
Abstract
The endoplasmic reticulum (ER), which plays important roles in apoptosis, is susceptible to oxidative stress. Because reactive oxygen species (ROS) are robustly produced in the ischemic brain, ER damage by ROS may be implicated in ischemic neuronal cell death. We induced global brain ischemia on wild-type and copper/zinc superoxide dismutase (SOD1) transgenic rats and compared ER stress and neuronal damage. Phosphorylated forms of eukaryotic initiation factor 2α (eIF2α) and RNA-dependent protein kinase-like ER eIF2α kinase (PERK), both of which play active roles in apoptosis, were increased in hippocampal CA1 neurons after ischemia but to a lesser degree in the transgenic animals. This finding, together with the finding that the transgenic animals showed decreased neuronal degeneration, indicates that oxidative ER damage is involved in ischemic neuronal cell death. To elucidate the mechanisms of ER damage by ROS, we analyzed glucose-regulated protein 78 (GRP78) binding with PERK and oxidative ER protein modification. The proteins were oxidatively modified and stagnated in the ER lumen, and GRP78 was detached from PERK by ischemia, all of which were attenuated by SOD1 overexpression. We propose that ROS attack and modify ER proteins and elicit ER stress response, which results in neuronal cell death.
Reactive oxygen species (ROS) are implicated in neuronal cell death in various situations. They play important roles in the brain after ischemia and reperfusion, in particular, because a number of events that predispose the formation of ROS takes place after ischemia and reperfusion (Chan, 1996). Indeed, there have been many reports showing that overproduction of ROS increased, but scavenging of them decreased, ischemic brain injury (Lewén et al., 2000). Meanwhile, the type of molecule (e.g., lipid, protein, nucleic acid) that ROS attack and to what degree are not fully understood. In addition, the organelle most susceptible to ROS attack is still not fully elucidated. This issue is under intense investigation, but of note is that a number of in vitro studies have demonstrated that the calcium pump in the endoplasmic reticulum (ER) is quite sensitive to oxidative damage (Dreher et al., 1995;Racay et al., 1995;Viner et al., 1996). Because the ER is one of the organelles that produces ROS, it may be an important target of ROS in neurons (Halliwell and Gutteridge, 1999).
Ample evidence suggests that ER damage is involved in neuronal cell death in various situations (Paschen and Doutheil, 1999). In the brain under ischemia, accumulation of unfolded proteins in the ER lumen (Hu et al., 2000), phosphorylation of eukaryotic initiation factor 2α (eIF2α) and RNA-dependent protein kinase-like ER eIF2α kinase (PERK) (Althausen et al., 2001;Kumar et al., 2001), inhibition of protein synthesis (Degracia et al., 2002;Nowak et al., 1985), and calcium depletion from the ER lumen (Paschen and Frandsen, 2001) all occur exclusively in the ischemic-vulnerable region, which strongly indicates the active role of the ER in neuronal cell death. Induction of the ER molecular chaperone after ischemia also suggests ER stress (Kitao et al., 2001). In contrast, the molecular mechanisms of ER damage in ischemic neurons have not been made clear. Taking all these facts together, we hypothesized that ROS produced by ischemia/reperfusion damaged the ER, thereby causing ischemic neuronal cell death.
Superoxide is the first of the ROS produced in cells. With superoxide dismutases (SODs), it is protonated to form hydrogen peroxide, which is subsequently scavenged by enzymes such as glutathione peroxidase and catalase (Halliwell and Gutteridge, 1999;McCord, 1985). Unlike immature cells, mature neurons possess enough glutathione peroxidase and catalase activity, and SODs are the critical enzymes that define oxidative neuronal damage (Ditelberg et al., 1996;Fullerton et al., 1998). Therefore, transgenic (Tg) overexpression or knockout of SOD genes are pertinent models for investigating the role of ROS in neuronal injury (Chan, 1994). This study was performed to provide evidence to support our hypothesis that ROS cause ER injury, which is implicated in ischemic neuronal cell death. Using copper/zinc SOD (SOD1) Tg and wild-type (Wt) rats, we induced transient global brain ischemia and compared ER stress and neuronal degeneration in hippocampal CA1 neurons, which are vulnerable to ischemic insults. Furthermore, to clarify the molecular mechanisms of oxidative damage to the ER, we observed oxidative protein modification in the ER of neurons after ischemia.
MATERIALS AND METHODS
SOD1 Tg rats
Heterozygous SOD1 Tg rats of the SOD1 with a Sprague-Dawley background, carrying human SOD1 genes with a four- to sixfold increase in copper/zinc SOD, were derived from the founder stock described previously (Chan et al., 1998). They were further bred with Wt Sprague-Dawley rats to generate heterozygous rats. The SOD1 Tg rats were identified by isoelectric focusing gel electrophoresis as described (Chan et al., 1998). There were no observable phenotypic differences, including in the cerebral vasculature, between the Tg rats and Wt littermates, as reported previously (Chan et al., 1998).
Transient global ischemia
Transient global brain ischemia was induced as previously reported, with slight modification (Smith et al., 1984). In brief, the animals were anesthetized with 1.5% isoflurane, 68.5% nitrous oxide, and 30% oxygen using a face mask, and the femoral artery was exposed and catheterized with a PE-50 catheter (427410; Becton Dickinson, San Diego, CA, U.S.A.) for continuous recording of arterial blood pressure. A midline neck skin incision was made, and the right jugular vein and both common carotid arteries were exposed. After intravenous injection of 150 IU/kg heparin, blood was quickly withdrawn via the jugular vein. When the MABP became 30 mmHg, both common carotid arteries were clamped with surgical clips. Blood pressure was maintained at 30 to 35 mmHg by withdrawing or infusing blood through the jugular vein during the ischemic period. After 5 minutes of ischemia, the clips were removed, and the blood was reinfused. Body temperature was monitored with a rectal probe and controlled at 37°C with a homeothermic blanket. Sham-operated animals underwent exposure of vessels without blood withdrawal or clamping of carotid arteries.
Assessments of neuronal degeneration
One and 4 hours and 1 and 2 days after 5 minutes of ischemia, the deeply anesthetized animals were perfused with 5 U/ml heparinized saline through the left cardiac ventricle until colorless fluid was obtained (n = 3 each time point, as well as the sham-operated group). Subsequently, 4% paraformaldehyde in 0.1 mol/L phosphate buffer was perfused, and the brains were removed. After postfixation in 4% paraformaldehyde, the brains were washed with water, dehydrated with ethanol, immersed in xylene, and embedded in paraffin. Coronal brain sections 5 μm thick at the hippocampal level were prepared and placed on glass slides. Neuronal cell damage was histologically evaluated by cresyl violet staining, as previously reported (Murakami et al., 1998). To investigate DNA fragmentation, we performed a terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) study (commercial kit S7100; Intergen, Norcross, GA, U.S.A.). Briefly, after pretreatment with 0.2% pepsin, the sections were incubated with terminal deoxynucleotidyl transferase and digoxigenin-conjugated nucleotides at 37°C for 1 hour. The sections were then incubated with peroxidase-conjugated antidigoxigenin antibody, and cells undergoing apoptosis were visualized using diaminobenzidine as a color substrate.
Immunohistochemical analysis
We performed immunohistochemical analysis for the phosphorylated forms of PERK and eIF2α both of which are ER stress markers (Harding et al., 1999;Rao et al., 2002). The sections were prepared as those for histologic and TUNEL studies. After deparaffinization, endogenous peroxidase activity was quenched with 30% methanol and 0.3% hydrogen peroxide in phosphate-buffered saline (PBS). The slides were boiled in citrate buffer with microwaves when necessary. After blocking nonspecific binding with 1% to 3% bovine serum albumin (BSA), the slides were incubated with primary antibodies at room temperature for 2 hours. The primary antibodies used and the dilutions for each were as follows: rabbit polyclonal anti-phospho-eIF2α antibody (#9721; Cell Signaling, Beverley, MA, U.S.A.) at 1:100 and rabbit polyclonal anti-phospho-PERK antibody (#3191; Cell Signaling) at 1:100. The primary antibodies used were the same as those used in our previous report (Hayashi et al., 2003). The slides were washed and then incubated with biotinylated mouse monoclonal anti-rabbit immunoglobulin G (IgG) (B3275; Sigma, St. Louis, MO, U.S.A.) at a 1:200 dilution. They were subsequently incubated with avidin-biotin-peroxidase complex (PK-6100; Vector Laboratories, Burlingame, CA, U.S.A.) for 30 minutes and then developed using vector VIP peroxidase substrate (SK-4600; Vector Laboratories). Methyl green was used for counterstaining. To ascertain specific binding of the antibody, a set of sections was stained in a similar way without the primary antibody. Furthermore, a set of sections was reacted with another anti-phospho-eIF2α antibody (#07-149; Upstate Biotechnology, Charlottesville, VA, U.S.A.) to confirm the specificity of the result.
Double fluorescent study of superoxide production and PERK phosphorylation
The production of superoxide anions during cerebral ischemia was investigated by in situ detection of oxidized hydroethidine (HEt), as described previously (Sugawara et al., 2002). HEt (D-11347; Molecular Probes, Eugene, OR, U.S.A.) is taken up by living cells and oxidized specifically by superoxide to a red fluorescent dye, ethidium (Bindokas et al., 1996). HEt solution (1 mL; 1 mg/mL in 1% dimethylsulfoxide with saline) was administered intravenously 15 minutes before ischemia induction. One hour after ischemia, the animals were deeply anesthetized, and the brains were perfused with heparinized saline and 4% paraformaldehyde in 0.1 mol/L phosphate buffer (n = 3). In the sham-operated animals, HEt was administered 75 minutes before the animals were perfused (n = 2). After postfixation in 4% paraformaldehyde, the brains were cut on a vibratome into slices 50 μm thick at the level of the hippocampus and placed on glass slides. Using these brain slices, we performed immunohistochemistry for the phosphorylated form of PERK. In brief, after incubation with primary and then secondary antibodies as described above, they were incubated with avidin-conjugated fluorescein isothiocyanate at a dilution of 1:40 (A-2001; Vector Laboratories) and covered with VECTASHIELD mounting medium with 4,6 diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Fluorescence was observed at excitation of 495, 510, and 360 nm and emission of greater than 515, 580, and 460 nm for fluorescein isothiocyanate, ethidium, and DAPI, respectively.
Western blot analysis
We performed Western blot analysis for the phosphorylated form of PERK, total PERK, and the phosphorylated form of eIF2α. The animals were deeply anesthetized 1 and 4 hours and 1 and 2 days after transient ischemia, and the CA1 subregion of the hippocampus was quickly removed (n = 3 each time point, as well as the sham-operated group). For protein extraction, tissue was homogenized with approximately seven volumes of protein extraction buffer (20 mmol/L HEPES potassium hydroxide [pH 7.5], 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L EGTA, 0.7% protease inhibitor cocktail [P8340; Sigma], 1% phosphatase inhibitor cocktails [P2850 and P5726; Sigma]). The homogenate was centrifuged at 10,000 g for 15 minutes at 4°C, and the supernatant was used for this study. Assays to determine the protein concentration were performed with comparison to a known concentration of BSA with the use of a kit (23227; Pierce, Rockford, IL, U.S.A.). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to previous reports (Hayashi et al., 1997;Noshita et al., 2001). In brief, the lysate equivalent of 5 μg of protein from each brain was run on the gel for 120 minutes at 120 V together with a biotinylated protein ladder size marker (#7727; Cell Signaling). In addition, positive controls were obtained by exposing rat hippocampal cultured cells (Palmer et al., 1999) to an ER toxin, dithiothreitol (DTT), which disturbs ER function and elicits strong ER stress responses (Harding et al., 2000). The protein on the gel was subsequently transferred to a polyvinylidene difluoride membrane (LC2002; Invitrogen, Carlsbad, CA, U.S.A.) in a buffer containing methanol, glycine, Tris base, and SDS. After the transfer, the membrane was placed in 5% to 10% powdered milk in PBS with 0.1% Tween 20 for 1 hour to block nonspecific binding and was then incubated with primary antibodies for 12 hours at 4°C. The primary antibodies used and each dilution were as follows: rabbit polyclonal anti-phospho-eIF2α antibody (#9721; Cell Signaling) at 1:2500, rabbit polyclonal anti-phospho-PERK antibody (#3191; Cell Signaling) at 1:2000, and goat polyclonal anti-total-PERK antibody (sc-9477; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) at 1:1000. The anti-phospho-eIF2α and anti-phospho-PERK antibodies are the same as those used in our previous report (Hayashi et al., 2003). After washing, the membrane was incubated with horseradish peroxidase conjugated anti-rabbit IgG (7074; Cell Signaling) or horseradish peroxidase conjugated anti-goat IgG (PI-9500; Vector Laboratories), together with horseradish peroxidase conjugated anti-biotin antibody (#7727; Cell Signaling). Then the signal was detected with a chemiluminescent kit (RPN2132; Amersham).
Immunoprecipitation study
We did a coimmunoprecipitation study to investigate the change in glucose-regulated protein 78 (GRP78) binding with PERK. GRP78 binds with PERK and inhibits its phosphorylation, thereby inhibiting ER stress-induced cell death (Bertolotti et al., 2000;Yu et al., 1999). The protein extract was prepared in the same manner as for the Western blot analysis (n = 3). To prevent nonspecific precipitation, 300 μg of protein from each brain sample were preincubated with 30 μL of protein G sepharose (71-7083-00; Pharmacia Biotech, Wikstroms, Sweden) and centrifuged, and the supernatant was used for the subsequent immunoprecipitation study. The supernatant sample was incubated with 5 μg of goat polyclonal anti-total-PERK antibody (sc-9477; Santa Cruz Biotechnology) for 1 hour at 4°C, and then 30 μL of protein G sepharose solution were added. After 2 hours of incubation, the samples were centrifuged and the precipitates were washed five times with protein extraction buffer. Samples obtained in this way were then used for the GRP78 Western blot analysis. Electrophoresis, blotting, and detection were essentially the same as described above. Briefly, the membrane was incubated in 10% powdered milk in PBS with 0.1% Tween 20 for 1 hour to block nonspecific binding. The anti-GRP78 antibody was purchased from Stressgen (#SPA-926; Stressgen, Victoria, Canada) and was diluted as 1:6000 for reaction. The secondary antibody and the dilution were the same as described above.
After the signal for GRP78 was evaluated, the membrane was used for PERK immunoblotting to confirm that the change in PERK-bound GRP78 was not caused by a change in the PERK expression level. It was placed in 10% powdered milk in PBS with 0.1% Tween 20 to block nonspecific binding and was subsequently incubated with an anti-PERK antibody (sc-9477; Santa Cruz Biotechnology) for 12 hours at 4°C. It was incubated with horseradish peroxidase conjugated anti-goat IgG (PI-9500; Vector Laboratories), and then the signal was detected. In this coimmunoprecipitation study, we also used a biotinylated protein ladder size marker (#7727; Cell Signaling) and a positive control from DTT-treated cell lysates to confirm the specificity of the antibody.
Detection of oxidative protein damage to the ER
To investigate ER oxidative protein damage, we extracted protein from the ER fraction as described previously, with modification (Ghribi et al., 2001). In brief, brain tissue from the hippocampal CA1 subregion, which was obtained 1 and 4 hours and 1 day after transient ischemia (n = 3), was gently homogenized with a glass homogenizer in seven volumes of the above-mentioned protein extraction buffer. Samples from the sham-operation were also obtained. The homogenate was first centrifuged at 750 g for 10 minutes and then at 10,000 g for 20 minutes at 4°C. The supernatant was further centrifuged at 100,000 g for 1 hour at 4°C to separate the cytosolic fraction from the ER fraction. We performed assays to determine protein concentration in comparison with a known concentration of BSA (kit, 23227; Pierce).
We observed three kinds of molecules as oxidative protein damage indicators: the carbonyl groups, 4-hydroxynonenal (HNE), and nitrotyrosine, all of which are in the proteins. Oxidation of proteins introduces the carbonyl groups at lysine, arginine, proline, and threonine residues. By reaction with 2,4-dinitrophenylhydrazine, they are derivatized to 2,4-dinitrophenylhydrazone, and this is detected by a specific antibody (Stadtman, 1993). For this procedure, we used a commercial kit (S7150; Intergen), and 2 μg of the ER fraction protein were studied. Protein can be damaged by superoxide, not only directly but also indirectly. Once arachidonic acid in the lipid bilayer is peroxidated by superoxide, it forms HNE, which easily conjugates with proteins (Toyokuni, 1999). The ER is a highly membranous organelle; therefore, ER proteins would be the targets for oxidative damage (White et al., 1993). After 5 μg of the ER protein were electrophoresed in SDS-polyacrylamide gel, the samples were transferred to polyvinylidene difluoride membranes (LC2002; Invitrogen) and incubated with a mouse monoclonal anti-HNE-conjugated protein antibody (24325; Oxis, Portland, OR, U.S.A.). The procedure was the same as that described for Western blotting. Because superoxide reacts quickly with nitric oxide and produces peroxynitrite, we speculated that tyrosine nitration plays an active role in oxidative protein damage. Using an anti-nitrotyrosine antibody (24312, Oxis), we performed a Western blot analysis as described above. We investigated immunoreactivity for nitrotyrosine from 2 μg of the ER protein. In all of these studies, the samples were electrophoresed together with a rainbow molecular weight (MW) marker (RPN800; Amersham, Piscataway, NJ, U.S.A.).
Quantification and statistical analysis
To evaluate the results of the Western blot and oxidative protein damage studies, the film was scanned with an imaging densitometer (GS-700; Bio-Rad, Hercules, CA, U.S.A.), and the optical density (OD) was quantified using Multi-Analyst software (Bio-Rad) as previously reported (Noshita et al., 2001). In the protein oxidation studies, the OD of the whole lane was investigated. Statistical significance between the two groups was established with an F test followed by an unpaired Student's t-test. P values less than 0.05 were considered statistically significant.
RESULTS
Overexpression of SOD1 protected hippocampal CA1 pyramidal cells from delayed neuronal death
The cresyl violet staining and TUNEL studies confirmed no neuronal degeneration in the hippocampal CA1 region 1 or 4 hours or 1 day after transient ischemia in either the Wt or Tg animals. At 2 days, however, most of the CA1 neurons were degenerated and TUNEL-positive in the Wt brains, known as delayed neuronal death (Kirino, 1982). In the Tg animals, in contrast, only a few neurons were degenerated and TUNEL-positive, but most of the cells remained morphologically intact, as previously demonstrated (Chan et al., 1998;Sugawara et al., 2002).
PERK phosphorylation after ischemia was less pronounced in the SOD1 Tg animals
PERK is localized on the ER membrane and is phosphorylated when stress is imposed on the ER (unfolded protein response) (Harding et al., 1999). It then phosphorylates eIF2α which inhibits protein synthesis and activates cell death programs (Harding et al., 2002;Kumar et al., 2001). Therefore, phosphorylation of PERK could be the initial step in cell death signaling from the ER. We investigated changes in PERK phosphorylation with Western blot and immunohistochemical analyses using an anti-phospho-PERK antibody (Fig. 1). In addition, we performed a Western blot study using an anti-total-PERK antibody. PERK shows slower mobility by SDS-PAGE when phosphorylated (Kumar et al., 2003), and this study could confirm the result of the study with the anti-phospho-PERK antibody. With the anti-phospho-PERK antibody (Fig. 1A, upper panel), a faint band with a MW of 170 kDa was observed in the sham-operated Wt and Tg brains with Western blot analysis. The MW of this band is compatible with phospho-PERK. Furthermore, the mobility was the same as the positive control obtained from DTT-treated cell lysates. In the Wt animals, the density of the band increased 1 hour after transient ischemia, but became less dense at 4 hours and even weaker at 1 day. At 2 days, the band for phospho-PERK became almost undetectable. In the Tg animal brains, there was a slight increase in the band at 1 hour, then a gradual weakening, but the degree of increase was less than in the Wt animals. Western blot analysis using the anti-total-PERK antibody (Fig. 1A, lower panel) also showed that PERK became phosphorylated after ischemia, and the temporal profile and difference between the Wt and Tg groups were compatible with the results of the study with the anti-phospho-PERK antibody. Of note was that the total PERK level was a bit decreased at 2 days in the Wt animals, which might be because of the neuronal degeneration observed at this time point. We carried out quantitative analysis for the Western blot with the anti-phospho-PERK antibody (Fig. 1B) and found that SOD1 overexpression significantly inhibited PERK phosphorylation 1 and 4 hours and 1 day after ischemia (∗∗ P < 0.01 compared with Wt animals at the same time point). Immunohistochemical analysis showed that hippocampal CA1 neurons were only faintly stained in the Wt and Tg control brains (Fig. 1C). In the Wt brains, immunoreactivity became very intense 1 hour after ischemia with the strongest immunoreactivity in the perinuclear region, which is compatible with the distribution in the ER (Nakagawa et al., 2000;Peters et al., 1991). At 4 hours, only some neurons retained strong immunoreactivity (arrowheads). The number of strongly stained neurons was further decreased at 1 day (arrowheads), and the neurons were degenerated and immunoreactivity could not be evaluated at 2 days. In the Tg brains, only some neurons showed a strong immunoreactivity 1 hour after ischemia (arrowheads). Thereafter, any strongly stained cells almost disappeared. Brain sections without the primary antibody showed no immunoreactivity (not shown).
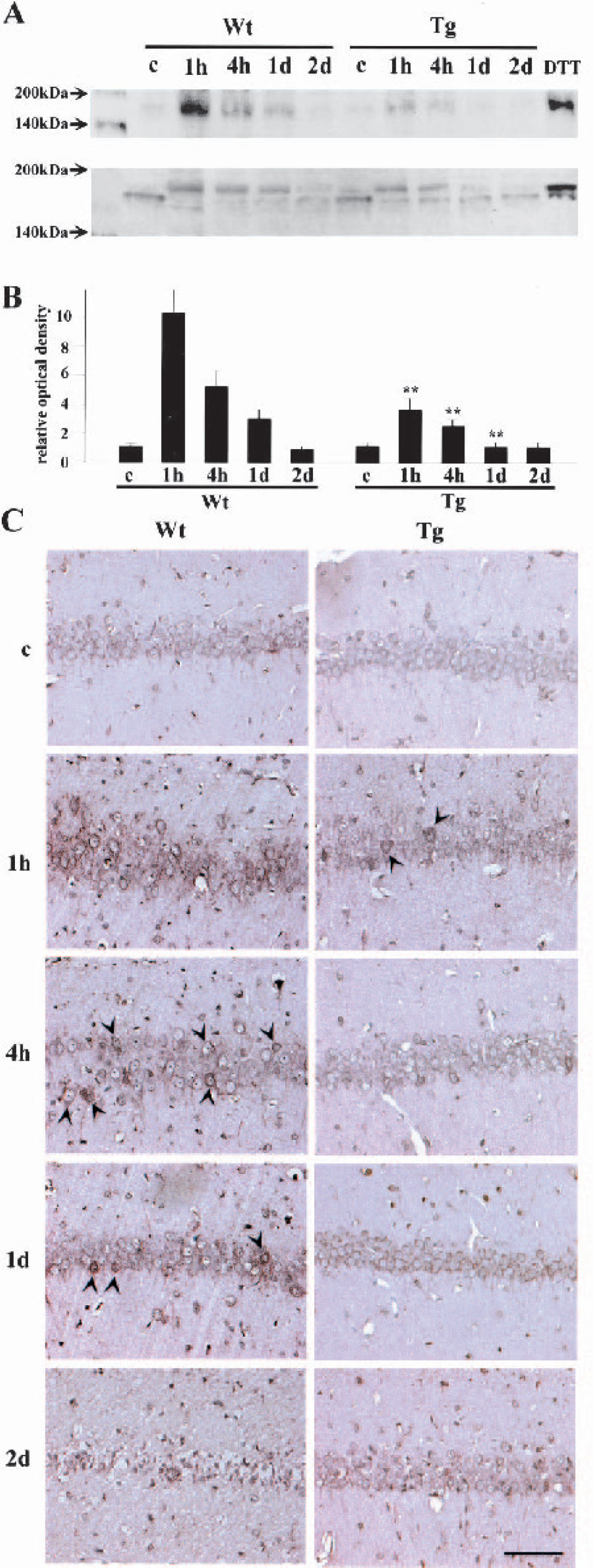
Changes in phosphorylation of PERK at the hippocampal CA1 pyramidal cell layer.
Prevention of PERK phosphorylation in Tg animals was accompanied by decreased superoxide production
In the sham-operated Wt and Tg brains, the ethidium signal (red), which reflects superoxide production, was only weakly detected as small particles (Fig. 2, arrowheads). In addition, almost no signals for phospho-PERK (green) were detected. In the Wt brains, however, a marked increase in the ethidium signal was observed 1 hour after transient ischemia. A small punctate signal, as well as a diffuse signal, was dramatically increased. In these cells, the signal for the phosphorylated form of PERK was also very strong. In the Tg brains at 1 hour, however, production of superoxide was far less pronounced and phosphorylation of PERK was much milder than in the Wt brains.
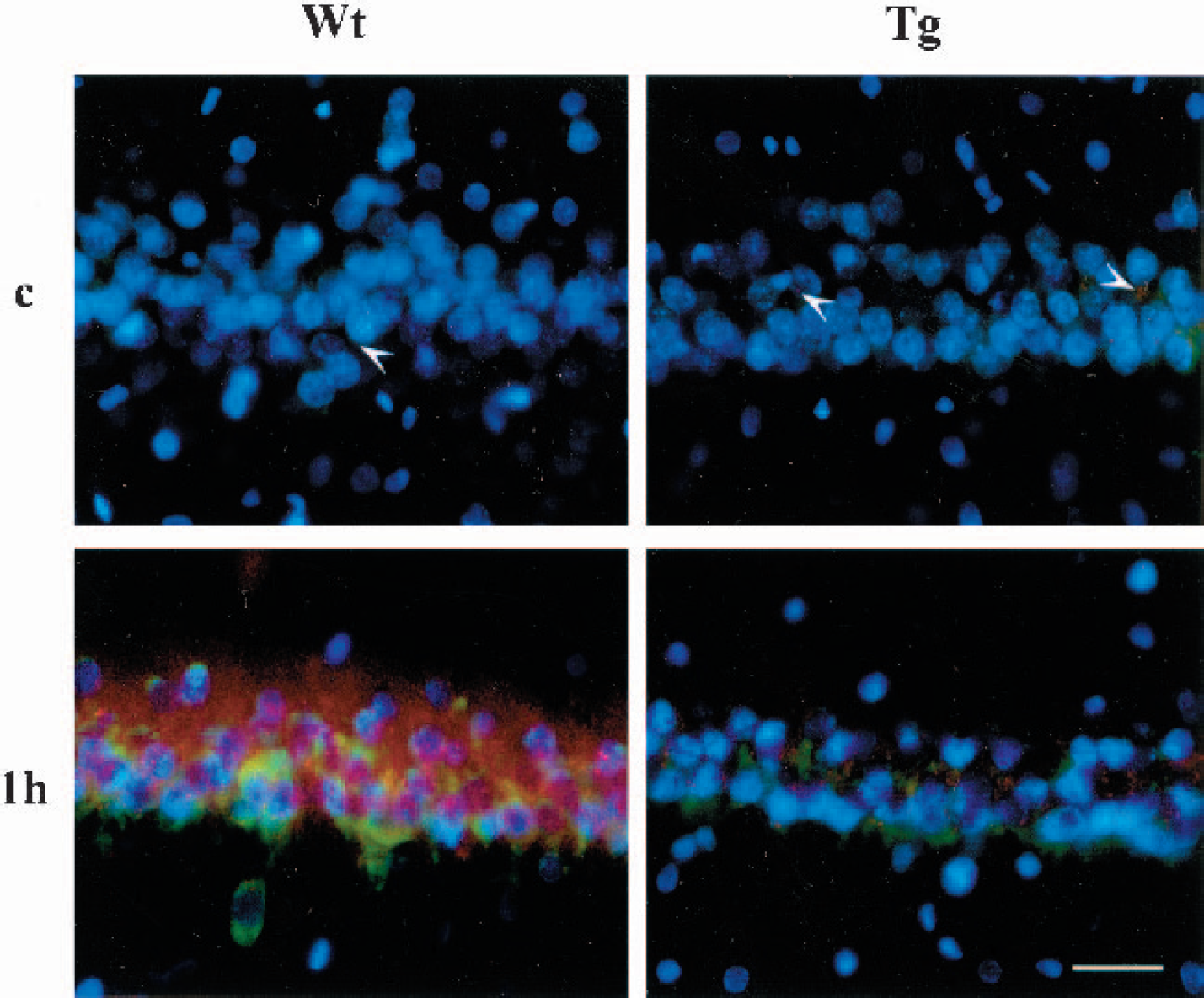
Fluorescent double staining for the ethidium signal (red) and the phosphorylated form of PERK (green). Nuclei were counterstained with DAPI (blue). In the sham-operated Wt and Tg brains (c), the ethidium signal was only weakly observed as small particles (arrowheads). One hour after 5 minutes of ischemia, a marked increase in the ethidium signal was observed in the Wt brains. The phosphorylated form of PERK was also significantly increased. In the Tg brains, in contrast, the signals for both ethidium and the phosphorylated form of PERK were increased 1 hour after ischemia, but the degrees of increase were much milder than those in the Wt brains. Bar = 30 μm.
Phosphorylation of eIF2α after ischemia was less pronounced in the SOD1 Tg animals
EIF2α plays a pivotal role in the initiation of protein synthesis. Once it is phosphorylated, it becomes inactive and inhibits new protein synthesis (Degracia et al., 2002;Harding et al., 2002). Furthermore, a recent report showed that phospho-eIF2α activates a certain transcription factor and induces proapoptotic molecules (Kaufman, 1999). Therefore, phosphorylation of eIF2α could be the key step in the ER stress-induced cell death program. We investigated changes in eIF2α phosphorylation with Western blot and immunohistochemical analyses using an anti-phospho-eIF2α antibody (Fig. 3). A weak band with a MW of 40 kDa was observed in the sham-operated Wt and Tg brains by Western blot analysis (Fig. 3A). The MW of this band is compatible with phospho-eIF2α. Furthermore, the mobility in the gel was the same as in the positive control obtained from DTT-treated cell lysates. In the Wt animals, the band became quite dense 1 hour after transient ischemia and remained dense at 4 hours but became weaker at 1 day. At 2 days, the band for phospho-eIF2α returned to the control level. In the brains of the Tg animals, the band became strong at 1 hour and then gradually weakened, but the degree of increase was smaller than in the Wt animals. Using a quantitative analysis (Fig. 3B), we found that SOD1 overexpression significantly inhibited phosphorylation of eIF2α 1 and 4 hours and 1 day after ischemia (∗∗ P < 0.01 compared with Wt animals at the same time point). Immunohistochemical analysis showed that hippocampal CA1 neurons were only faintly stained in the Wt and Tg control brains (Fig. 3C). In the Wt brains, immunoreactivity became quite intense 1 hour after ischemia. At 4 hours, only some neurons retained a strong immunoreactivity (arrowheads). The number of strongly stained neurons was further decreased at 1 day (arrowheads); the neurons degenerated, and immunoreactivity could not be evaluated at 2 days. In the Tg brains, however, immunoreactivity became strong at 1 hour but was less so in the Wt animals. At 4 hours, only a small number of neurons had a dense immunoreactivity (arrowheads), and they became close to the control level at 1 and 2 days. The same results were obtained when an anti-phospho-eIF2α antibody from another source was used, which strongly suggests that the immunoreactivity observed was a specific one (not shown). In addition, brain sections without the primary antibody showed no immunoreactivity (not shown).
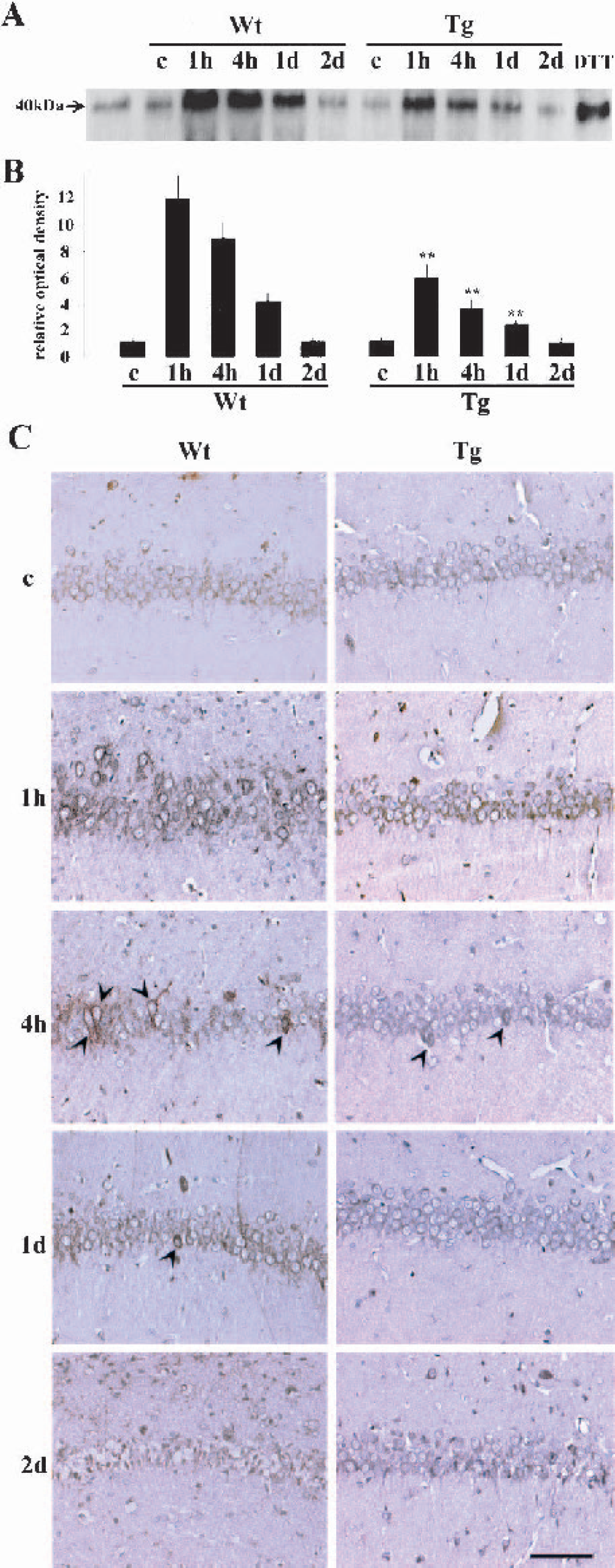
Changes in phosphorylation of eIF2α at the hippocampal CA1 pyramidal cell layer.
Release of GRP78 from PERK by ischemia was diminished by SOD1 overexpression
GRP78, an ER resident molecular chaperone, normally binds with PERK and inhibits its phosphorylation. When unfolded proteins increase in the ER lumen, GRP78 becomes attached to the unfolded proteins and is released from PERK, thus allowing PERK to be phosphorylated (Bertolotti et al., 2000;Harding et al., 1999;Liu et al., 1998). In this study, protein lysates were immunoprecipitated with an anti-PERK antibody and then analyzed by Western blot for GRP78. The membrane was also reacted with anti-total-PERK antibody to confirm that the change in PERK-bound GRP78 was not due to the change in the PERK expression level. The results are shown in Figure 4A and the OD analysis in Figure 4B. By the immunoblotting for GRP78, a dense band with a MW of 78 kDa was confirmed in the Wt and Tg sham-operated brains, indicating that a substantial amount of GRP78 was bound with PERK (Fig. 5A, upper panel). The MW of the band was the same as in the positive control obtained from DTT-treated cell lysates. In the Wt brains, the band became quite weak 1 hour after transient ischemia, which indicates that most of the GRP78 was detached from PERK. In the Tg brains, the band became weak 1 hour after ischemia, but was still stronger than in the Wt brains, indicating that release of GRP78 by ischemia was partially prevented by Tg SOD1 overexpression. In contrast, there were no changes in total PERK expression levels (Fig. 5A, lower panel). Therefore, this indicates that the change in PERK-bound GRP78 was not caused by the change in the PERK expression level. Of note is that the phosphorylated form of PERK showed slower mobility in the gel than the unphosphorylated form, which is compatible with the results shown in Figure 1. A quantitative analysis for GRP78 revealed that ischemia significantly decreased GRP78 and PERK binding in both groups, but overexpression of SOD1 significantly prevented its decrease (∗∗ P < 0.01 compared with the sham-operated group of the same genotype; ##P < 0.01 compared with the Wt animals 1 hour after ischemia) (Fig. 4B).
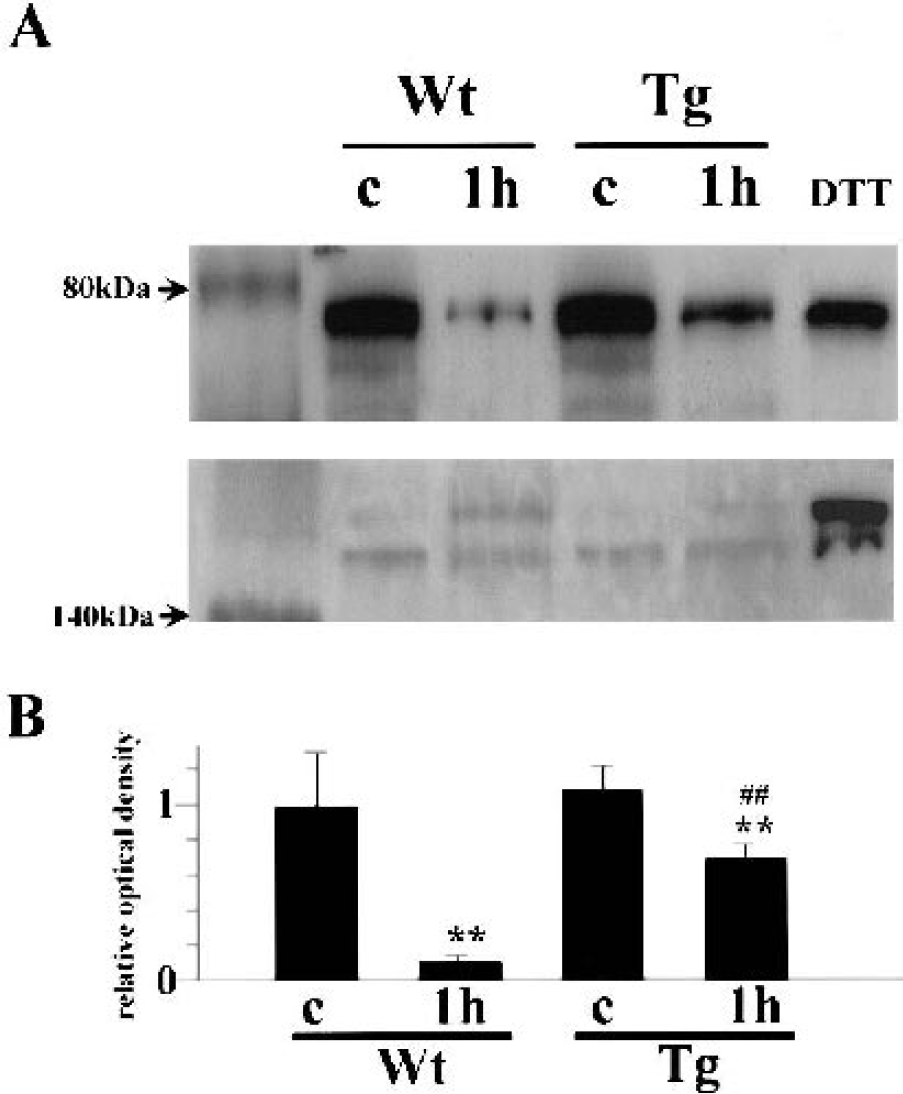
Coimmunoprecipitation study of GRP78 with PERK.
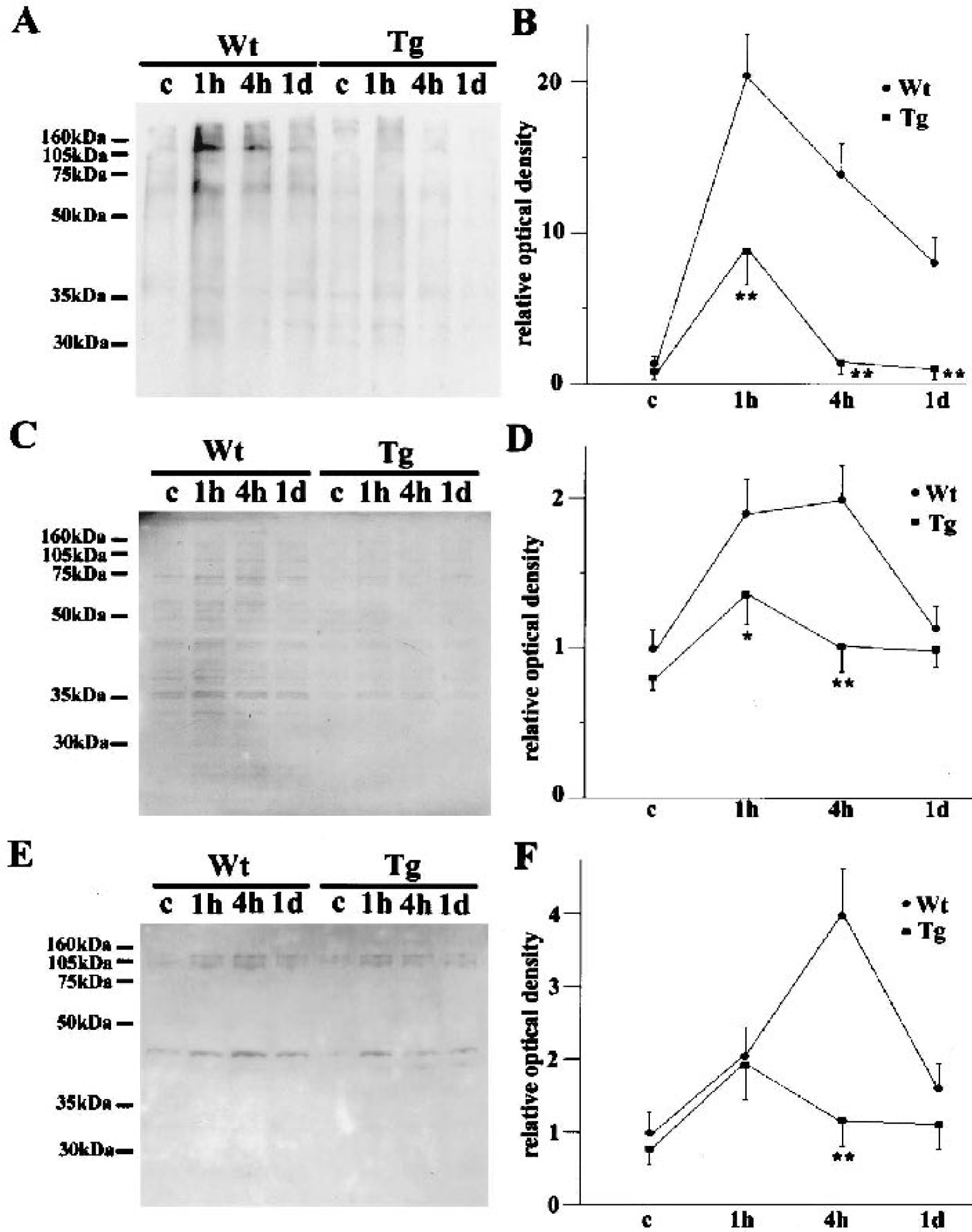
Changes in oxidatively damaged protein in the hippocampal CA1 region.
Oxidative protein damage to the ER was prevented by SOD1 overexpression
Detachment of GRP78 from PERK indicated an increase in unfolded proteins in the ER lumen. Because SOD1 overexpression attenuated this detachment, oxidative protein damage may be the cause of this protein malfolding. To confirm this, we investigated oxidatively damaged protein in the ER fraction. We used three strategies, one that detected protein carbonyl group formation, another that detected protein-HNE conjugation, and a third that detected nitrotyrosine formation. Carbonyl group formation indicates direct oxidative protein damage, and the latter two show indirect damage (Lipton et al., 1993;Stadtman, 1993;Toyokuni, 1999). We suspected that both mechanisms of protein damage were possible; direct damage could easily occur because the ER is one of the organelles that produces many ROS, and indirect damage could occur through lipid peroxidation, which probably occurs because lipids are easily peroxidated by superoxide (Halliwell and Gutteridge, 1999), and through nitrotyrosine formation, which could easily take place because the ER produces nitric oxide (Martinelli et al., 2002). The results are summarized in Figure 5. In the sham-operated Wt brains, only a small amount of the carbonyl group was detected (Fig. 5A). One hour after 5 minutes of ischemia, it was markedly increased, but then it decreased at 4 hours and 1 day. In the Tg animals, however, it was increased at 1 hour and decreased thereafter, but the degree of increase was far milder than in the Wt animals. A quantitative analysis (Fig. 5B) showed that Tg overexpression of SOD1 significantly prevented carbonyl group formation by ischemia from 1 hour to 1 day after ischemia (P < 0.01 compared with Wt animals at the same time point). A small amount of HNE-conjugated protein (Fig. 5C) was seen in the sham-operated Wt and Tg brains. In the Wt brains, it was substantially increased 1 and 4 hours after ischemia but was decreased at 1 day. In the Tg animal brains, it was increased at 1 hour but decreased at 4 hours and 1 day. The degree of increase was milder than in the Wt animals. A quantitative analysis (Fig. 5D) showed that Tg overexpression of SOD1 significantly prevented formation of HNE-conjugated protein by ischemia at 1 and 4 hours (P < 0.05 at 1 hour; P < 0.01 at 4 hours compared with Wt animals at the same time point). In peroxynitrite-induced protein damage, only a small amount of nitrotyrosine was confirmed in the sham-operated brains (Fig. 5E). It was increased at 1 hour and peaked at 4 hours but decreased at 1 day in the Wt animals. In the Tg animals, it was increased at 1 hour, but it is noteworthy that it was decreased as early as 4 hours. A quantitative analysis (Fig. 5F) showed that the amount of nitrotyrosine was significantly smaller in the Tg animals 4 hours after ischemia (P < 0.01 compared with Wt animals at the same time point).
DISCUSSION
In this study, we demonstrated that Tg overexpression of SOD1 reduced ER damage and delayed neuronal death of hippocampal CA1 neurons after ischemia. These findings are compatible with previous reports showing that the ER is sensitive to oxidative stress (Dreher et al., 1995;Oyadomari et al., 2001;Racay et al., 1995;Viner et al., 1996;Yu et al., 1999). The studies for carbonyl group formation, HNE-protein conjugation and nitrotyrosine formation revealed that many kinds of proteins in the ER fraction are nonspecifically damaged by oxidative stress; the results of these studies show many bands or a smear-like appearance (Fig. 5). This does not conflict with reports that the ER calcium pump is quite sensitive to oxidative damage because some proteins might be functionally intact even when oxidatively modified (Cabiscol and Levine, 1995). Functionally important domains of the ER calcium pump might be more vulnerable to oxidative stress than those of other proteins. Rather, our results may give us insights into the mechanisms of ER damage by ROS. Massive production of protein carbonyl groups indicates that a direct attack of ROS on proteins took place in the ER (Fig. 5A and Fig. 5B). This is compatible with the fact that the ER is the organelle that produces ROS (Halliwell and Gutteridge, 1999). The results of this study are noteworthy because direct oxidative damage to proteins has received less attention than damage to lipids or DNA. Krause et al. (1992) reported that proteins and RNA are not the targets of ROS in the dog brain after cardiac arrest. This may be because of the difference in animal species and ischemic models. In rats exposed to hyperbaric oxygen, however, more protein damage than lipid peroxidation was revealed in the brain (Chavko and Harabin, 1996). Hence, proteins may be more susceptible to oxidative stress than is generally considered. In contrast, the HNE-conjugation analysis showed that lipid peroxidation and secondary protein damage also occurred in the ER (Fig. 5C and Fig. 5D). ROS produced in the ER could be responsible for this damage, but we cannot rule out other possibilities. The ER and mitochondria are in such close proximity that they can influence each other's function with diffusible molecules (Rizzuto et al., 1998). This fact led us to consider the possibility that ROS produced in mitochondria peroxidated lipids of the ER membrane and then damaged ER proteins. Because the ER is a highly membranous organelle, lipid peroxidation and HNE formation may easily occur with the existence of ROS. Another possible mechanism of oxidative protein damage to the ER is tyrosine nitration; superoxide reacts very quickly with nitric oxide and produces the highly toxic molecule peroxynitrite. This molecule, in turn, attacks tyrosine residue and causes its nitration. Because nitric oxide is produced in the ER in neurons, protein modification through this mechanism may play an active role in ER stress under ischemia (Martinelli et al., 2002). To our surprise, we found that the amount of nitrotyrosine was almost the same in the Wt and Tg animals 1 hour after ischemia (Fig. 5E and Fig. 5F). This may indicate that nitric oxide, but not superoxide, was the limiting substrate that produced peroxynitrite. Because the reaction of superoxide with nitric oxide is three times faster than the rate at which SOD1 scavenges superoxide, Tg overexpression of SOD1 might not be sufficient to prevent peroxynitrite formation (Beckman et al., 1996). Of note was that nitrotyrosine was further increased in the Wt rats, but decreased in the Tg animals at 4 hours (Fig. 5E and Fig. 5F). This might be because the ER protein degradation system was impaired in the Wt but not in the Tg animals. To investigate the relevance of superoxide to the ER-associated degradation system, however, another study is required.
In this study, we investigated three possible mechanisms of oxidative ER protein damage, but other mechanisms of ROS-caused ER damage cannot be excluded. For example, calcium depletion because of dysfunction of the ER calcium pump caused by an ROS attack can inhibit normal protein folding and subsequently elicit the ER stress response (Verkhratsky and Petersen, 2002). In addition, protein isomerase, which is involved in protein folding in the ER, requires free sulfhydryl residues (Freedman, 1984). Oxidative stress may attack these residues and then prevent ER protein folding.
Once damaged proteins are produced in the ER, they tend to remain as unfolded or malfolded proteins (Hu et al., 2000). GRP78, an ER resident molecular chaperone, is normally bound with PERK, which is an ER resident protein kinase (Harding et al., 1999). When the amount of unfolded protein is increased, however, GRP78 is released from PERK and binds with the unfolded protein. PERK without GRP78 binding is dimerized and then becomes phosphorylated (Bertolotti et al., 2000;Harding et al., 2002;Rao et al., 2002). Phospho-PERK, in turn, phosphorylates eIF2α (summarized in Fig. 6). When ER stress is not severe, phosphorylation of eIF2α is cytoprotective, because it decreases the load for the ER. When ER stress is severe, however, it activates the cell death program (DeGracia et al., 2002;Kumar et al., 2001;Paschen and Doutheil, 1999). This is one of the features of the unfolded protein response. Our coimmunoprecipitation study (Fig. 4) showed that GRP78 was detached from PERK after ischemia, indicating that it became attached to unfolded proteins in the ER. The finding that the Tg rats had a decrease in GRP78, and PERK binding to a lesser degree, suggests that less oxidative protein damage occurred because of SOD1 overexpression, which is compatible with studies for carbonyl group formation, HNE-conjugation, and nitrotyrosine formation (Fig. 5). A double fluorescence study for ethidium and phospho-PERK (Fig. 2) also suggests that production of ROS causes PERK phosphorylation. A Western blot analysis showed that the total PERK expression level was decreased at 2 days in the Wt animals (Fig. 1), which may be because of the loss of neurons. However, the total PERK expression level was not decreased in the early phase after reperfusion (Fig. 1 and Fig. 4). Therefore, a change in PERK-bound GRP78 cannot be explained by the change in the total PERK expression level. In contrast, it was recently reported that the total PERK expression level was decreased after ischemia (Kumar et al., 2003). A decrease in phospho-PERK 2 days after ischemia in our model might be caused partially by a decrease in total PERK.
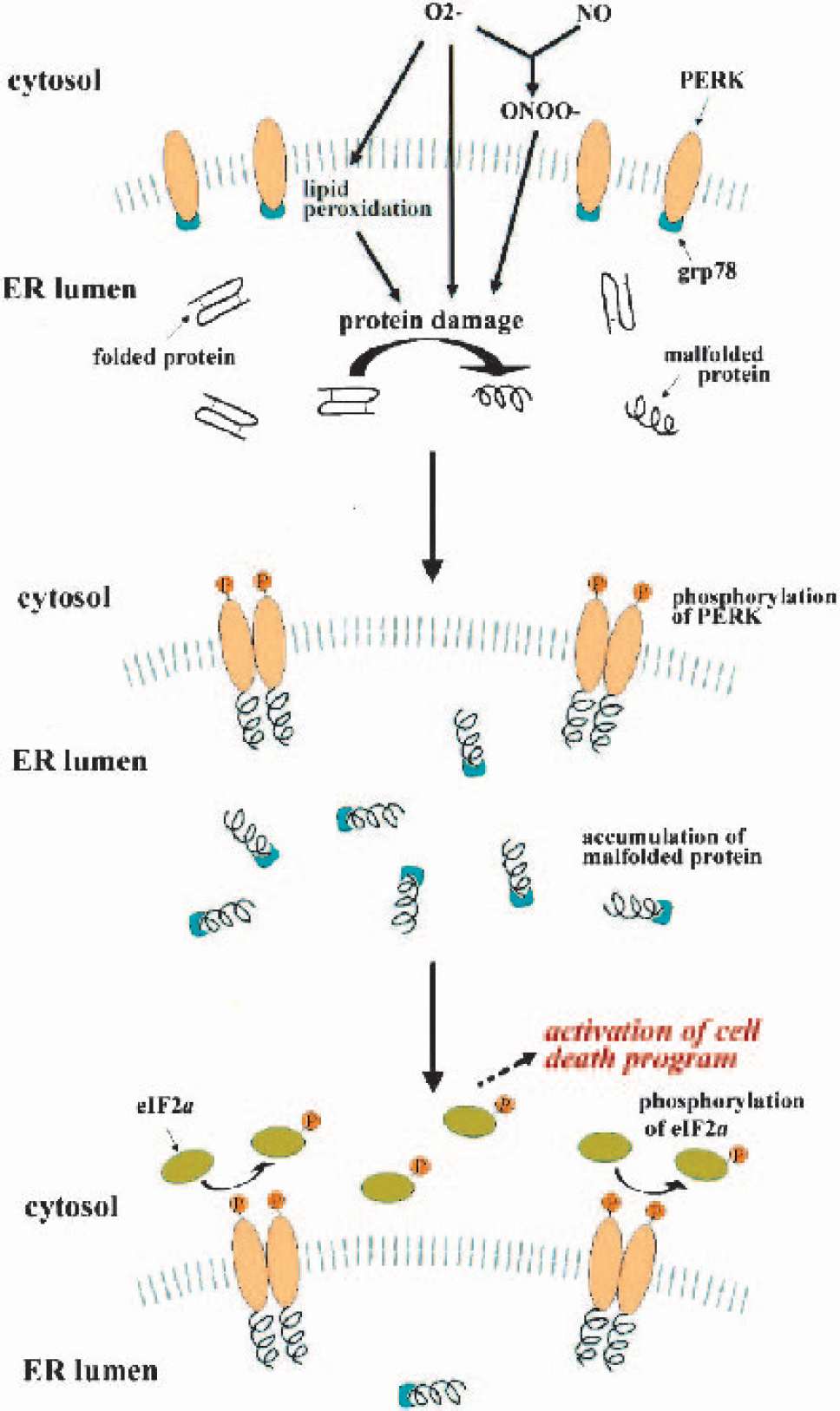
Schematic diagram of proposed mechanisms of neuronal cell death induced by oxidative ER damage. Superoxide anion (O2−) causes damage to the ER resident proteins, both directly and indirectly, through formation of peroxidated lipid or peroxynitrite (ONOO−). Damaged proteins become malfolded and accumulate in the ER lumen, which then detaches GRP78 from PERK and binds with GRP78 or PERK. PERK without GRP78 binding becomes dimerized and phosphorylated and causes phosphorylation of eIF2α. The phosphorylated form of eIF2α then elicits activation of cell death programs. NO, nitric oxide.
Burda et al. (1994) reported that the level of phospho-eIF2α was increased to approximately four times that in the sham-control in the neocortex after ischemia, although our results revealed that it was increased up to twelve times that in the sham-control in the hippocampus. This might be because of the difference in the ischemic models, but we can speculate that the ischemic-vulnerable neurons showed a particularly prominent eIF2α phosphorylation. Indeed, eIF2α phosphorylation is often associated with cell death (Sherman and Goldberg, 2001). How phospho-eIF2α activates the subsequent cell death program is, however, not precisely clear. Although the phosphorylated form of eIF2α decreases total protein synthesis, it increases some proapoptotic proteins, such as activating transcription factor-4 and C/EBP-homologous protein, by “bypass scanning” (Jousse et al., 1999;Paschen et al., 1998). There is also a report that showed that the phosphorylated form of eIF2α is cytotoxic by itself (Srivastava et al., 1998). Another possibility is the involvement of caspase-12, which is activated by severe ER stress (Nakagawa et al., 2000). Active caspase-12 causes cytochrome c release from mitochondria and activation of caspase-3 and caspase-9, which are observed in the ischemic brain and inhibited by SOD1 overexpression (Asahi et al., 1997;Fujimura et al., 2000;Nakagawa et al., 2000;Siman et al., 2001;Sugawara et al., 2002).
ROS are ubiquitous small molecules. They are probably not as selective of their targets as enzymes or antibodies are. Which molecules are attacked by ROS may be dependent upon the chemical characteristics and topographic positions of the ROS and their possible targets. It seems unlikely that ROS selectively attack antiapoptotic molecules; however, ample evidence suggests that specific cell death-related proteins are activated by ischemia, and this activation is inhibited by scavenging ROS (Lewén et al., 2000). Therefore, we can envision some mechanisms that underlie this biologic convergence. The ER, which is susceptible to oxidative stress and is important for activation of cell death pathways, could be the organelle responsible for this biosignal convergence. In addition, oxidative stress may act as a switch for this biologic system.
Footnotes
Acknowledgment
We thank Liza Reola, Bernard Calagui, and Ghezal Omar for technical assistance, Hiroki Toda for cultured cell preparation, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.