ethanesulfonic acid pH3.5, 1 mM DTT, and 1 mM EDTA for 15 minutes at 37°C. Cell homogenates (100 μL each) were then added to the assay buffer/substrate mixture and incubated at 37°C for 10 minutes. Reactions were terminated by addition of 50 μL 1 N HCl in 2% Triton X-100. Fast Blue B (1 mM, Sigma) was added and the color developed for 10 minutes before reading at 550 nm. Cathepsin activities were normalized and reported as mU/μg protein.
In situ Tissue Cathepsin B and L Activity
Fluorescent enzyme histochemistry was performed on non-human primate striatal tissues by modification of the procedure of Manship et al. 24 Serial 10 μm cryosections of striatum were preincubated for 1 hour at 20°C in 100 μL 0.1 M 2-(N-morpholino)-ethanesulfonic acid buffer (containing 1.3 mM EDTA, 10 mM DTT, and 10% (w/v) polyvinyl alcohol (pH6.0 for Z-arg-arg-MNA and pH3.5 for Z-phe-arg-MNA)). To verify that cysteine protease activity was observed, sections were pre-incubated in the buffer containing 10 μM E64d for 1 hour. The enzymatic reaction was initiated by adding 100 μL buffer containing 0.5 mM Z-arg-arg-MNA or Z-phe-arg-MNA and 4 mM 5-nitrosalicyladehyde. The sections were incubated in a damp chamber at room temperature for 24 hours, in the absence of light. Sections were then rinsed in distilled water to remove reagents. The slides were finished with Vectashield mounting medium (Vector, Burlingame, CA, USA). The fluorescence produced by the coupling reaction between MNA and 5-nitrosalicyladehyde was visualized under a Zeiss Axioskop photomicroscope (emission 420 nm, excitation 590 nm) (Zeiss, Oberkochen, Germany).
Western Immunoblots
For electropheretic protein separation, cell lysates or concentrated supernatants were reduced with DTT and loaded (with equal protein content or volume, respectively) on 10% Tris-glycine gels run at 110 V for 1 hour. Proteins were electroblotted for 1 hour onto polyvinylidene fluoride membranes (Millipore), then blocked for 1 hour with 5% milk solids/TBS. 15 The membranes were probed with rat anti-human/mouse cathepsin B MoAb or goat anti-mouse cathepsin L (both R&D Systems, Minneapolis, MN, USA) at 4°C overnight. In the ex vivo protease secretion assays, the membranes were probed with mouse MoAbs to perlecan (Millipore) or perlecan domain V (R&D Systems), or a rabbit polyclonal antibody to the collagen IVα2 chain at 4°C overnight, then incubated with appropriate secondary antibodies at room temperature for 1 hour. The membranes were washed in TBS/Tween 20 and the proteins identified with an enhanced chemiluminescence detection system (ECL Plus, GE Healthcare, Buckinghamshire, UK), according to the manufacturer's instructions. In the instances when vehicle control samples provided a baseline band, the amount of the band was subtracted from those of the test samples before analysis (see Figure 8).
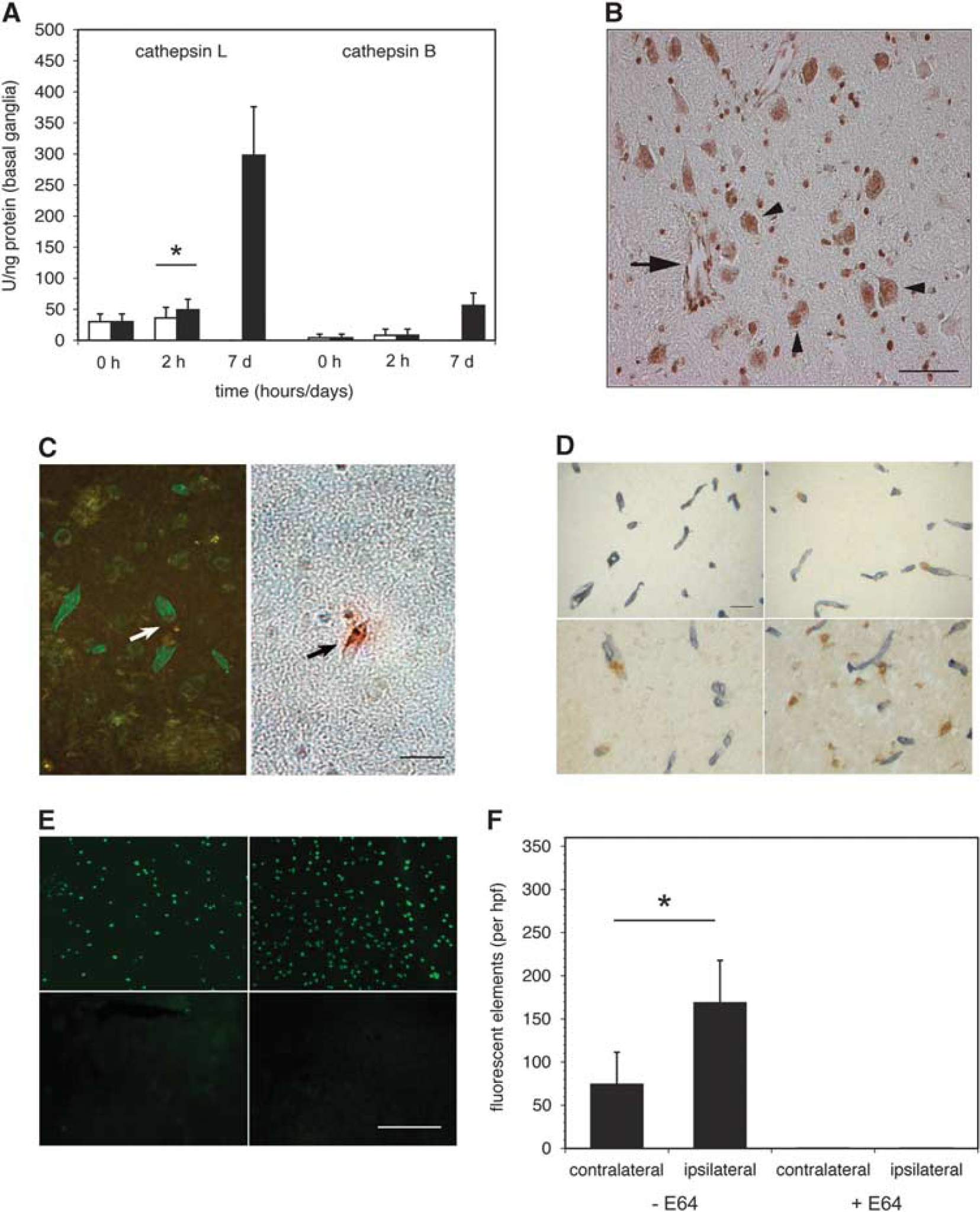
Cerebral cathepsin B and L expression during middle cerebral artery occlusion (MCAO) in the non-human primate. (
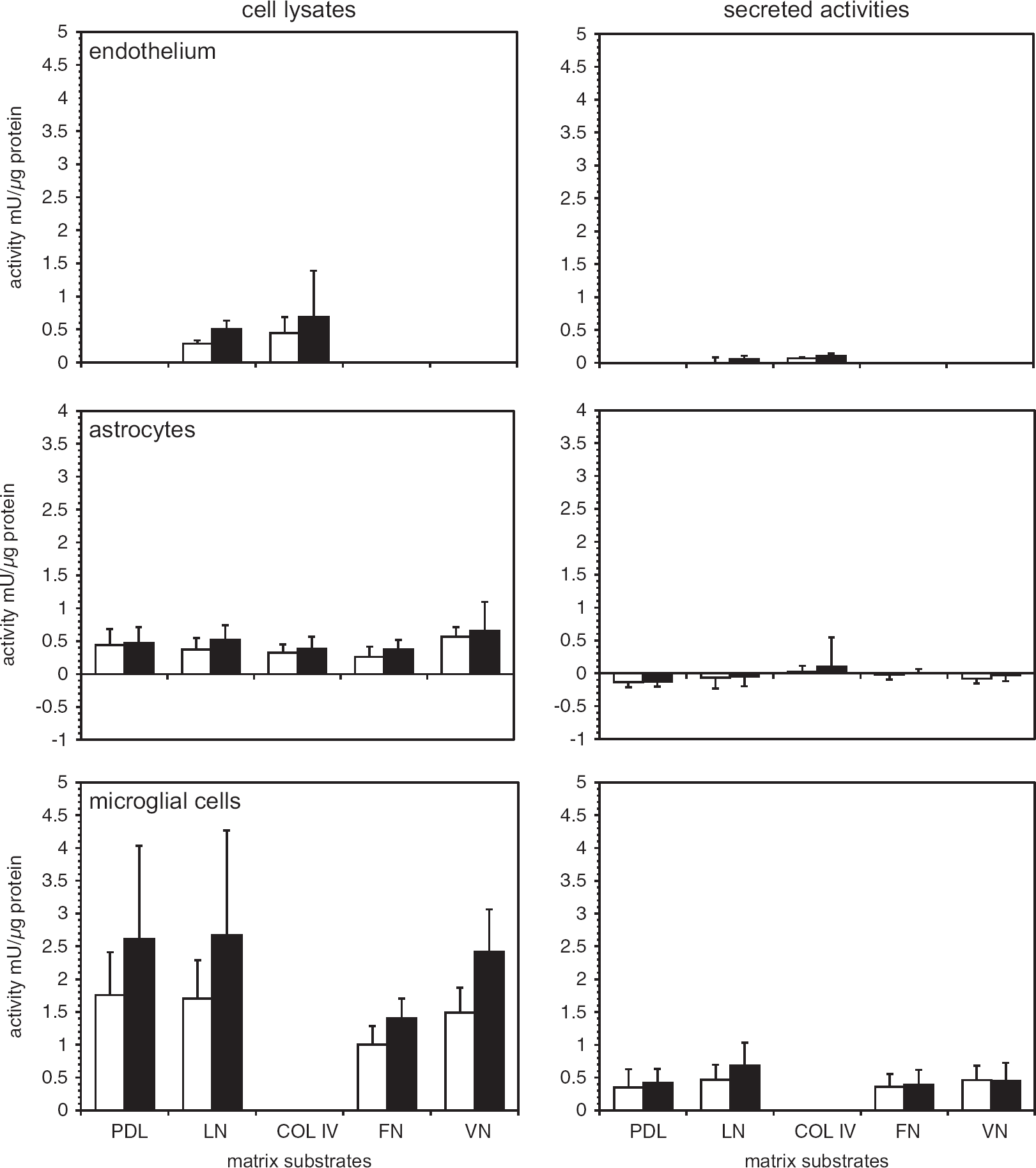
Cellular and secreted cathepsin L activities and extracellular matrix (ECM) exposure. Murine primary cerebral endothelium, astrocytes, and microglia grown on various ECM substrates under normoxia (open bars) and oxygen-glucose deprivation (OGD) (filled bars). Substrates were poly-D-lysine (PDL), laminin (LN), collagen type IV (COL IV), fibronectin (FN), and vitronectin (VN). Cathepsin L activities of cell lysates and conditioned media were determined based on standard curves of the activity of purified cathepsin L from human liver, and reported as relative cathepsin L activity per protein content (mU/μg protein). Only microglia displayed significant secretion of cathepsin L under normoxia and OGD at neutral pH. Data are mean activity ± s.d. (n = 6 samples per condition in endothelium; n = 9 samples per condition in astrocyte and microglia cells).
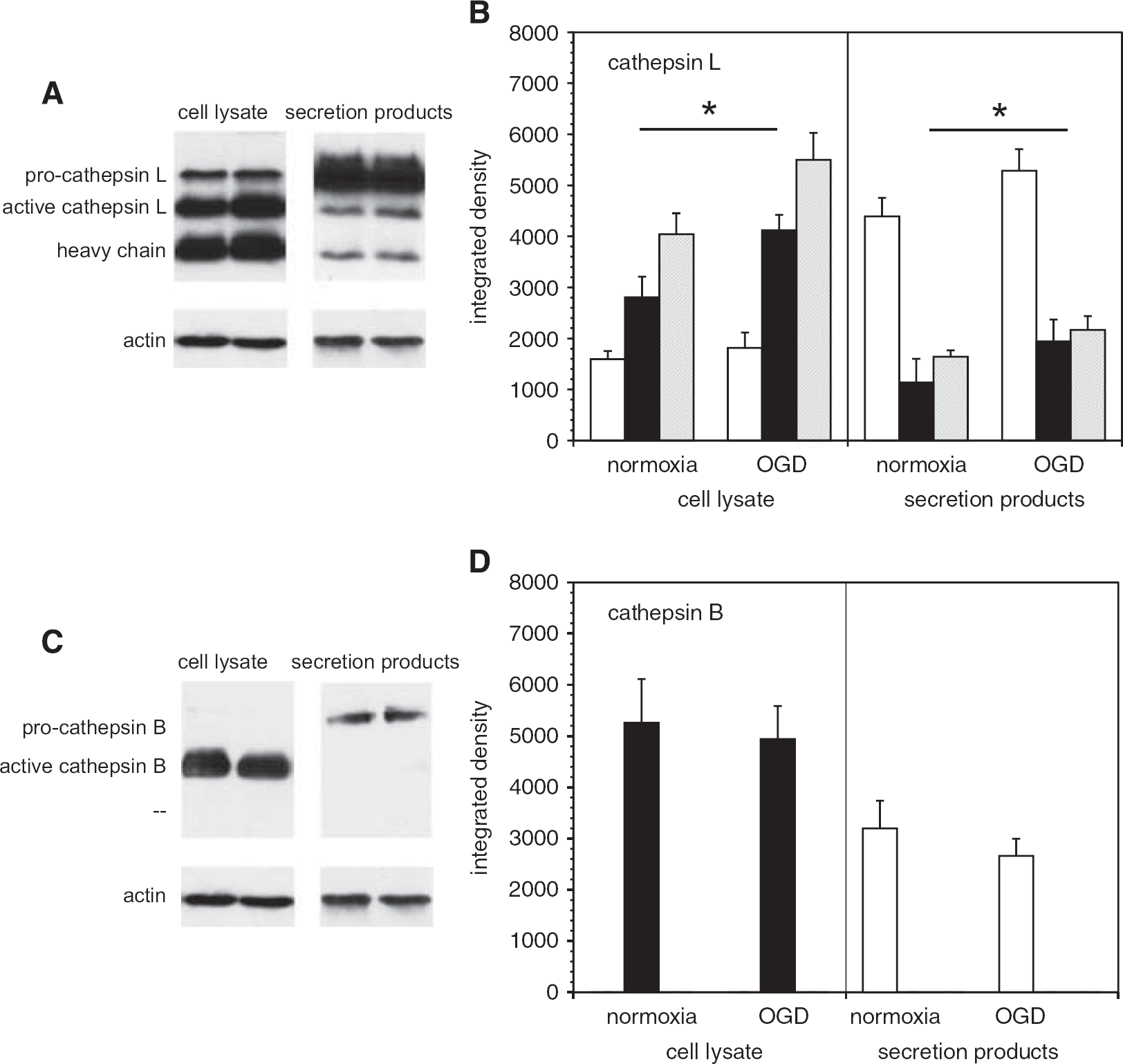
Cathepsin B and L expression in microglia. Western immunoblots of cathepsin B and L expression performed in murine cerebral microglia subject to normoxia or oxygen-glucose deprivation (OGD), before preparation of cell lysates and conditioned media. (
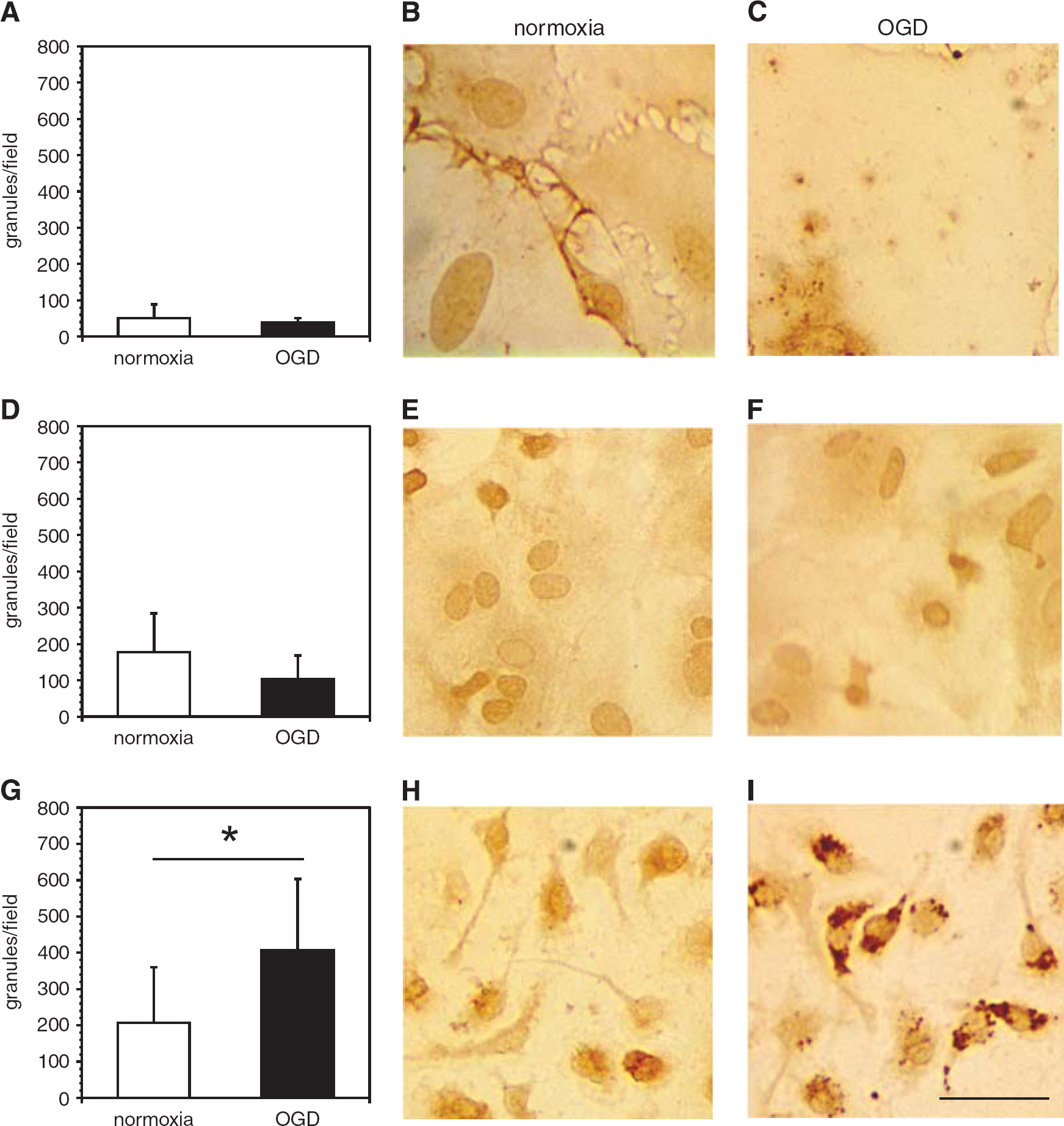
Association of immunoreactive cathepsin L with murine primary endothelial cells, astrocytes, and microglial cells. Immunoreactive cathepsin L is significantly associated with microglia under oxygen-glucose deprivation (OGD). Murine primary endothelial cells (
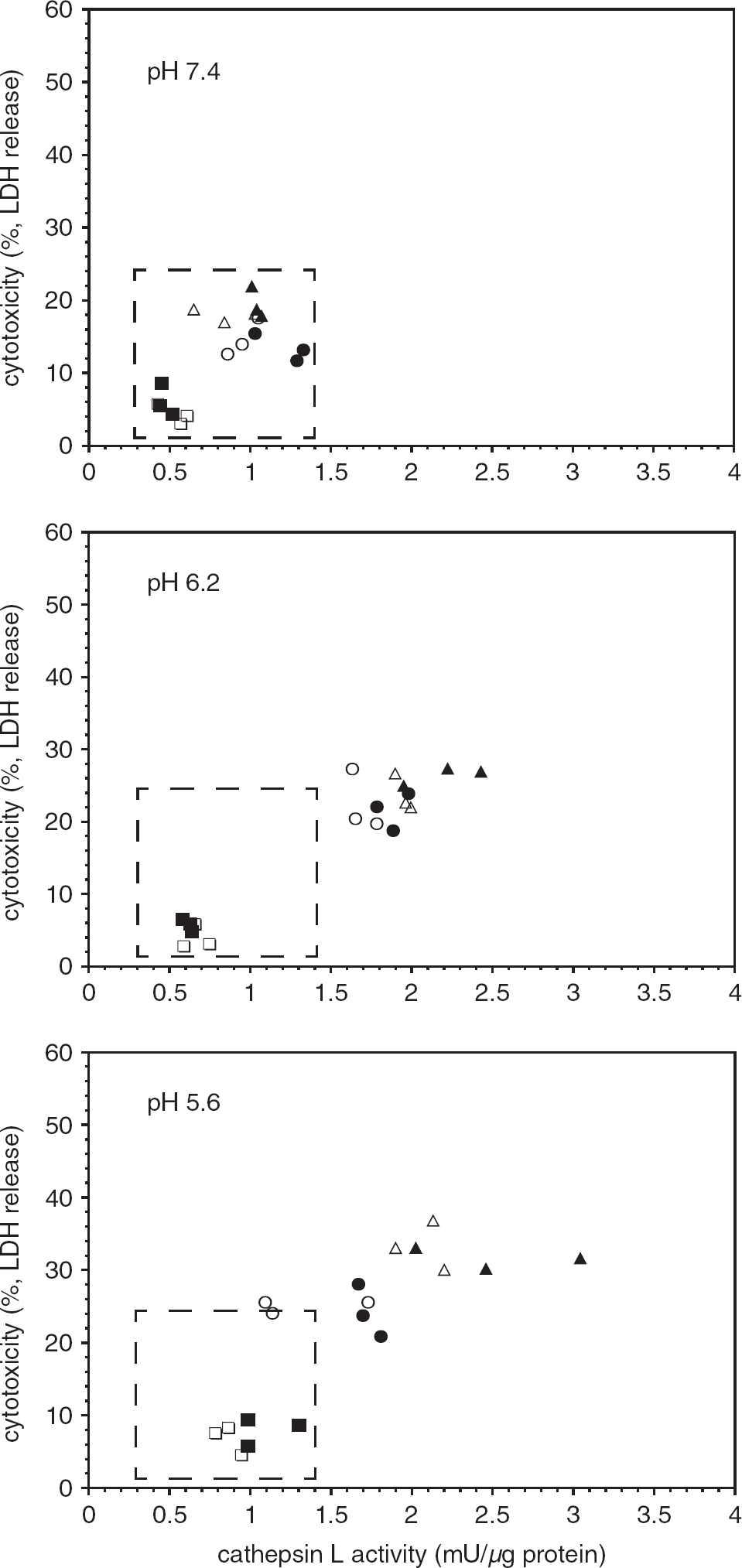
Effect of pH on microglial secretion of cathepsin L activity and cell survival (lactate dehydrogenase (LDH) release) under normoxia or oxygen-glucose deprivation (OGD). Decrease in pH resulted in both increased cathepsin L secretion under normoxia (open symbols) and matched OGD (filled symbols). Generally, at lower pH, secretion under OGD exceeded that under normoxia (same symbols). Cathepsin L release was tied to microglial demise, which was also pH-dependent. Data represent n = 9 (or n = 3, each of three separate experiments (different symbols)). See Supplementary Figure 4.
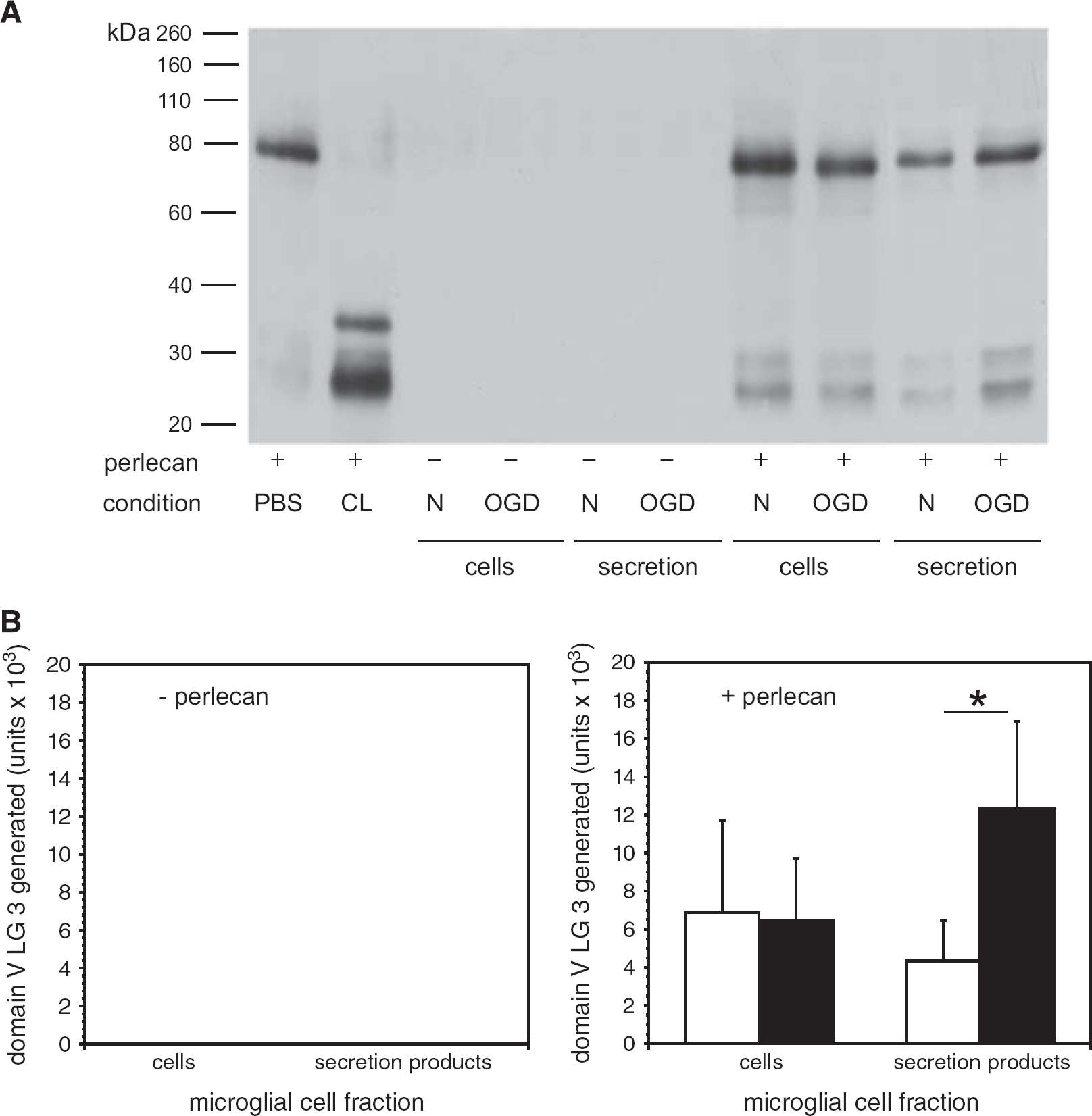
Degradation of purified perlecan by secretion products of murine microglia subject to normoxia or oxygen-glucose deprivation (OGD). (
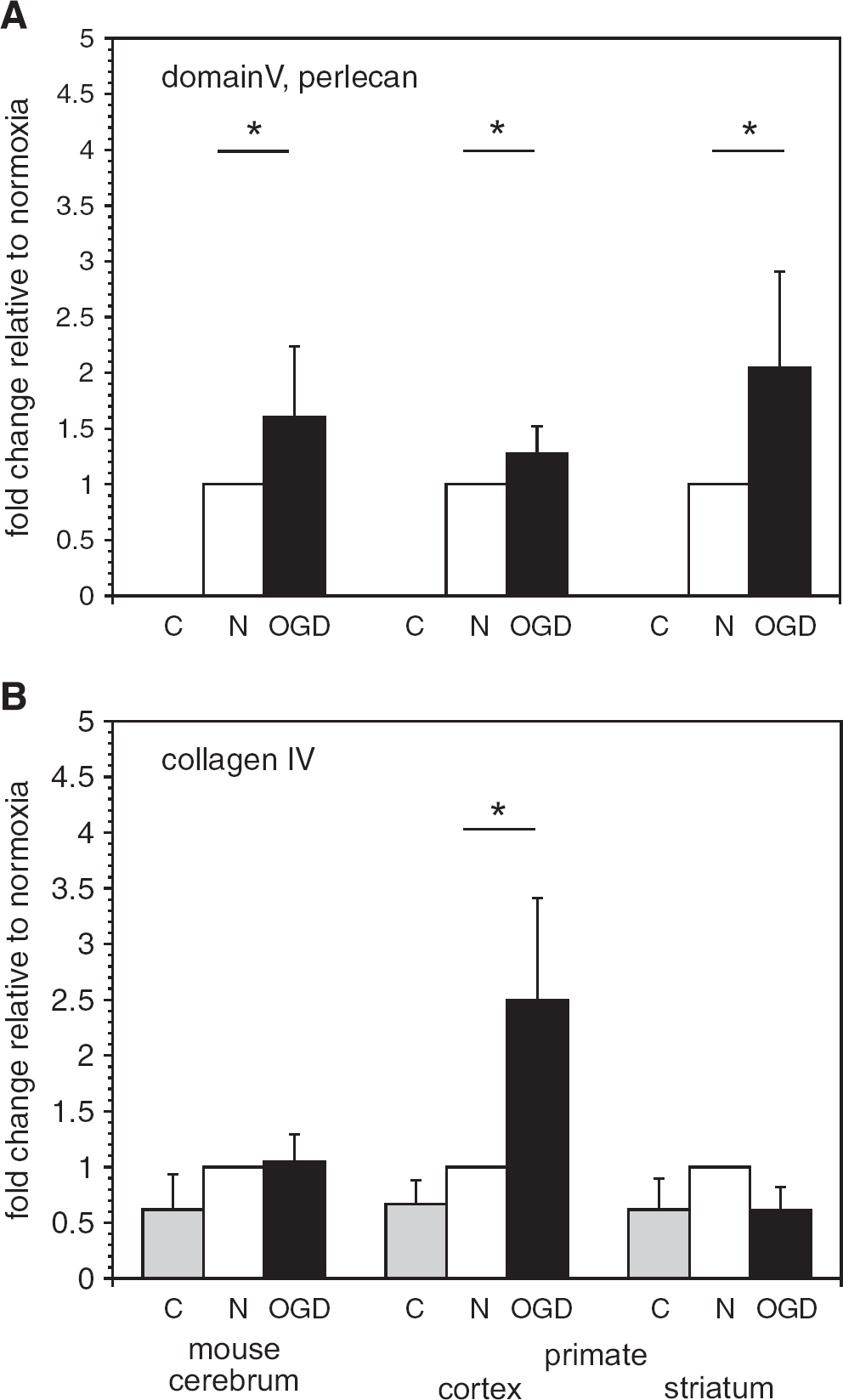
Degradation of vascular extracellular matrix (ECM) by microglial secretion products. Degradation of perlecan domain V (26 kDa) (
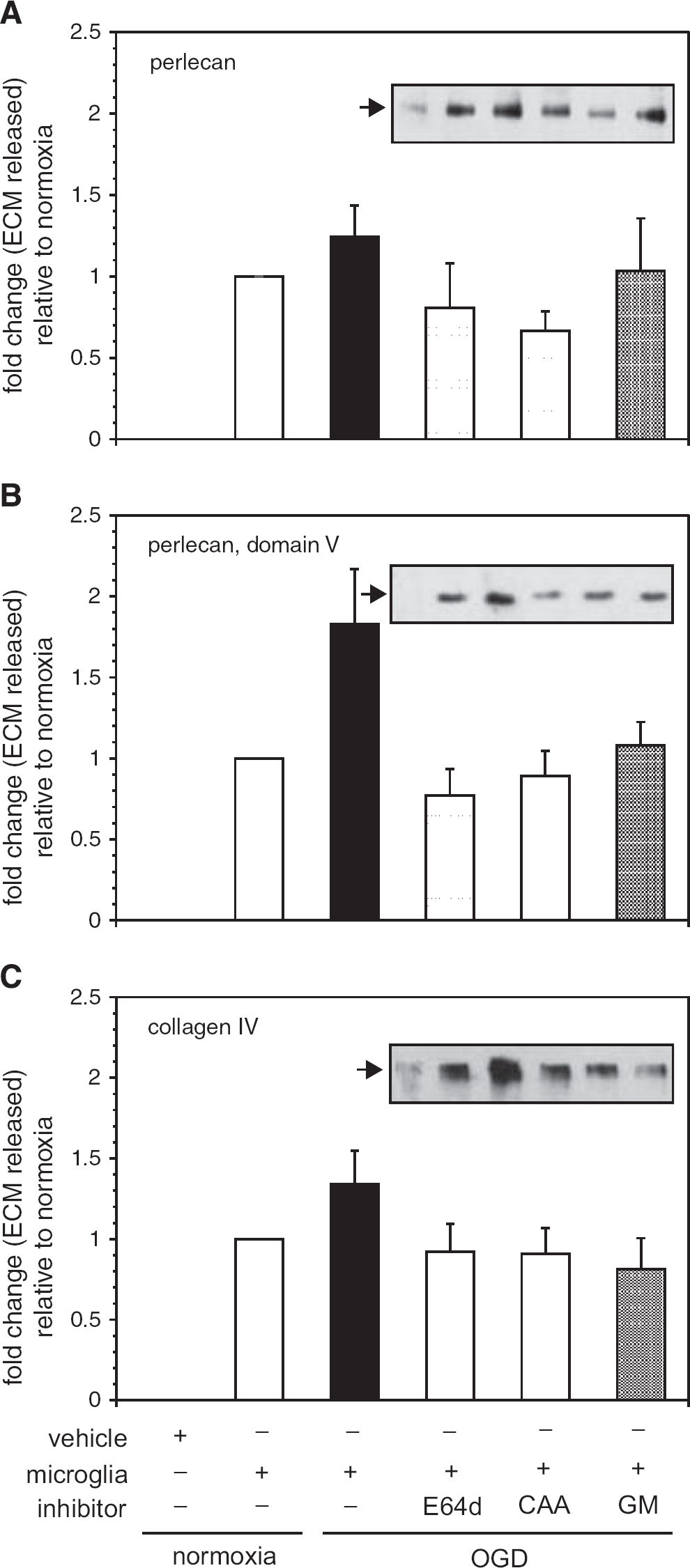
Identification of extracellular matrix (ECM) degradation activity derived from microglia that were subject to normoxia or oxygen-glucose deprivation (OGD). The impact of inhibitors of cysteine protease activity (E64d), cathepsin L activity (CAA0225, CAA), or matrix metalloproteinase (MMP)-like activities (GM6001, GM) on the ability of ECM proteolytic activities secreted from microglia to degrade ECM in murine cortex ex vivo is shown. Microglia subject to OGD generate activity that significantly increases the release of perlecan (
Immunohistochemistry/Immunofluorescence
Antigens of interest in cerebral microvessels were developed on 10 μm frozen sections as previously described. 2 Both single label and dual label studies were performed. Where two MoAbs were tested against each other (e.g., perlecan vs. cathepsin L), the immunohistochemical process (cathepsin L) preceded the immunofluorescent process (perlecan). To determine in situ sources of cathepsin L, the rat MoAb to cathepsin L (R&D Systems) was applied to murine primary endothelium, astrocytes, and microglia (Supplementary Table 1).
Ex vivo Protease Secretion Assay
For the secretory protease assay, media were thoroughly removed from the cell cultures and replaced with Ringer's solution (pH7.4). After incubation for 2 hours at 37°C, the supernatants were collected. Purified perlecan (0.8 μg) was incubated with 1.0 μg purified cathepsin L (control) and cell extracts were incubated with 10 μL Ringer's solution (to 20 μL total) for 24 hours at 37°C. The samples were then separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis for immunoblotting.
Ex vivo Extracellular Matrix Degradation Assay
ECM substrates were obtained from perfused mouse brain, and homogenization of dissected cortex was performed with a glass tissue grinder at 4°C. Ringer's solution containing 0.86% NaCl, 0.03% KCl, and 0.033% CaCl2 was added to the homogenate, which was heated for 60 minutes at 70°C, and then centrifuged for 10 minutes at 6,000 g to remove soluble material. 25 For primate cortex and striatal samples, 10 cryosections (10 μm/each) in PBS were homogenized with a Dounce homogenizer and centrifuged at 6,000 g for 10 minutes. The supernatants were discarded and the pellets stored at −80°C.
For the assay, the pellet was resuspended in PBS, and aliquoted equally into Eppendorf tubes, each centrifuged at 6,000 g for 10 minutes. To the individual pellets, 500 μL microglia-conditioned medium (normoxia or OGD) was added, and the mixture vortexed, then incubated at 37°C for 2 hours. The supernatants were concentrated 20-fold, separated on 10% SDS–PAGE, and examined by immunoblot.
Quantitative Real-Time RT-PCR
Total RNA was extracted from microglia and astrocytes, subjected to normoxia or OGD, using the RNeasy Plus Mini kit (QIAGEN, Valencia, CA, USA) as previously described. 15 Eluted RNA was treated with RNase-free DNase (Ambion, Austin, TX, USA). The RT-PCR reaction was set up according to the SuperScript III Platinum One-Step Quantitative RT-PCR System protocol (Invitrogen, Carlsbad, CA, USA). The final reaction conditions were 200 nM per primer, 100 nM per probe, 200 μM of each dNTP, 5 mM MgSO4, and 100 ng total RNA. The PCR cycling conditions were 45 cycles at 95°C for 30 seconds and 60°C for 30 seconds. Primers and TaqMan probes were designed with primer3 software (Supplementary Table 2). The expressions reflect duplicate studies for each culture and the means and standard deviations of four to six separate cultures.
Statistical Analyses
Summary data are reported as mean ± standard deviations (s.d.). For the cell-based studies, each OGD experiment was performed in triplicate, and replicates of three or more studies were performed on separate days with different primary cultures. For real-time RT-PCR, each matrix condition was tested in duplicate with three or more studies performed on separate days. Fisher's exact test, Student's t test, and analyses of variance were performed where appropriate on non-transformed data. After significant analyses of variance, pair-wise group comparisons were undertaken with Tukey's HSD procedure. Analyses were performed in Systat 12 (Systat Software, 2007, San Jose, CA, USA) or using Prism (GraphPad Software, La Jolla, CA, USA) software. We report two-sided P-values throughout.
With the exception of preliminary studies, the analyses and critical parts of the research team were masked to assignment of animals and precise culture interventions.
RESULTS
Differential Generation of Cathepsin B and L Activities
On the basis of the rapid appearance of cysteine protease activity after MCAO in non-human primate striatal tissue, 2 cathepsin B and L activities within those tissues were assayed. Baseline tissue cathepsin B and L activity levels were low (Figure 1A), in keeping with the absence of their immunoreactivity in non-ischemic tissue. 2 At 2 hours after MCAO, cathepsin L activity had significantly increased in the ischemic territory (over the contralateral non-ischemic striatum, 49 ± 16.5 vs. 36 ± 17 U/ng; P = 0.01; Figure 1A), whereas no change in cathepsin B activity was detected. By 7 days after ischemia onset, cathepsin L activity within the ischemic territory had greatly increased, while cathepsin B activity increased more modestly.
Relationship of Microvessel to Neuron Cathepsin L Expression
Acutely, both microvessels and nearby neurons displayed immunoreactive cathepsin L in ‘nests’ within the ischemic core (Figure 1B). Cathepsin L-immunoreactive microvessels were predominantly associated with dUTP+ neurons and were often surrounded by regions lacking cathepsin L antigen. However, within the regions of dUTP+ neurons, there was no detectable evidence of non-neuronal (glial) cell cathepsin L immunoreactivity.
Cathepsin L and the Microvessel Wall
In the ischemic core, cathepsin L expression within the microvessel wall was significantly associated with loss of perlecan from within the adjacent basal lamina (Figure 1C) (P < 0.00001). Dual immunohistochemistry (cathepsin L)/immunofluorescence (perlecan) at 2 hours MCAO showed that perivascular cathepsin L expression was associated with loss of microvessel perlecan only in the ischemic territory (Figure 1D).
In situ cell-specific cathepsin L activity significantly increased within the ischemic striatum at 2 hours MCAO, above baseline cathepsin L activity (Figures 1E and 1F). This activity was completely abolished by the cysteine protease inhibitor E64 (10 mM). Cathepsin B activity did not change compared with non-ischemic tissue (data not shown).
Identification of Cell Source(s) of Cathepsin L in the Neurovascular Unit
To more precisely determine the non-neuronal source(s) of cathepsin L within the neurovascular unit, its expression and secretion by murine primary cerebral microvessel endothelial cells, astrocytes, and microglia in culture were quantitated under normoxia and experimental ischemia at neutral pH.
Endothelium. A moderate increase in cathepsin L activity was shown when primary endothelial cells grown to confluence on laminin or collagen IV were subject to experimental ischemia (OGD) (Figure 2). Secreted cathepsin L activity was very low during normoxia and OGD, and did not appear to be matrix-dependent (Figure 2). There was no apparent change in cathepsin B activity to OGD (n = 5, data not shown).
Astrocytes. Primary astrocytes displayed variably increased cathepsin L activity during OGD compared with normoxia when the cells were grown on PDL, laminin, collagen IV, VN, or FN (n = 9 each) (Figure 2). Minimally detectable cathepsin L activity was released (Figure 2). There was no significant change in cathepsin B expression or release.
Microglia. In contrast, primary microglia grown on PDL or laminin showed significantly increased cathepsin L activity under OGD relative to normoxia (n = 9 each) (Figure 2). Microglia displayed 3.99 ± 1.48-fold higher cathepsin L activity than that detected in astrocytes, and 6.08 ± 2.25-fold higher cathepsin L activity than endothelial cells under non-ischemic conditions. Oxygen-glucose deprivation stimulated a 39.4–62.5% mean increase in microglial cathepsin L activity, for cells grown on PDL, laminin, VN, or FN (Figure 2). At neutral pH, OGD did not stimulate a further increase in cathepsin L release compared with normoxia, regardless of the matrix substrate. There was no change in cathepsin L activity secreted based on the matrix exposure, although laminin produced a modest non-significant increase under OGD. (Figure 2). No difference in cell viability under conditions of normoxia or OGD was observed. Astrocytes did not modulate microglial cathepsin L generation (data not shown).
Cathepsin B and L Gene Expression
OGD stimulated primary microglia to increase cathepsin L, but not cathepsin B, gene expression above normoxic levels (Supplementary Figure 1). However, primary astrocytes did not express either cathepsin B or L transcripts above normoxic levels when the cells were grown separately on PDL.
Microglial Cell Expression and Release of Cathepsins B and L
Cathepsin B and L expression in murine microglia grown on PDL were analyzed by Western immunoblot. Both the inactive pro-form and the active form of cathepsin L were detected in microglial cells. Oxygen-glucose deprivation stimulated a 47.0 ± 21.5% increase in active cathepsin L within the cells (Figures 3A and 3B), and a 71.4 ± 37.6% increase in active cathepsin L secretion (P = 0.005 each, n = 6). Active cathepsin B was detected in microglia; however, its expression was not altered by OGD. Only inactive pro-cathepsin B was secreted, and this did not change with OGD (Figures 3C and 3D).
Granule-like structures containing cathepsin L significantly associated with microglia were revealed under conditions of OGD, but not with endothelium or astrocytes (Figure 4). The in situ cathepsin L immunofluorescence assay also showed that primary microglia significantly increased cathepsin L expression under OGD compared with normoxia (data not shown). Astrocytes and endothelia did not display changes in cathepsin L with OGD. Treatment with 2 μM E64 for 1 hour fully inhibited the cathepsin L activity in both cell types.
Decreased pH Increases Cathepsin L Activity Release
Given the known pH optimum for cathepsin L activity, and the reduction of the pH to 6.2 and below in the ischemic core, the effects of altering pH on the release and activity of cathepsin L in astrocytes and microglia were determined. Reduction to pH5.6 had no effect on the activity of cathepsin L in astrocytes or the secreted release products (Supplementary Figure 2). However, a pH5.6 environment increased the appearance of cathepsin L activity within microglia and its release over the time of the assay (Figure 5, Supplementary Figure 3). In part, this could be explained by a substantial increase in cell demise (Figure 5). At low pH, OGD did not have a role in the production of cathepsin L, but did in its release.
Microglial Cathepsin L Release Alters Matrix Integrity after Oxygen-Glucose Deprivation
An ex vivo assay developed to detect the products of matrix protease-mediated digestion of microvascular ECM in cerebral tissue was devised (see Methods). Purified cathepsin L cleaved the 80 kDa domain V from purified perlecan to generate the LG3 fragment (Figure 6A). Cathepsin L activity was shown when
Microglia subjected to OGD secreted activity that significantly increased the release of perlecan domain V from
Inhibitors of cysteine protease (E64d) and cathepsin L (CAA0225) prevented perlecan release from
DISCUSSION
Remodelling of the cerebral microvasculature in acute response to focal ischemic injury involves changes in the basal lamina ECM. Within 1–2 hours after MCAO, in the non-human primate, significant changes in laminin, collagen IV, FN, and perlecan occur within microvessel basal lamina matrix in the ischemic core.1,2 Knowing how acute changes in microvessel matrix architecture occur quantitatively, the contributions of individual matrix proteases, and their specific cell sources are central to devising methods for preservation of microvessel and neurovascular unit integrity, and for reducing hemorrhagic transformation.1,26 These studies were prompted by the observation in the non-human primate of acute cathepsin L protein and activity expression in the neurovascular unit coincident with the simultaneous disappearance of its substrates perlecan and collagen IV in the microvasculature. 2 The loss of perlecan also coincides with the acute appearance of (pro-)MMP-2. 2 Previous work was interpreted to show that cathepsin L is only generated by (cortical) neurons in response to ischemia. 10 Preliminary immunohistochemical studies failed to show any association of cathepsin L with other non-neuronal non-vascular cell populations within the ischemic striatum, an observation not inconsistent with secretion of cathepsin L (see Figure 1B, data not shown).
Here, we show that cathepsin L, an acidic cysteine protease, also appears on select microvessels proximate to select (dUTP+) neurons in the ischemic core. The rapid simultaneous appearance of immunoreactive cathepsin L by neurons and nearby microvessels suggests that these microvessel-neuron events are coordinated. 11 Cathepsin L expression and perlecan loss are associated with the same microvessels. Microglia appear to be a major non-vascular source of cathepsin L under experimental ischemia. The experiments here extend the findings of Fukuda et al, 2 who first showed the vascular association of cysteine protease activity (cathepsin L) and its impact on perlecan, to non-neuronal sources of cathepsin L activity within the neurovascular unit. These experiments also extend observations of ischemia-related alterations in matrix integrity to specific matrix protease subsets and cell sources.
Among cysteine proteases, cathepsins B, L, D, and E are expressed in cerebral tissue. 27 Cathepsin L production has been attributed to neurons, microglia, astrocytes, fibroblasts from various origins, and other cell types.28–30 These experiments confirm that cysteine protease activity generated acutely during focal ischemia in the striatum is predominantly cathepsin L, not cathepsin B, which responds much later (Figure 1). 2 They further show that (i) non-neuronal cathepsin L derives from microglia, not endothelial cells or astrocytes; (ii) the active form of cathepsin L is secreted predominantly; and (iii) cathepsin L activity released during ischemia can cause the release, degradation, and loss of vascular perlecan and collagen IV from cortical tissues. In contrast, although microglia are a source of cathepsin B, experimental ischemia does not generate and release active cathepsin B acutely in the setting here. Hence, the active cathepsin L generated by microglia can potentially contribute to acute microvessel basal lamina matrix degradation. These processes may also involve a collagen-degrading MMP-like activity, also derived from microglia. 15
Perlecan is a minor, but important functional component of the cerebral microvessel basal lamina. 2 Endothelial cells interact with the perlecan core protein in cerebral microvessels through β1 integrins.31,32 Through its heparan sulfate side chains, perlecan retains growth factors, including basic fibroblast growth factor bound to perlecan domain V, that are released on perlecan cleavage. 4 Purified perlecan can be cleaved by plasmin, MMP-1, or MMP-3 under controlled conditions. 4 Purified cathepsin L also decreases the antigenicity of perlecan core protein in primate cerebral microvasculature. 2 Recently, Bix et al showed that the perlecan domain V cleavage fragment has biological properties in the setting of experimental focal ischemia in rodents. 33 However, it is not known whether processing of other cerebral microvessel ECM substrates in the basal lamina, including type IV collagen, potentiates the release of perlecan or perlecan-degrading proteases. The inhibition studies here suggest that this may be the case (Figure 8).
Two in vivo findings in the non-human primate support a role for cathepsin L in the acute degradation of the microvessel matrix: (i) the significant acute increase in cathepsin L activity in the ischemic core, and (ii) the significant temporal and topographical relationship between the perivascular appearance of cathepsin L and perlecan loss from the microvessel ECM. Inhibition of cathepsin L activity secreted from microglia prevents both perlecan and collagen IV degradation in vitro and ex vivo from cerebral tissues in these studies.
Inhibition of MMP-like activities derived from microglia (with GM6001) also prevents perlecan and collagen IV degradation from cerebral tissues. Note that these experiments do not unequivocally explain the impact of cathepsin L on both matrix substrates acutely during focal ischemia, but highlight the probability that more than one matrix protease system could be simultaneously involved and potentially interact. Plausible scenarios during focal cerebral ischemia are that: (i) microglia release active cathepsin L during ischemia, as well as MMP-like activities (e.g., pro-MMP-9, 15 and/or others) and these interact in tissue, (ii) microglia release cathepsin L, which is responsible for the release of specific inactive matrix proteases from the ECM, that in the active form themselves lead to perlecan and collagen IV degradation, (iii) microglia release proteases that interact with proteases generated from vascular cells (e.g., astrocytes 15 ), or (iv) a combination of these scenarios. Importantly, cathepsin L activity derived from microglia under experimental ischemia (Figures 2, 3, 4, and 5), but also normoxia, indicating that MMP-like activity released during ischemia in a matrix-dependent manner was not the predominant matrix protease released. 15 This indicates that acute microvessel matrix degradation under focal ischemia cannot be explained by (pro-)MMP-9 activation exclusively.
Evidence exists in other settings that other MMP-like activities, including ADAMTS1, may be involved in cathepsin L-like activities. 34 The nature of the interactions of matrix proteases within the microvasculature during cerebral ischemia and development is poorly understood, but is important because of the potential roles of cathepsin L in tissue destruction, neovascularization, and remodeling.
Local tissue pH is relevant to cathepsin L activity and to the course of tissue injury. These in vitro pH studies mimic the ischemic core and peripheral tissue, as in focal ischemia in the non-human primate. 16 Strong showed that the pH of the ischemic core in the primate attains pH6.2. In vitro pHs 6.2 and 5.6 promote exposure and significantly increased release of cathepsin L activity by isolated microglia under OGD (Figure 8). The acidic milieu is also associated with progressive microglial demise, not seen at neutral pH.
Interestingly, both cathepsins L and S regulate antigen presentation by both astrocytes and microglial cells. These are under modulation by tumor necrosis factor-α and interferon-γ, 30 cytokines also generated during focal cerebral ischemia. 35 In innate inflammatory responses within the central nervous system (CNS), cathepsin L participates in MHC class II expression. 36 However, in certain chronic neurodegenerative disorders, excessive cathepsin L generation can be damaging. For instance, in ß-amyloid deposition disorders Aß peptides increase cathepsin L activity as a potential upstream event in Alzheimers-type neuron degeneration. 37 Furthermore, in brain tissue from Alzheimers-diseased patients, pro-cathepsin L generation accompanies full activation of microglial cells, but not resting or moderately activated cells. 38 Hence, in the CNS, cathepsin L production during acute injury appears to be reactive.
In the context of the neurovascular unit, these experiments suggest that cathepsin L may have roles in neuron-microvessel biology. Deletion of cathepsin B alone has no obvious neuronal phenotype. 29 However, the absence of cathepsin L is associated with apoptosis of select cortical neurons and neurons of the cerebellar granule and Purkinje cell layers, and with reactive astrocytosis. 29 Outside the CNS, in addition to a defect in epidermal differentiation, mice deficient in cathepsin L (nackt, nkt) developed significant left ventricular hypertrophy and valvular insufficiency as an adaptive response to interstitial fibrosis. 39 Ctsl–/– mice developed worse cardiac dysfunction and greater scar formation than Ctsl+/+ littermates after left coronary artery ligation. 40 The impact on cardiac or cerebral microvessels is not reported. It follows that the prolonged absence of the ability to generate active cathepsin L allows the accumulation of materials that can render injury to cardiac function, although the presence of cathepsin L itself is not apparently toxic. The impact in the CNS beyond this is not reported.
Several technical limitations of these studies must be mentioned: (i) sections of frozen brain tissue can ‘spill’ cathepsin L and MMPs and can thereby cause unanticipated ECM degradation and protease activity even under normoxia (Figure 1D), hence the need for isolated cells, (ii) cells cannot be cultured at acidic pH, therefore the in vitro design of the pH studies here, (iii) the lower pH has no impact on the matrix assay(s), and (iv) the specific association of cathepsin L release with specific cells was determined at neutral pH. Also, some release of latent proteases from the tissue slice(s) was possible that could have been increased by the cathepsin L and MMP-like activities generated in the matrix assay (e.g., see normoxia, Figure 7). Those observations have prompted further experiments into the interaction of cathepsin L and MMP-like activities that are beyond the scope of this report, that begin to address mechanisms of these interactions.
Striatal ischemia generates cathepsin L within/around select microvessels that coincides with the loss of basal lamina perlecan. While microglia release cathepsin L during focal ischemia, microvessel-associated expression of cathepsin L could also derive from activated pericytes, vessel wall histiocytes, and/or macrophages activated during ischemia, although identification of these potential sources was not in the scope of this study, but is of interest. 28 Also unclear is whether the increased cathepsin L activity within the ischemic regions contributes to loss of the microvessel permeability barrier integrity, although this is likely via degradation of the basal lamina ECM. The circulating matrix proteins VN and FN, found only in plasma and not in the CNS, significantly stimulate (pro-)MMP-9 generation/release by microglia with extravasation during ischemia, 15 however, they do not appear to stimulate microglial cathepsin L release (Figure 2). Astrocytes do not appear to participate in postischemic cathepsin L release, nor do they modulate microglial cathepsin L release. Here, microvessel cathepsin L release occurs under conditions of hypoxia and acidosis generated during cerebral ischemia.
These studies show for the first time that non-neuronal cathepsin L expression generated acutely during focal ischemia also involves activated microglia, but not endothelial cells or astrocytes. In the ischemic striatum, no evidence of microglial cathepsin L production or release was evident by immunohistochemistry in the primate, 2 however, the tissue produced the activity. Perivascular expression of cathepsin L in vivo is associated with the disappearance of perlecan. 2 These studies also confirm the acute and coordinated appearance of cathepsin L antigen on microvessels in close proximity to neurons with the simultaneous significant appearance of cathepsin L activity in the ischemic striatum acutely after MCAO. This activity is shared by microglia that can rapidly release active cathepsin L in response to ischemia compatible with conditions of the acidic ischemic core.
The appearance of cathepsin L together with select MMPs, 3 serine proteases, 18 and other proteases in the ischemic regions indicates the rapidly changing microvessel milieu triggered by focal ischemia. These studies confirm the potential roles that microglia activated during ischemia can have in microvessel damage in the ischemic core.15,41 Importantly, these studies indicate that active cathepsin L derived from microglia is damaging to microvessel structure in the ischemic core, and emphasizes that MMP-like activities are not the sole matrix protease family acutely generated after focal ischemia that can potentially degrade basal lamina ECM in cerebral tissue. Indeed, active cathepsin L alone could account for the matrix alterations observed during ischemia. Furthermore, the coincident increase in microvessel permeability implies that cathepsin L together with MMP-like activity (to be identified) could accentuate microvessel barrier permeability. Although this remains unexplored at this time. These studies indicate that other matrix proteases generated by activated microglia also have roles with cathepsin L in vascular matrix destruction under conditions of focal ischemia, and that their mechanisms of interaction should be determined to establish a basis for potential successful treatment interventions.
Footnotes
Each co-author contributed to this work in multiple ways. All co-authors were masked to sample assignments during the conduct of the various experiments, and simple randomization schemes were applied for those assignments. Conception of the hypothesis and the design of the work was led by Drs del Zoppo and Gu. Specific experiment sets included cell culture preparations (Y-HG, MK, SYH), Western immunoblot analysis (Y-HG, MK, SYH), mRNA transcription analysis (XW), and protease identification and activity inhibition studies (SF, Y-HG), with masked statistical analysis (JAK). The development of the matrix degradation assays was overseen by Drs Gu and del Zoppo. All co-authors participated in the preparation of the manuscript to its final form, which was supervised by the senior author (GJdZ).
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Ms. G.I. Berg for her able assistance in the preparation of this manuscript. The authors also thank Mr L Eggleston for his expert technical assistance in the conduct of the original activity measurements.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.