
Abstract
Stroke triggers an inflammatory cascade which contributes to a delayed cerebral damage, thus implying that antiinflammmatory strategies might be useful in the treatment of acute ischaemic stroke. Since two unrelated peroxisome proliferator-activated receptor-γ (PPARγ) agonists, the thiazolidinedione rosiglitazone (RSG) and the cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), have been shown to possess antiinflammatory properties, we have tested their neuroprotective effects in experimental stroke. Rosiglitazone or 15d-PGJ2 were administered to rats 10 mins or 2 h after permanent middle cerebral artery occlusion (MCAO). Stroke outcome was evaluated by determination of infarct volume and assesment of neurological scores. Brains were collected for protein expression, gene array analyses and gene shift assays. Our results show that both compounds decrease MCAO-induced infarct size and improve neurological scores. At late times, the two compounds converge in the inhibition of MCAO-induced brain expression of inducible NO synthase and the matrix metalloproteinase 9. Interestingly, at early times, complementary DNA microarrays and gene shift assays show that different mechanisms are recruited. Analysis of early nuclear p65 and late cytosolic IκBα protein levels shows that both compounds inhibit nuclear factor-κB signalling, although at different levels. All these results suggest both PPARγ-dependent and independent pathways, and might be useful to design both therapeutic strategies and prognostic markers for stroke.
Introduction
Stroke is one of the leading causes of death and disability worldwide. In spite of this, its acute therapeutic management is almost limited to thrombolysis. As the inflammatory cascade triggered by the ischaemic injury in both occluded blood vessels and brain parenchyma is an important feature of the pathophysiological response to the ischaemic injury, antiinflammatory strategies might be a useful therapy for acute stroke treatment.
The compounds rosiglitazone (RSG) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) are known to exert antiinflammatory actions (rev. in Straus and Glass, 2001; Martens et al, 2002; Stumvoll, 2003). They are agonists of the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ; Willson et al, 2001; Berger and Moller, 2002), a ligand-dependent nuclear transcription factor belonging to the nuclear hormone receptor superfamily of nuclear receptors, that has been proposed to participate in a broad range of cellular functions that include adipocyte differentiation, glucose homeostasis, apoptosis and antiinflammatory actions (rev. in Willson et al, 2001; Berger and Moller, 2002). Rosiglitazone is a member of the thiazolidinedione (TZD) class of compounds used for its antidiabetic properties, which has shown a high selectivity for PPARγ with a Kd of 40 nmol/L, whereas 15d-PGJ2, a naturally occurring derivative of prostaglandin D2, is a high-affinity ligand for PPARγ and has been proposed as one of the natural agonists of this receptor.
Peroxisome proliferator-activated receptor-γ agonists have been shown to inhibit proinflammatory and inflammatory mediators (Jiang et al, 1998; Marx et al, 1998; Ricote et al, 1998) and to exert antiinflammatory effects in several settings, including the central nervous system (CNS) (rev. in Landreth and Heneka, 2001; Feinstein, 2003; Kielian and Drew, 2003). Indeed, both RSG and 15d-PGJ2 antagonise the expression of inflammatory mediators; however, these effects seem to be accomplished through both PPARγ receptor-dependent and -independent mechanisms (Castrillo et al, 2000; Li et al, 2000; Rossi et al, 2000; Straus et al, 2000; Chawla et al, 2001; Moore et al, 2001; Takata et al, 2002; Park et al, 2003; Pérez-Sala et al, 2003; Welch et al, 2003).
Regardless of the mechanism involved, the antiinflammatory actions of these compounds could be beneficial in stroke. We have therefore investigated the effects of RSG and 15d-PGJ2 on stroke outcome in a rodent model of cerebral ischaemia by permanent occlusion of the middle cerebral artery (MCAO).
Materials and methods
Materials
Rosiglitazone was from Calbiochem (San Diego, CA, USA) and 15d-PGJ2 was from Cayman Chemical (Ann Arbor, MI, USA). The rest of the reagents were from Sigma (Madrid, Spain), or as indicated in the text.
Animals
Adult male Fischer rats weighing 250 to 275 g were used. All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense (following EU directives 86/609/CEE and 2003/65/CE). Rats were housed individually under standard conditions of temperature and humidity, and a 12 h light/dark cycle (lights on at 08:00), with free access to food and water.
Middle Cerebral Artery Occlusion (MCAO)
Animals were anaesthetised with 1% to 1.5% halothane in a mixture of 70% nitrogen/30% oxygen, and body temperature was maintained at 37°C using a heating pad throughout the surgery procedure and during postsurgery recovery. Permanent focal cerebral ischaemia was induced by occlusion of the ipsilateral middle cerebral artery (MCA) as described (De Cristóbal et al, 2001; Hurtado et al, 2003). Briefly, for the CCA ligature, a midline ventral cervical incision was made, and the CCA was isolated and permanently occluded with a silk ligature. For the MCA occlusion, a 1 cm incision perpendicular to the line connecting the lateral canthus of the left eye and the external auditory canal was made to expose and retract the temporalis muscle. A 2-mm burr hole was drilled and the MCA was exposed by cutting and retracting the dura. The MCA was elevated and cauterised. Rats in which the MCA was exposed but not occluded served as sham-operated controls (SHAM). After surgery, subjects were returned to their cages and allowed free access to water and food.
Experimental Groups
Several groups were used for determinations of infarct size, neurological assessment, and determination of biochemical and molecular parameters: MCAO 10 mins before an intraperitoneal injection of saline (MCAO; n = 23) or dimethyl sulfoxide (DMSO) (vehicle; 10% in saline; n = 2l), and MCAO 10 mins before an intraperitoneal injection of either RSG 1 to 3 mg/kg (MCAO + RSG1/3; n = 21) or 15d-PGJ2 1 mg/kg (MCAO + 15d-PGJ2; n = 21). For infarct size and neurological assesment, additional groups of animals were administered either 3 mg/kg RSG or 1 mg/kg 15d-PGJ2 2 h after the occlusion, or a combination of both 1 mg/kg RSG and 1 mg/kg 15d-PGJ2 10 mins after the occlusion (n = 6). An additional control group consisted of SHAM-operated animals 10 mins before an intraperitoneal injection of saline (SHAM; n = 18). Injection volume was ≤ 0.5 mL/250 g body weight.
Infarct Size
Infarct outcome was assessed at 48 h, a time at which we have observed that infarct lesions have reached their maximal degree (unpublished results). Animals were killed, brains were removed, and a series of 2 mm of coronal slices were obtained and stained in 1% 2,3,5-triphenyl-tetrazolium chloride (TTC) in 0.1 mol/L phosphate buffer, and infarct size was determined as described (Mackensen et al, 2001).
Neurological Characterisation After Middle Cerebral Artery Occlusion
Before the killing, a neurological test was made by two unaware independent observers, as described previously (Hunter et al, 2000). According to the test, animals were scored as: 0 points – no deficit; 1 point – failure to extend right forepaw fully; 2 points – decreased grip of right forelimb while tail pulled; 3 points – spontaneous circling or walking to the contralateral side; 4 points – walks only when stimulated with depressed level of consciousness; 5 points – unresponsive to stimulation.
Protein Expression in Brain Homogenates
Brain cortical and striatal tissue was collected from the infarcted and surrounding areas. For determination of inducible NO synthase (iNOS), cyclooxygenase (COX)-2 and matrix metalloproteinase 9 (MMP-9) protein expression levels, rats were killed 18 h after MCAO. Brain areas corresponding to the infarct and surrounding areas were collected. Brain samples were homogenised by sonication for 10 secs at 4°C in 4 volumes of homogenisation buffer containing 320 mmol/L sucrose, 1 mmol/L DL-dithiothreitol, 10 μg/mL leupeptin, 10 μg/mL soybean trypsin inhibitor, 2 μg/mL aprotinin and 50 mmol/L Tris, brought to pH 7.0 at 20°C with HCl. The homogenate was centrifuged at 4°C at 12,000 g for 20 mins and the pellet was discarded.
Protein Expression in Cytosolic and Nuclear Extracts
Cytosolic and nuclear extracts were prepared as described (Cárdenas et al, 2000). Determination of p65 was performed in nuclei obtained from brains of rats killed 1 h after MCAO, whereas, for PPARγ, rats were killed 2 and 18 h after MCAO. For IκBα, cytosolic extracts were obtained from brains of rats killed 5 h after MCAO.
Gene Microarray Analysis
Rats were decapitated 5 h after MCAO and ipsilateral cortices were used immediately for total RNA extraction with the TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA). The RNA was then cleaned (Rneasy Mini Kit, Qiagen, Valencia, CA, USA) and analysed in a 2100 Bioanalyzer following the instructions of the manufacturer of the array (Affymetrix). Fragmentation of biotin-labelled complementary RNA (cRNA) was performed at 94°C using 40 mmol/L Tris-acetate, pH 8.1, 100 mmol/L KOAc and 30 mmol/L MgOAc. Samples were hybridised to Affymetrix Genechip Test3 Array and then to Affymetrix GeneChip Rat expression probe array 230A. The data were analysed with the GeneChip Analysis Suite software.
Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction (QRT-PCR)
The mRNA levels of selected genes that were found to be modified in the array (see Supplemental material) were quantified by real-time QRT-PCR amplification. Total RNA was extracted from isolated brain slices of four animals, using Trizol solution (Gibco BRL, NY, USA). RNA quantity was determined spectrophotometrically and the purity was confirmed by the relative absorbance at 260 nm versus 280 nm. RNA samples were stored in diethyl pyrocarbonate-treated water at −80°C. Complementary DNA (cDNA) synthesis was performed with 4 μg of total RNA and M-MLU reverse transcriptase (Invitrogen) in accordance with the manufacturer's instruction, using oligo-dT as primer. Complementary DNA was stored at −20°C.
Specific primer sequences can be found in Supplemental Table 1. Primers and probes against the selected genes of interest were designed from rat sequences available in GenBank using Primer Express Software (PE Applied Biosystems). Polymerase chain reaction amplification and quantification were performed using SyBr Green PCR kit (PE Applied Biosystem) in a model 7700 Sequence detector (PE Applied Biosystems), and the results were analysed using sodium dodecyl sulphate (SDS) 1.9 Software (PE Applied Biosystems). The absence of genomic contamination in the RNA samples and of DNA contamination in RNA mimics was confirmed with reverse transcriptase-negative controls for each experiment.
PPARγ ligands improve the neurological status of rats exposed to MCAO
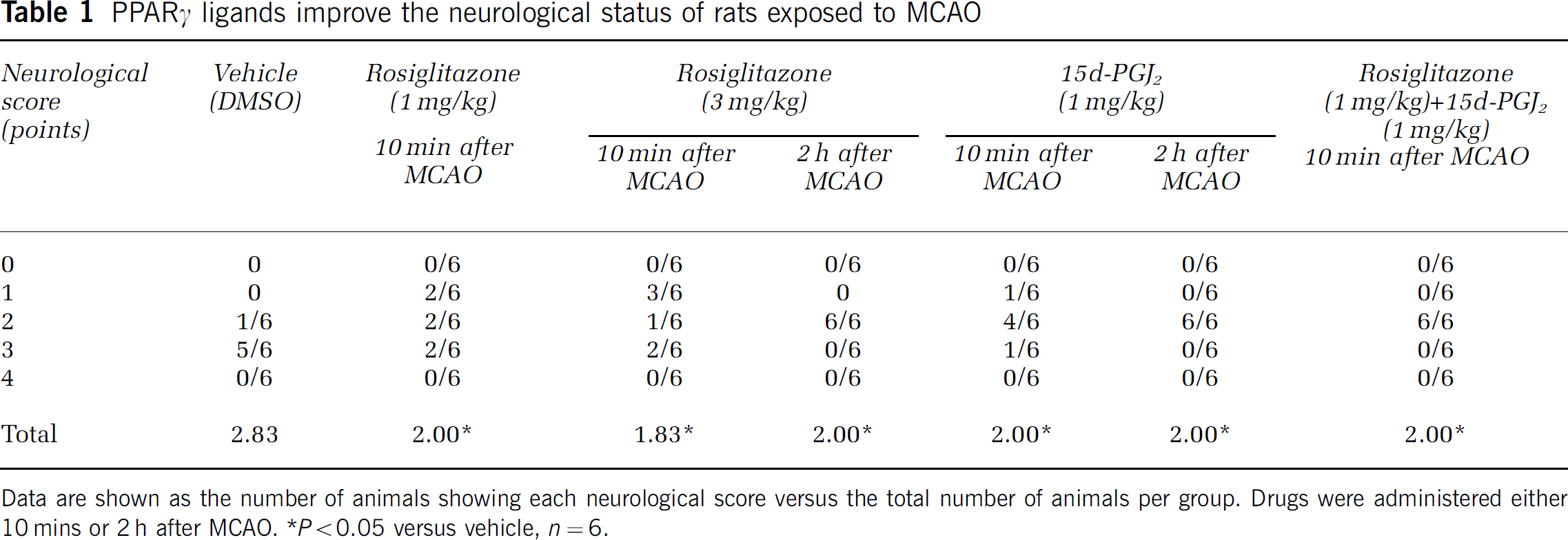
Data are shown as the number of animals showing each neurological score versus the total number of animals per group. Drugs were administered either 10 mins or 2 h after MCAO.
P < 0.05 versus vehicle, n = 6.
Western Blot Analysis
Samples containing 20 μg of protein were loaded and the proteins size-separated in 7% to 10% SDS-polyacrylamide gel electrophoresis (110 mA). Proteins were blotted onto a polyvinylidene fluoride (PVDF) membrane (Hybond™-P, Amersham Biosciences Europe GmbH, Friburg, Germany) and incubated with specific primary antibodies against PPARγ (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1000), iNOS (Santa Cruz; 1:500), COX-2 (Santa Cruz, 1:1000), MMP-9 (Chemicon, Temecula, CA, USA; 1:2000), p65 (Santa Cruz; 1:1000) and IκBα (Santa Cruz; 1:1000). Proteins recognised by the antibody were revealed by ECL™-kit following the manufacturers' instructions (Amersham Biosciences Europe GmbH, Friburg, Germany). β-Actin and Sp1 levels were used as loading controls for total-cytosolic and nuclear protein expression, respectively.
Gel Mobility Shift Assays
A double-stranded oligonucleotide corresponding to PPAR response element (PPRE), composed of GGGGACCAGGA-CAAAGGTCA, which is located in the 5′ upstream of the acyl-CoA oxidase gene (Tugwood et al, 1992), was used as probe. Labelled probe (5 × 105cpm) was incubated with nuclear extracts (5 μg) in a 20-μL reaction consisting of 2 μg poly dI:dC, 10 mmol/L Hepes, pH 7.9, 50 mmol/L KCl, 5% glycerol, 2% Ficoll, 0.05% NP-40, and 1 μL rabbit serum. Reactions were incubated for 10 mins on ice, radiolabeled probe was added, and reaction was continued for another 10 mins on ice. Reactions were run on a 5% nondenaturing Polyacrylamide gel in 0.5 × TBE buffer.
NO−x (NO−2 and NO−3) Assay
NO release was estimated from the amounts of nitrite (NO−2) and nitrate (NO−3) in brain homogenates. NO−3 was calculated by first reducing NO−3 into NO−2 in the presence of Cd, and NO−2 was determined by a colorimetric assay based on the Griess reaction as described (Cortas and Wakid, 1990).
Statistical Analysis
Results are expressed as mean ± s.e.m. of the indicated number of experiments; statistical analysis involved one-way analysis of variance (ANOVA, or the Kruskal–Wallis test when the data were not normally distributed), followed by individual comparisons of means (Student–Newman–Keuls or Dunńs method, when the data were not normally distributed). P < 0.05 was considered statistically significant.
Results
Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2 on Infarct Outcome After Permanent Middle Cerebral Artery Occlusion
The administration of RSG or 15d-PGJ2 10 mins after the occlusion caused a decrease in MCAO-induced infarct size determined 48 h after the ischaemic injury (Figure 1). In addition, the animals treated with these compounds also showed better scores in a neurological assessment scale after MCAO (Table 1). When these drugs were administered 2 h after the occlusion, the decrease in infarct size persisted (94.9 ± 0.8 or 101.8 ± 2.9 mm3 after 3 mg/kg RSG or 1 mg/kg 15d-PGJ2, respectively, n = 6). When RSG (1 mg/kg) and 15d-PGJ2 (1 mg/kg) were coadministered 10 mins after the occlusion, infarct size was further decreased (77.4 ± 4.8 mm3, n = 6, P < 0.05 versus MCAO + RSG1 or MCAO + 15d-PGJ2).
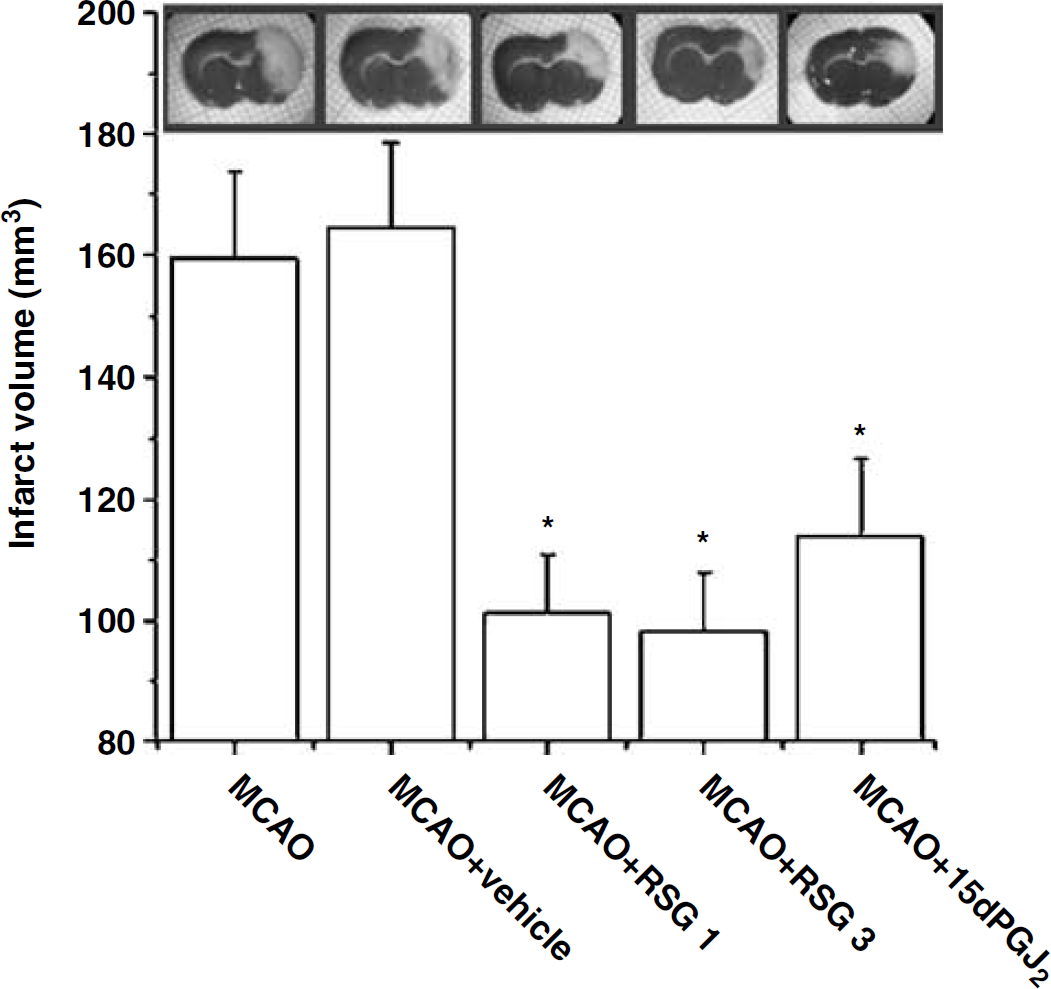
Neuroprotective effect of RSG and 15d-PGJ2 after experimental stroke. The TZD RSG (1 to 3 mg/kg) and the cyclopentenone prostaglandin (15d-PGJ2, 1 mg/kg) reduce infarct volume after permanent MCAO. Data are mean ± s.e.m., n = 6 to 8, *P < 0.05 versus MCAO (see Materials and methods for details). Photographs of brain slices from representative experiments.
Expression of Inducible NO Synthase and cyclooxygenase-2, and Nitrite/Nitrate (NO−x) Levels After Middle Cerebral Artery Occlusion. Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
Expression of the enzymes iNOS and COX-2 was studied, as both are described to mediate inflammatory damage after stroke (del Zoppo et al, 2000). Occlusion of the MCA caused the expression of the inflammatory enzymes iNOS and COX-2 in rat brain, as shown by the levels of these proteins found 18 h after the ischaemic insult (Figures 2A and 2B). The administration of either RSG or 15d-PGJ2 inhibited MCAO-induced expression of iNOS, but did not affect COX-2 levels at the time examined (Figures 2A and 2B).
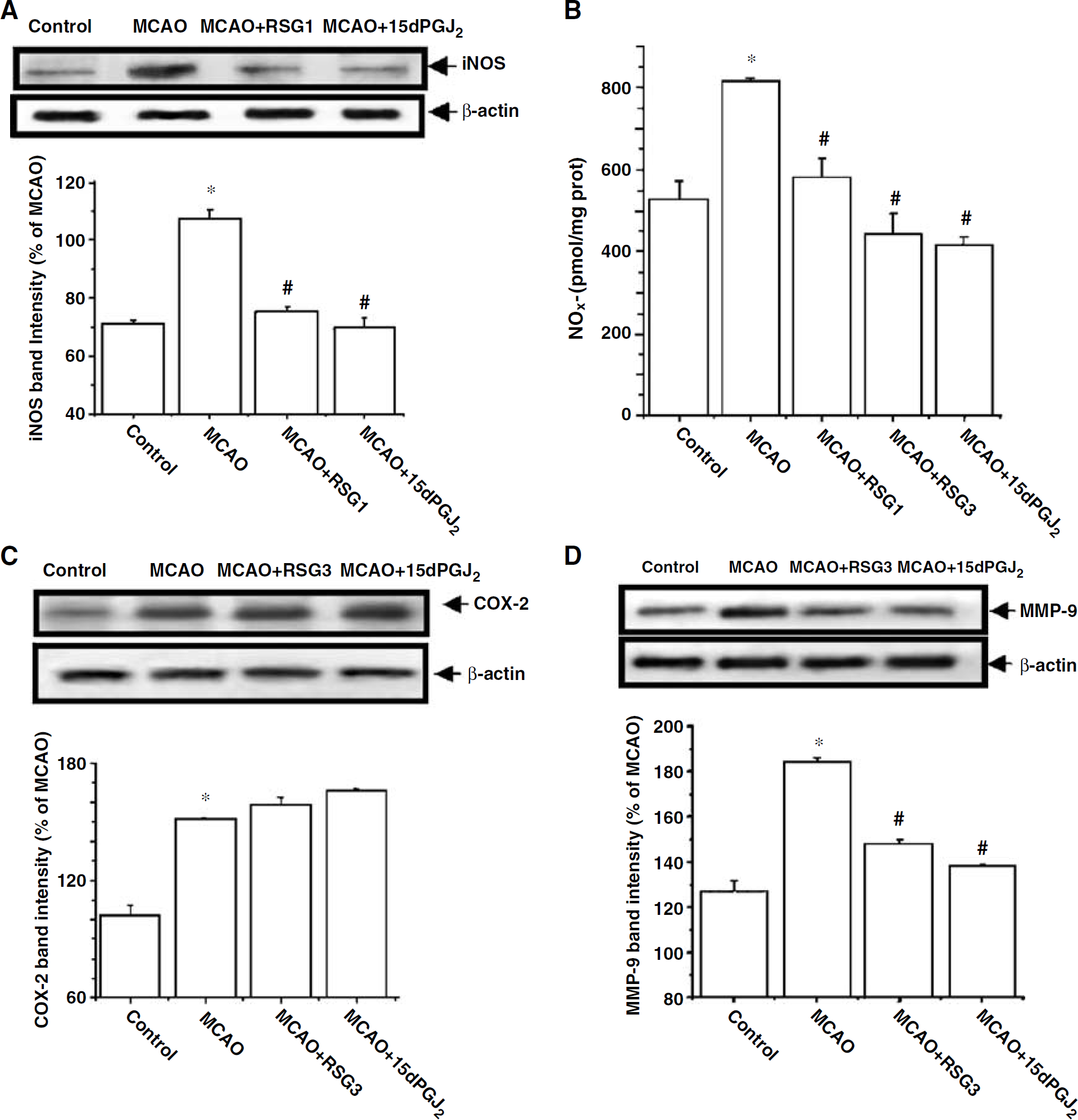
Rosiglitazone and 15d-PGJ2 inhibit MCAO-induced iNOS and MMP-9, but not COX-2, expression after experimental stroke. Effect of RSG (1 to 3 mg/kg) and 15d-PGJ2 (1 mg/kg) on iNOS (
Levels of NO−x 18 h after MCAO were determined as an indicator of NO synthesis. Permanent MCAO caused an increase in brain NO−x, which was decreased by the two compounds tested (Figure 2C).
Expression of Matrix metalloproteinase 9 After Middle Cerebral Artery Occlusion. Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
Occlusion of the MCA caused an increase in the levels of MMP-9, a matrix metalloproteinase which is induced and mediates damage caused by proinflammatory stimuli, including cerebral ischaemia (Mun-Bryce and Rosenberg, 1998). Both RSG and 15d-PGJ2 decreased the levels of this MMP after experimental stroke (Figure 2D).
Nuclear Levels of Nuclear Factor-κB (NF-κB) after Middle Cerebral Artery Occlusion. Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
Nuclear factor-κB is a transcription factor that plays a key role in the expression of a variety of genes involved in inflammatory responses (Gosh et al, 1998). As a sign of its activation, the nuclear levels of its subunit p65 were determined 1 h after MCAO. Experimental ischaemia caused activation of NF-κB, as revealed by the nuclear translocation of the NF-κB subunit p65. The cyclopentenone prostaglandin 15d-PGJ2 decreased MCAO-induced p65 translocation. Conversely, RSG did not modify p65 nuclear levels after MCAO (Figure 3A).
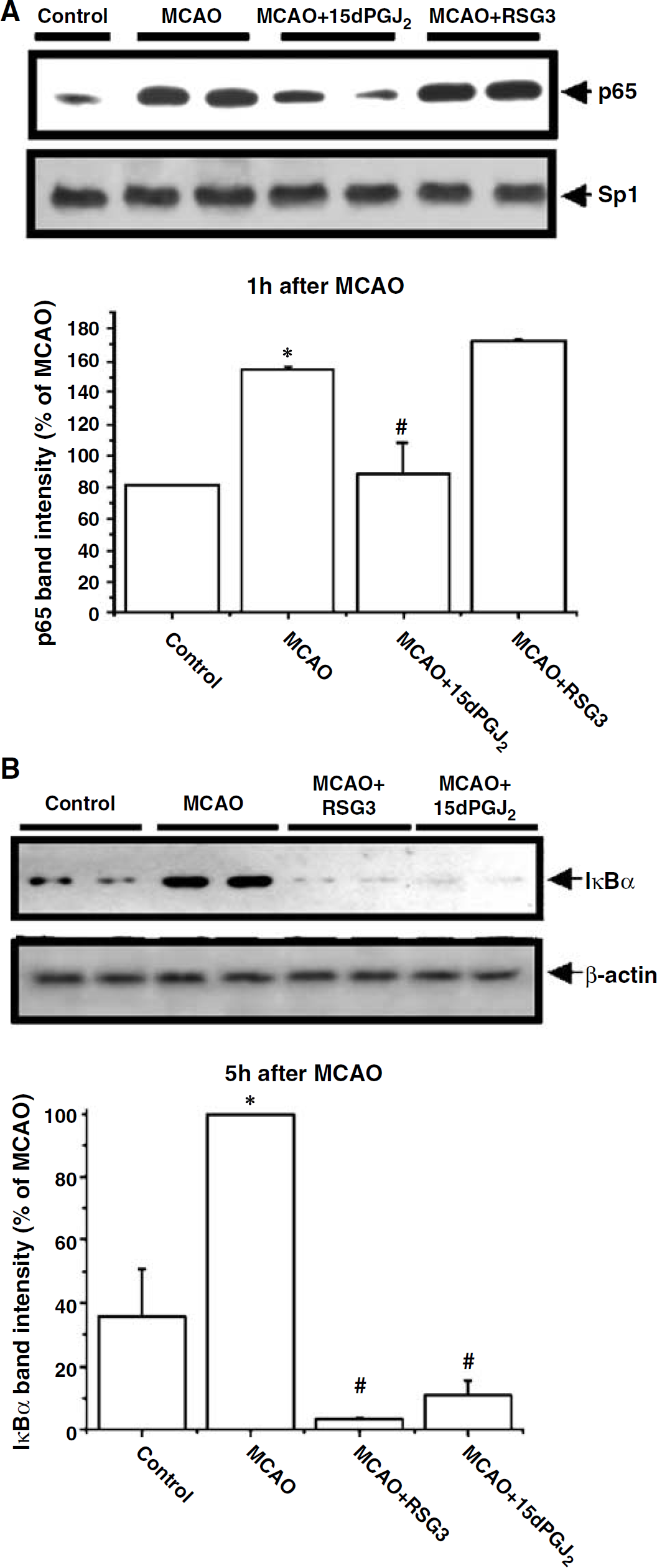
Effect of RSG and 15d-PGJ2 on NF-κB activation and transcriptional activity. Effect of RSG (3 mg/kg) and 15d-PGJ2 (1 mg/kg) on nuclear p65 levels at 1 h (
Cytosolic Levels of IκBα after Middle Cerebral Artery Occlusion. Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
As IκB itself is an NF-κB-dependent gene, late IκB levels were determined as an indicator of NF-κB transcriptional activity induced after its nuclear translocation. Cytosolic IκBα levels increased 5 h after the ischaemic insult when compared with control brains (Figure 3B). Both RSG and the cyclopentenone prostaglandin 15d-PGJ2 abolished the increase in IκBα levels induced 5 h after MCAO.
Expression of Peroxisome Proliferator-Activated Receptor-γ in Nuclear Extracts After Middle Cerebral Artery Occlusion. Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
Western blot analysis showed the presence of PPARγ in nuclear extracts obtained from control animals (Figure 4A). Middle cerebral artery occlusion did not significantly modify the expression of this receptor when measured 2 and 18 h after the ischaemic insult (Figure 4A). In the presence of either RSG (3 mg/kg) or 15d-PGJ2 (1 mg/kg), PPARγ levels were significantly increased 2 h after MCAO, but this increase was maintained at 18 h only in the presence of 15d-PGJ2 (Figure 4A).
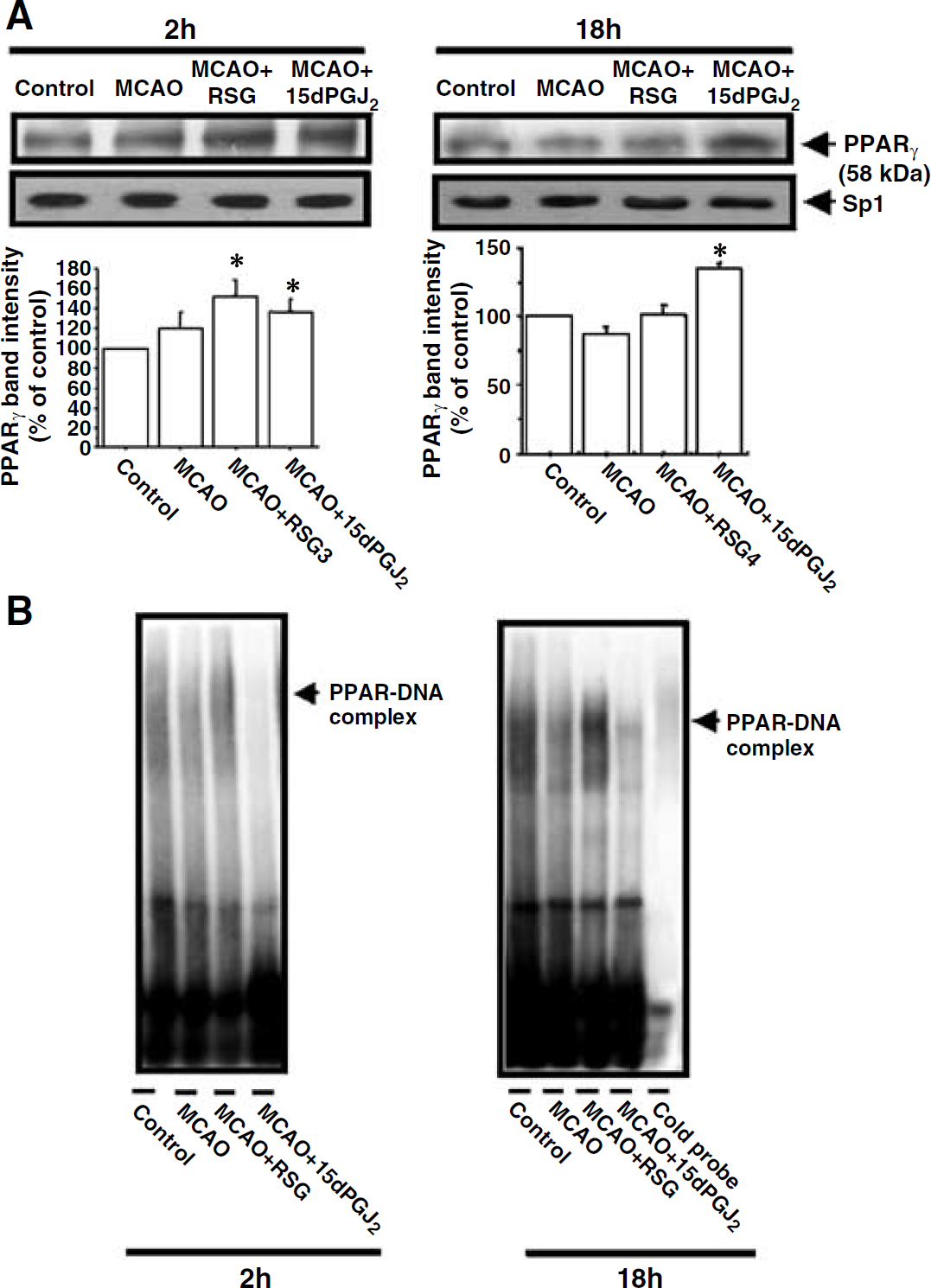
Effect of RSG and 15d-PGJ2 on the PPRE-binding activity of nuclear proteins. (
Effect of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2 on the Binding of Nuclear Proteins to the Peroxisome Proliferator-Activated Receptor Response Element
We studied by gel shift analysis whether RSG or 15d-PGJ2 increase the binding of PPAR isoforms to the PPRE. When studied 2 h after the MCAO, binding of nuclear elements to the probe containing the PPRE was essentially unaltered in most conditions tested, excluding nuclei from MCAO + 15d-PGJ2 animals, in which binding was inhibited (Figure 4B). At 18 h after the ischaemic insult, RSG (3 mg/kg) increased the amount of nuclear protein bound to the PPRE probe (Figure 4B), whereas 15d-PGJ2 (1 mg/kg) virtually abrogated the gel retardation band associated with PPRE/nuclear proteins (Figure 4B). The binding was specific, as addition of excess amount of cold oligonucleotide blocked the binding (Figure 4B).
DNA Microarray Assesment of Global Gene Expression After Middle Cerebral Artery Occlusion. Effects of Rosiglitazone and 15-Deoxy-Δ12,14-Prostaglandin J2
At 5 h after MCAO, RNA was extracted from the brain of animals treated with RSG (3 mg/kg), 15d-PGJ2 (1 mg/kg) or vehicle, and DNA microarray analyses were performed. This short time frame of treatment was chosen to identify predominantly direct transcriptional responses.
Regarding the transcripts corresponding to the inflammatory proteins studied in this work, at short times, MCAO did not affect iNOS or MMP-9 genes, the expression of which has been described to occur at later times after permanent focal ischaemia (Iadecola et al, 1995; Romanic et al, 1998) (Table 2). Permanent MCAO did cause the upregulation of IκBα, an effect that was inhibited by both RSG and 15d-PGJ2 (Table 2). Similarly, COX-2 mRNA was increased after MCAO, although it was differentially affected by the two drugs tested (Table 2).
Effect of rosiglitazone or 15d-PGJ2 on several transcripts 5 h after MCAO
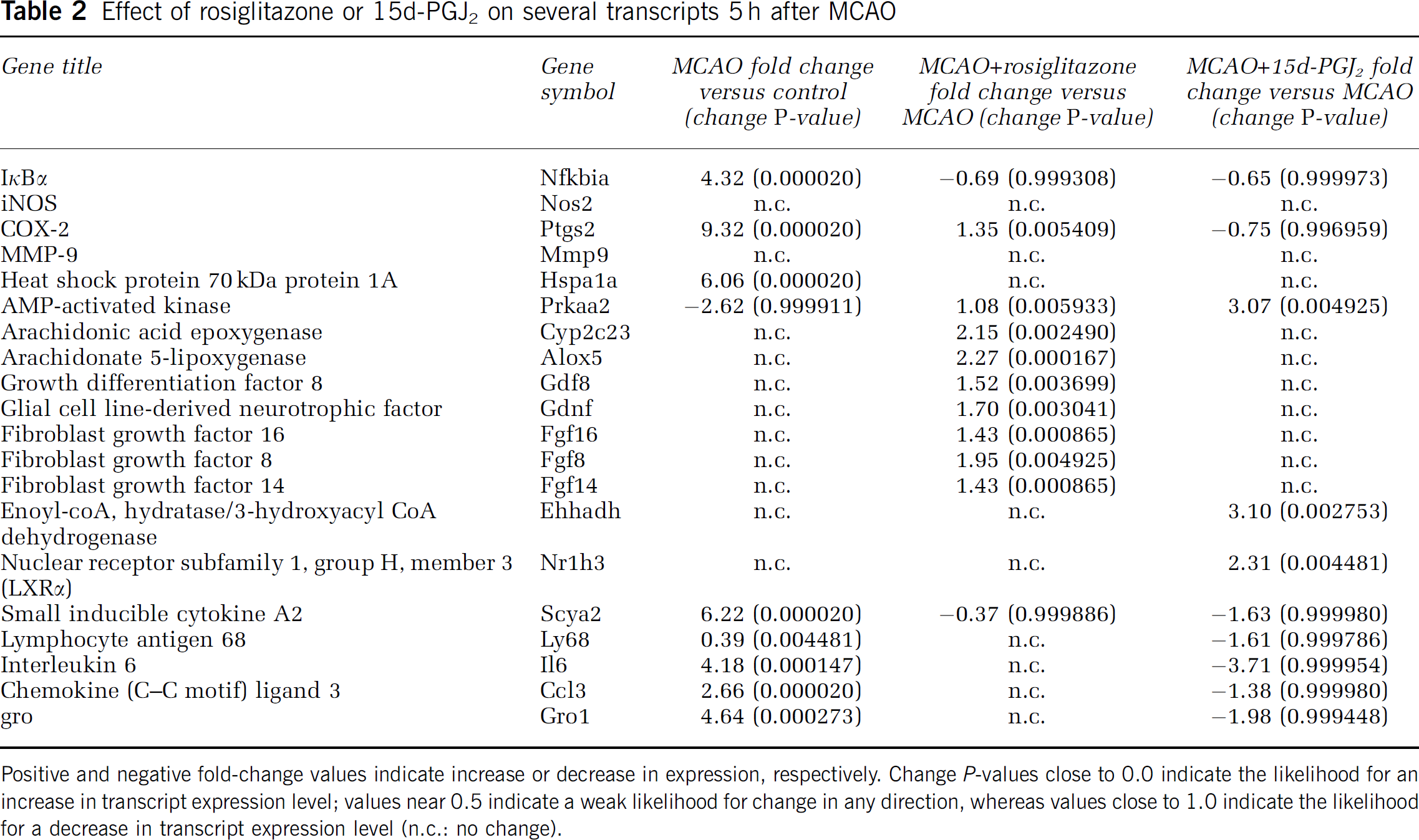
Positive and negative fold-change values indicate increase or decrease in expression, respectively. Change P-values close to 0.0 indicate the likelihood for an increase in transcript expression level; values near 0.5 indicate a weak likelihood for change in any direction, whereas values close to 1.0 indicate the likelihood for a decrease in transcript expression level (n.c.: no change).
In addition, the administration of RSG caused upregulation of several genes when compared with the MCAO group (Table 2), such as 5-lipoxygenase -5-LOX-, epoxygenase and several growth factors (fibroblast growth factors, growth differentiation factor 8, glial cell line-derived neurotrophic factor). However, the administration of 15d-PGJ2 caused upregulation of some genes when compared with the MCAO group, such as the nuclear receptor 1H3 (NR1H3, LXRα), and downregulation of several inflammmation-related genes such as inflammmatory cytokines and molecules involved in neutrophil Chemotaxis (interleukin-6, gro, small inducible cytokine A2, chemokine ligand 3) (Table 2).
Confirmation of Change in Expression
To validate the microarray results, we performed real-time RT-PCR on four of the differentially expressed genes (Supplemental Table 1). They were chosen because of their clear signal in the microarray and/or its modulation in the different experimental groups. There was a statistically significant correlation between these two different assays, with a P-value < 0.05, both in the up- and downregulated genes (Supplemental Figure 1).
Discussion
We hereby show that, albeit recruiting different signalling pathways, the TZD RSG and the cyclopentenone prostaglandin 15d-PGJ2 significantly ameliorate infarct outcome after MCAO in rats.
Both compounds significantly ameliorated stroke outcome, as shown by a remarkable reduction of the infarct volume and a substantial recovery in the neurological deficit induced by the MCAO, when administered either 10 mins or 2 h after the infarct, thus implying that these drugs might be therapeutically useful for stroke treatment. Some of these compounds could exhibit a limited CNS access in a healthy brain, but this is overcome due to the disruption of the blood–brain barrier that occurs in cerebral ischaemia (rev. in Rubin and Staddon, 1999). Proapoptotic actions of 15d-PGJ2 have been reported in cancer cells and, in CNS, in LPS-activated microglial cells and cortical neurons (Bernardo et al, 2003; Rohn et al, 2001). Although the microarray data did not show any major modification in apoptosis-related mediators after treatment with any of the compounds studied, the activation of the apoptotic pathway cannot be precluded; moreover, an action of 15d-PGJ2 at this level could be beneficial for resolution of inflammation via induction of apoptosis of activated microglia or infiltrated macrophages, as we have previously suggested (Hortelano et al, 2000).
In the search for mechanisms involved in this neuroprotective effect, we found that, at late times, both compounds inhibit both MCAO-induced iNOS expression and increase in the levels of NO−2 and NO−3, the stable metabolites of NO, in rat brain. Since iNOS mediates cytotoxicity in many cell systems including the ischaemic brain (rev. in del Zoppo et al, 2000), its inhibition may explain at least part of the neuroprotective effect of these compounds. Inhibition of iNOS expression with PPARγ ligands has been similarly described in CNS cells after an inflammatory challenge with lipopolysaccharide (Petrova et al, 1999; Bernardo et al, 2000; Heneka et al, 2000; Kim et al, 2002) and, recently, after experimental stroke (Sundararajan et al, 2005).
The inducible cyclooxygenase COX-2 also participates in the ischaemic inflammatory cascade (Planas et al, 1995; Collaco-Moraes et al, 1996; Ohtsuki et al, 1996). Our results confirm that COX-2 expression is induced in rat brain, but neither RSG nor 15d-PGJ2 affected its levels at late times. However, the microarray results indicate that, at earlier times, MCAO-induced increase in COX-2 transcript is partly increased or decreased, respectively, by RSG or 15d-PGJ2, effects that are likely to be vanished at the time that protein expression levels are determined, 18 h after MCAO, and reflect the controversy in the literature regarding the actions of these molecules. Indeed, whereas inhibition of COX-2 expression by PPARγ ligands has been shown (Inoue et al, 2000; Subbaramaiah et al, 2001), other reports also show that PPARγ activation does not affect (Staels et al, 1998), or even increases, the expression of COX-2 (Pontsler et al, 2002; Pang et al, 2003).
The MMP-9 (gelatinase B) is another inflammatory mediator that contributes to ischaemic cerebral damage (Mun-Bryce and Rosenberg, 1998), because it participates in extracellular matrix degradation (Asahi et al, 2001); we and others have shown that MMP-9 participates in the haemorrhagic transformation in acute ischaemic stroke in humans (Montaner et al, 2001; Castellanos et al, 2003). Our data confirm that experimental stroke increases the expression of this metalloproteinase. More importantly, both RSG and 15d-PGJ2 inhibit MCAO-induced MMP-9 expression. It has been reported that increased NO production is necessary for MMP-9 activation (Gu et al, 2002; Marcet-Palacios et al, 2003; Mayers et al, 2003), that might constitute a potential extracellular proteolysis pathway to neuronal cell death in cerebral ischaemia. Given the relevance of both interrelated signalling for cell damage, the dual inhibition of iNOS and MMP-9 expression after MCAO is likely to be one of the main mechanisms responsible for the neuroprotective effect of these compounds. However, we cannot discard that the reduced damage caused by these compounds on other targets may indirectly affect the levels of these mediators. The microarray studies did not show any change in iNOS or MMP-9 mRNA levels 5 h after MCAO, in agreement with previous findings showing that, whereas COX-2 mRNA is detectable 6 h after a permanent MCAO (Nogawa et al, 1997), iNOS mRNA and MMP-9 protein have not been detected before 12 h after permanent occlusions (Iadecola et al, 1995; Romanic et al, 1998). In contrast with our results, a report has described the proinflammatory actions of TZDs on epithelial cells (Desmet et al, 2005).
As commented above, both drugs have been shown to act as PPARγ agonists. In agreement with early work (Kainu et al, 1994; Cullingford et al, 1998), PPARγ receptor is present in rat brain in the different experimental conditions of our work. Interestingly, 15d-PGJ2 causes a sustained increase in the levels of this receptor after MCAO, which deserves further studies.
There is a great deal of controversy on the role of PPARγ in explaining the antiinflammatory actions of RSG and 15d-PGJ2. It has been proposed that transrepressional inhibition of the transcriptional activity of NF-κB, a transcription factor that plays a key role in the induction of inflammatory response genes such as iNOS and COX-2 (Gosh et al, 1998), mediates the antiinflammatory actions of PPARγ agonists (Jiang et al, 1998; Ricote et al, 1998; Li et al, 2000). Alternatively, it has been shown that the antiinflammatory actions of PPARγ agonists occur via PPARγ-independent mechanisms (rev. in Berger and Moller, 2002). In this context, we and others have shown that 15d-PGJ2 is able to inhibit NF-κB activation in a PPARγ-independent manner (Castrillo et al, 2000; Rossi et al, 2000; Straus et al, 2000) by obstructing its nuclear translocation. This is confirmed by our results showing that MCAO-induced NF-κB activation, shown by the translocation of p65 to the nucleus at early times, 1 h, is reduced by 15d-PGJ2. On the contrary, RSG did not alter p65 translocation, indicating that its neuroprotective actions are not exerted by a direct inhibition of NF-κB nuclear translocation, although this experiment does not discard whether it affects NF-κB transcriptional activity. To ascertain this point, we studied the levels of the inhibitory protein IκBα, which is rapidly induced after NF-κB activation (Le Bail et al, 1993; Sun et al, 1993), thus constituting an indicator of NF-κB transcriptional activity. Our results show that IκBα is remarkably increased in the brain of MCAO-exposed animals 5 h after the occlusion, indicating prior NF-κB activation, and that this effect is completely blocked by the molecules tested, showing that both inhibit NF-κB signalling, although at different steps. Microarray data obtained at 5 h exhibit a similar profile for IκBα, although the effect on the transcript is less robust, a situation which will likely correspond to IκBα protein levels recovery at later times. Indeed, IκBα is expected to return to normal levels to maintain normal functioning of NF-κB, and also because IκBα itself plays an important role in ischaemic penumbra (Aronowski et al, 2000).
To determine whether RSG or 15d-PGJ2 activates PPARγ in our setting, gel shift analyses were performed. Rosiglitazone increased the amount of protein bound to a PPRE mainly at the latest time studied, strongly suggesting that PPAR is activated and may mediate RSG-induced effects. A novel finding is that 15d-PGJ2, at the concentration tested, remarkably inhibits the binding of nuclear protein to the PPRE, indicating that, at the times studied, PPARγ activation might not be involved in its neuroprotective actions in this model. This inhibition could be because this prostaglandin is able to modify covalently critical cysteine residues in the DNA-binding domains of some transcription factors (Straus et al, 2000; Pérez-Sala et al, 2003). In any case, the differences in binding are not due to different levels of receptor in each group, as shown by our results, indicating that PPARγ is present at the times studied.
A global gene microarray survey was performed to explore gene expression responses after RSG and 15d-PGJ2. The study was performed 5 h after the MCAO, in an attempt to identify predominantly direct transcriptional responses elicited by these compounds. To our knowledge, most studies on gene expression induced by PPARγ agonists have been performed at later times, usually longer than 24 h. Interestingly, although the inhibitory effects of RSG and 15d-PGJ2 on NF-κB transcriptional activity are confirmed as indicated by reduced IκBα levels, other results regarding gene expression after MCAO were unrelated, thus confirming that the initial pathways recruited by each compound are not fully overlapping. Of note, a novel finding of our work is the potent induction of 5-LOX and epoxygenase after RSG. Since lipoxygenase (LOX) and epoxygenase products are polyunsaturated fatty acids which are able to activate PPARγ and α receptors (Vanden Heuvel, 1999), our results suggest that RSG could function by inducing the synthesis of PPAR agonists, and poses an interesting hypothesis suggesting that RSG is an indirect PPAR agonist. Another novel finding is the potent induction of several growth factors caused by RSG, that may participate in repair processes of the damaged cerebral tissue.
However, treatment of MCAO animals with 15d-PGJ2 caused a very significant decrease in several inflammation-related genes at early times, such as cytokines (IL-6), and molecules involved in neutrophil Chemotaxis, a result which may explain at least part of the antiinflammatory actions of this prostaglandin by inhibiting local inflammation, as well as that derived from leukocyte infiltration. Interestingly, 15d-PGJ2 also caused a potent upregulation of the mRNA for the nuclear receptor LXRα (Nr1h3), the activation of which has been shown to antagonise NF-κB signalling and to have antiinflammatory activity in vivo (Joseph et al, 2003).
All these results strongly suggest that, at short times, the mechanisms of RSG and 15d-PGJ2 do not overlap; this is supported by the fact that, when coadministered in combination, there is a potentiation of the effects achieved separately. However, at later times, both drugs converge in the inhibition of noxious inflammatory proteins such as iNOS and MMP-9. All these findings open new lines of investigation on the mechanisms to explain the antiinflammatory actions of these compounds.
Recent data have reported protective effects of TZDs such as pioglitazone and troglitazone on transient focal ischaemia (Shimazu et al, 2005; Sundararajan et al, 2005). Our present report is the first evidence of the differential actions of two unrelated PPARγ agonists on cerebral ischaemia, and constitutes a thorough study of the mechanisms implicated.
Given that these compounds were administered after the onset of the ischaemic damage, our findings may possess therapeutic repercussions in the management of acute ischaemic stroke. Furthermore, our results imply that the endogenous levels of 15d-PGJ2 could serve as a helpful prognostic marker in stroke patients, as we have recently shown (Blanco et al, 2005). In summary, our results imply that, regardless of the signalling involved, these compounds exert potent neuroprotective actions and might be useful as therapies or markers in acute-stroke patients.
Footnotes
Acknowledgements
The authors are greatly indebted to Professor Federico Gago (Universidad de Alcalá, Spain) for his continuous support, to Dr Paloma Martín-Sanz and Dr Magali Silberman for help with gene shift assays, and to Mr Rafael Mayoral and Mr Alfonso Moyano for assistance in the QPCR assays.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.