
Abstract
Increasing clinical and experimental evidence suggests that traumatic brain injury (TBI) elicits an acute inflammatory response. In the present study we investigated whether white blood cells (WBC) are activated in the cerebral microcirculation early after TBI and whether WBC accumulation affects the posttraumatic cerebrovascular response. Twenty-four anesthetized rabbits had chronic cranial windows implanted 3 weeks before experimentation. Animals were divided into four experimental groups and were studied for 7 hours (groups I, IIa, and III) or 2 hours (group IIb). Intravital fluorescence videomicroscopy was used to visualize WBC (rhodamine 6G, intravenously), pial vessel diameters, and blood–brain barrier (BBB) integrity (Na+-fluorescein) at 6 hours (groups I, IIa, and III) or 1 hour (group IIb) after TBI. Group I (n = 5) consisted of sham-operated animals. Groups IIa (n = 7) and IIb (n = 5) received fluid-percussion injury at 1 hour. Group III (n = 7) received fluid-percussion injury and 1 mg/kg anti–adhesion monoclonal antibody (MoAb) “IB4” 5 minutes before injury. Venular WBC sticking, intracranial pressure (ICP), and arterial vessel diameters increased significantly for 6 hours after trauma. IB4 reduced WBC margination and prevented vasodilation. Intracranial pressure was not reduced by treatment with IB4. Blood–brain barrier damage occurred at 1 hour but not at 6 hours after TBI and was independent of WBC activation. This first report using intravital videomicroscopy to study the inflammatory response after TBI reveals upregulated interaction between WBC and cerebral endothelium that can be manipulated pharmacologically. White blood cell activation is associated with pial arteriolar vasodilation. White blood cells do not induce BBB breakdown less than 6 hours after TBI and do not contribute to posttraumatic ICP elevation. The role of WBC more than 6 hours after TBI should be investigated further.
Keywords
Traumatic brain injury (TBI) is the major cause of death in young adults in the United States and Europe. One third of these are patients who die in the hospital as a result of secondary damage to the brain with intracranial hypertension as the major clinical manifestation (Miller et al., 1992). Early pathophysiologic signs of secondary brain damage such as brain edema and congestive brain swelling reflect cerebrovascular dysfunction and bear striking similarity to traditional inflammation observed in other organs. The precise mechanisms mediating secondary brain damage are not well understood.
Clinical and experimental studies during the past years have found elevated levels of proinflammatory and inflammatory mediators, such as platelet-activating factor, free radicals, and components of the arachidonic acid cascade, in serum, parenchyma, and CSF early after head injury (McIntosh, 1994). Interleukin-6, tumor necrosis factor, and other cytokines have been described in the CSF of severely head-injured patients peaking within the first days after trauma (Goodman et al., 1990; Kossmann et al., 1996; McClain et al., 1991; Ott et al., 1994). Experimental models of TBI have demonstrated that cytokines are found and produced in the brain in response to trauma (DeKosky et al., 1996; Fan et al., 1995; Shohami et al., 1996; Taupin et al., 1993; Woodroofe et al., 1991; Yan et al., 1992), and that inhibition of these factors may confer neuroprotection (Shohami et al., 1996). The stereotypic picture of posttraumatic cerebrovascular dysfunction supports the view that TBI initiates a cascade of neurochemical events in which inflammatory mediators may be important. White blood cells (WBC) are cytotoxic cells that may play a key role in this scenario; they produce many of the inflammatory mediators seen after TBI and become activated in response to these factors. Recently, WBC accumulation in brain parenchyma has been described within the first 4 to 48 hours after experimental TBI (Biagas et al., 1992; Carlos et al., 1997; Clark et al., 1996b; Clark et al., 1994; Horner et al., 1992; Kaczorowski et al., 1995; Schoettle et al., 1990; Soares et al., 1995; Uhl et al., 1994; Wilson et al., 1995; Zhuang et al., 1993). Only a little is known about the possible role of WBC after TBI, particularly whether WBC are passive responders to brain injury or actually modulate the posttraumatic microcirculatory response.
The goal of the present study was to shed light on the behavior of WBC early after TBI and to test the hypotheses that (1) TBI leads to WBC accumulation in the cerebral microcirculation, and (2) WBC in the cerebral microcirculation affect the cerebrovascular response to TBI.
MATERIALS AND METHODS
Implantation of chronic cranial windows
Twenty-four New Zealand rabbits weighing 2.5 to 3.5 kg were employed in this study, which was approved by the Cornell University Medical College Animal Use Committee. To avoid artifacts associated with the surgical procedure (skin incision, trephination, dura reflection, and exposure of the brain surface), cranial windows were implanted 2 to 4 weeks before the final experiments.
Rabbits were anesthetized with xylazine (5 mg/kg) and ketamine (35 mg/kg) subcutaneously and were placed prone in a stereotactic frame supporting head and body. The modified version of a chronic cranial window described previously by Levasseur et al. (1975) was used. A trephination (diameter, 8 mm) was prepared over the left cerebral hemisphere, the dura mater was reflected, and a sterilized glass disk was inserted into the cranial defect (Fig. 1). The window was sealed and held in place using dental cement and bone screws. The scalp tissues were then reapproximated with 3-0 silk sutures. Rabbits were monitored daily for general condition and condition of the surgical site.
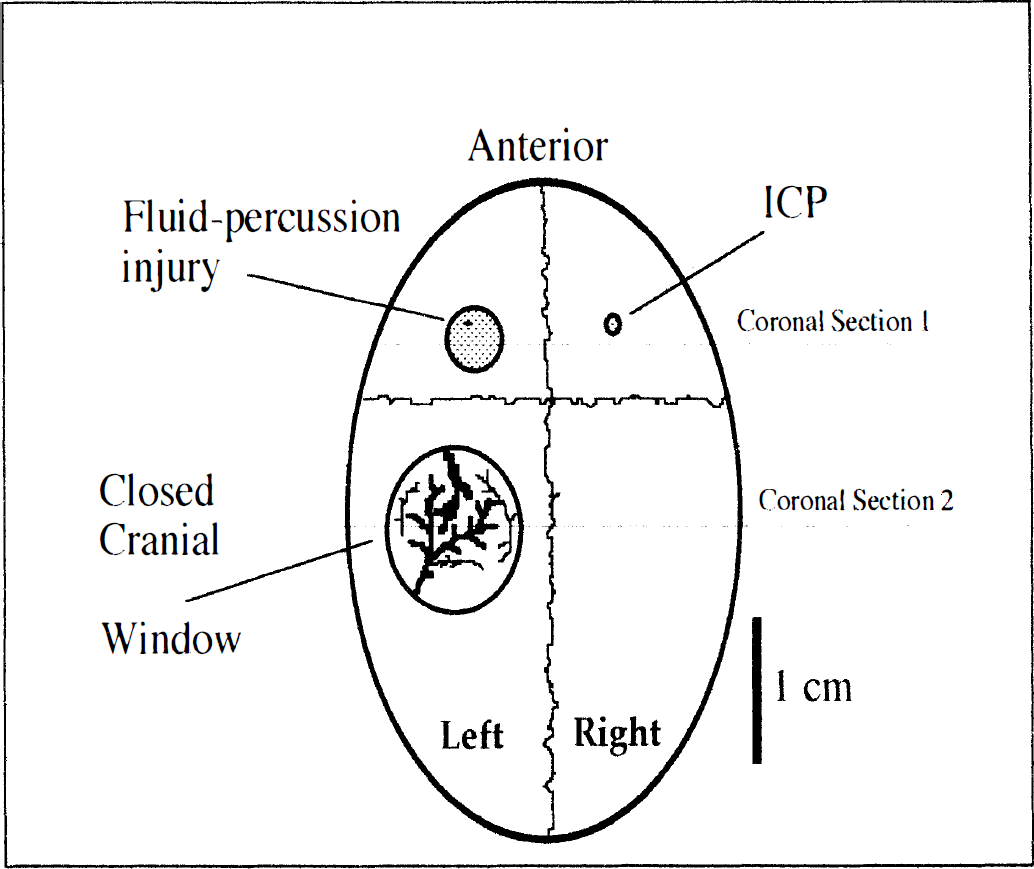
The skull preparation. Trauma and closed cranial window were located over the left hemisphere. Coronal sections 1 and 2 indicate where the brain slices for histologic analysis were taken from.
Final experiments
Anesthesia was induced by xylazine and ketamine intramuscularly. The animals were given a tracheotomy for artificial ventilation (Harvard Apparatus, South Natick, MA, U.S.A.). Anesthesia was maintained by intravenous bolus injections of 20% urethane. A femoral artery catheter allowed continuous monitoring of mean arterial blood pressure (MAP) and blood sampling. Intravenous access was achieved through an ear vein, and body temperature was controlled by a heating pad.
The animal's head was then immobilized in a sphinx position with a stereotactic holder. Intracranial pressure (ICP) was measured using a microsensor ICP transducer (Codman, Johnson & Johnson, New Brunswick, NJ, U.S.A.). A drill hole (diameter, 1.4 mm) was prepared over the right cerebral hemisphere, opposite to the trauma and the cranial window, for insertion of the microsensor into the epidural space (Fig. 1). Intracranial pressure and MAP (Tektronix 400 Recorder), end-tidal P
Brain injury
Experimental brain injury was produced using a modification of the lateral (parasagittal) fluid-percussion injury model originally described in rats by McIntosh et al. (1989). The device consisted of a plexiglass cylindrical saline reservoir closed at one end by a plexiglass cork. The opposite end of the reservoir was connected to a saline-filled hose fitted with a metal transducer housing and the hollow metal injury screw (inner diameter, 3 mm; outer diameter, 4 mm). A sterile piece of rubber (from a surgical glove) was securely fixed over the end of the injury screw to allow the transmission of the pressure wave onto the brain, while preventing the loss of saline into the intracranial cavity. A drill hole (diameter, 4 mm) over the left hemisphere anterior to the cranial window was prepared and the dura was left intact (Fig. 1). To produce an insult, the piston was struck by a 3.4-kg metal weight from a specific fall height. This resulted in a 3.5-atm barotraumatic injury delivered to the brain for 20 to 25 milliseconds. An electrically triggered pressure release valve guaranteed rapid deflation of the intracranial balloon. After the impact the injury screw was removed and the cranial defect was filled with sterile bone wax.
Intravital videomicroscopy
A custom-designed Leitz microscope for intravital videomicroscopy equipped with a 50-W mercury light source (Opti Quip 1200), Ploem-Pak illuminator filter block (Leitz, Wetzlar, FRG) for epi-illumination, 5× to 32× objectives, and an MTI VE1000 SIT camera with external control board was used for assessment of microcirculatory parameters. Images were recorded on a Sony U-Matic VCR, VO 9600, and were displayed on a 20-inch Sony videomonitor for off-line evaluation. An image analysis software program (NIH Image, version 1.55) on a Macintosh Quadra 700 computer was used to measure vessel diameters. The animal's position under the microscope could be controlled in two axes using manually driven micromanipulators attached to a custom-made x–y microscope stage.
WBC behavior in the cerebral microcirculation
White blood cells in six to eight randomly picked postcapillary pial venules per animal were visualized by intravenous injection of rhodamine 6G (0.3 ml/kg of a 0.05% solution R6G, Sigma Chenical, St. Louis, MO, U.S.A.) for in vivo labeling. Rhodamine 6G labels WBC and platelets, leaving endothelial cells and erythrocytes unstained (Dirnagl et al., 1994). Selective observation of rhodamine 6G-stained WBC was possible using epi-illumination with a Leitz N2 filter block (excitation, 530 to 560 nm; emission, >580 nm). Firmly adherent WBC were defined as cells that remained stationary for at least 30 seconds and are given as the number of WBC per mm2 of vessel wall. Thirty seconds appears to be the minimal duration of firm adhesion that is required as a prelude to emigration during an inflammatory response (Kubes and Granger, 1992).
Vessel surface (s) was calculated from inner vessel radius (r, maximum 30 μm) and segment length (l, 100 to 150 μm) using the formula:
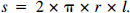
Vessel diameters
Mean diameters of six to eight pial arterioles (diameters between 40 and 120 μm) per animal were determined using NIH image analysis software.
Blood–brain barrier integrity
Blood–brain barrier integrity was assessed qualitatively by intravenous infusion of Na+-fluorescein 30 minutes before termination of the experiments, i.e., at 5.5 hours in groups I, IIa, and III and at 0.5 hour in group IIb. Microvascular leakage was indicated by extravasation of Na+-fluorescein (molecular weight, 376), appearing initially as fluorescent spots from individual postcapillary pial venules and later as gross and diffuse accumulation of dye underneath the window. The initial dose of Na+-fluorescein was 1 mL/kg body weight of a 2% solution followed by continuous intravenous infusion at a rate of 1 mL/kg body weight to maintain stable plasma concentration (Unterberg et al., 1988). In pilot experiments positive tests of barrier opening were carried out by inducing osmotic BBB disruption with intraarterial infusion of 7.5% NaCl. The response of the cerebral microcirculation was very similar with gross and diffuse opening of the BBB within 10 minutes after infusion.
Histology
After termination of the experiments brains were removed and fixed in 10% formaldehyde. Coronal sections from the lesion site (coronal section 1, Fig. 1) and from underneath the window (coronal section 2, Fig. 1) from both hemispheres were prepared and stained with hematoxylin and eosin. White blood cells in cortex vessels (surface and parenchymal) and WBC that were extravasated into cortical tissue were counted by a histologist blinded to the animal groups in three anatomically similar high-power fields (400×) per hemisphere and section under a light microscope. Results are given in Table 1 as mean number of WBC per high-power field per hemisphere.
White blood cells detected in brain tissue and inside cerebral vessels
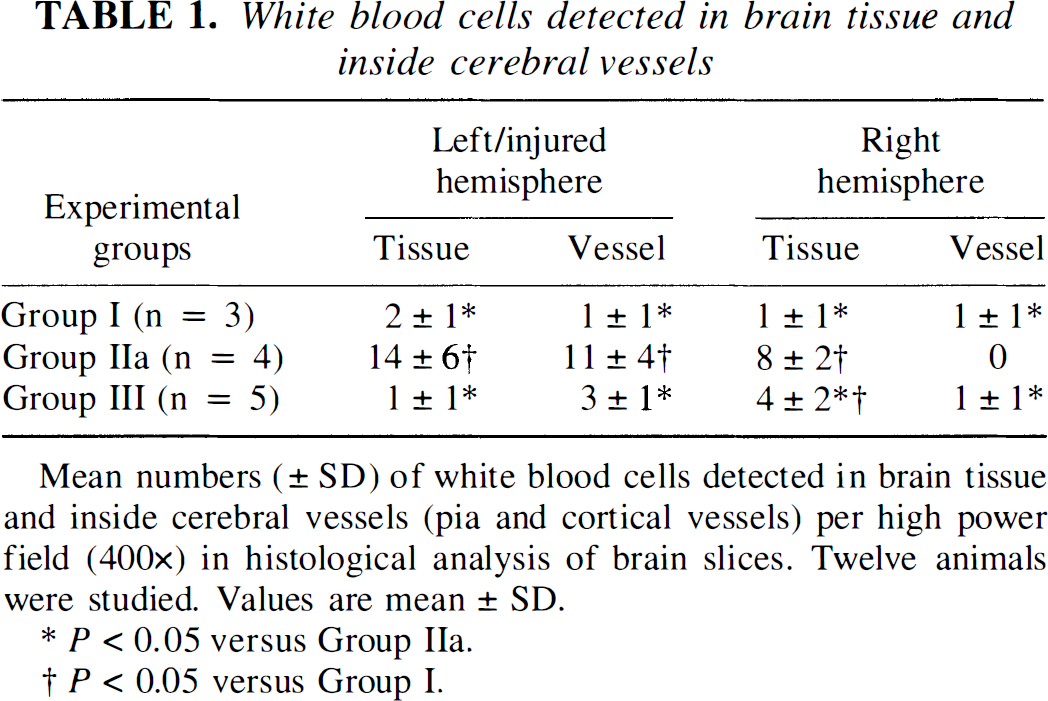
Mean numbers (± SD) of white blood cells detected in brain tissue and inside cerebral vessels (pia and cortical vessels) per high power field (400×) in histological analysis of brain slices. Twelve animals were studied. Values are mean ± SD.
P < 0.05 versus Group IIa.
P < 0.05 versus Group I.
Experimental protocol
Animals with chronic cranial windows were assigned to one of four experimental groups as follows.
Group I. Group I served as a sham-operated control group and did not undergo TBI. After a baseline period of 60 minutes, the animals were studied for 6 hours and were killed by intravenous injection of euthanasia solution. Arteriolar vessel diameters and WBC sticking were determined at specified intervals. Brains were rapidly removed for histology.
Groups IIa and III. After a baseline period of 1 hour, animals in groups IIa and III underwent fluid-percussion brain injury and were monitored for 6 hours. Five minutes before injury animals received either 1.5 mL/kg body weight of normal saline (group IIa) or 1 mg/kg body weight of the well-characterized anti-CD 18 MoAbIB4 (group III) (gift from Samuel Wright, M.D., Rockefeller University, New York, NY, U.S.A.). Arteriolar vessel diameters and WBC sticking were determined at specified intervals. Blood–brain barrier function was tested 5.5 to 6 hours after TBI and animals were killed by intravenous injection of euthanasia solution. Brains were rapidly removed for histology.
Group IIb. After a baseline period of 1 hour animals underwent fluid-percussion injury and were monitored for 1 hour. Blood–brain barrier function was tested at 0.5 to 1 hour after TBI and animals were killed by intravenous injection of euthanasia solution.
Data analysis
One-way analysis of variance was used to compare data in Tables 1 and 2. For Table 3 and Figs. 2 and 3 a two-factor repeated measures analysis of variance with Fisher's post-hoc test for multiple comparisons was used to compare specific time points to baseline. The two-factor randomized analysis of variance with Fisher's post-hoc test was used to test for sigificant differences between treatment groups (P value < 0.05). All variables are depicted as means ± SD.
Blood-brain barrier permeability and WBC margination to venular endothelium after fluid-percussion injury
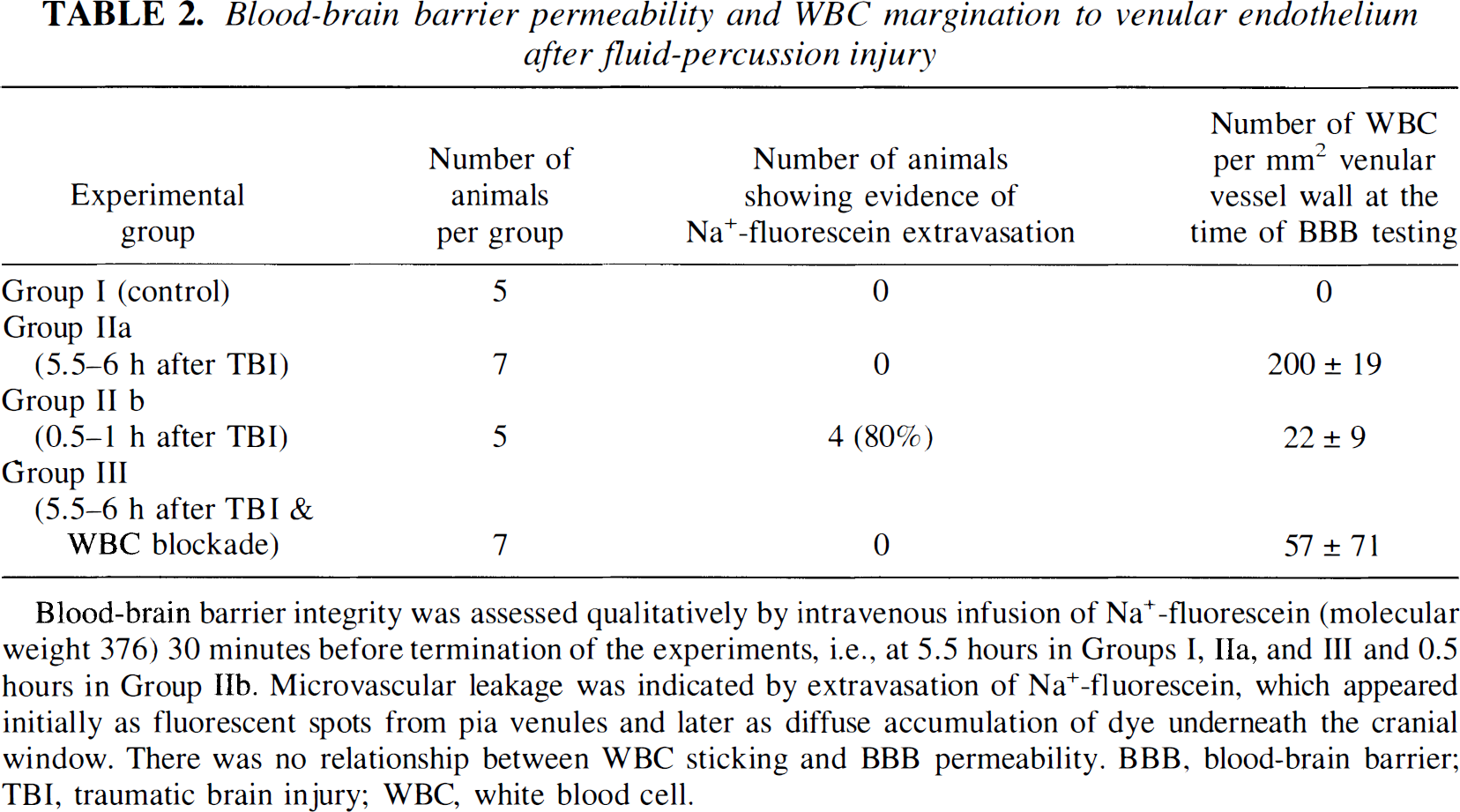
Blood-brain barrier integrity was assessed qualitatively by intravenous infusion of Na+-fluorescein (molecular weight 376) 30 minutes before termination of the experiments, i.e., at 5.5 hours in Groups I, IIa, and III and 0.5 hours in Group IIb. Microvascular leakage was indicated by extravasation of Na+-fluorescein, which appeared initially as fluorescent spots from pia venules and later as diffuse accumulation of dye underneath the cranial window. There was no relationship between WBC sticking and BBB permeability. BBB, blood-brain barrier; TBI, traumatic brain injury; WBC, white blood cell.
Mean arterial blood pressure, intracranial pressure and endtidal P
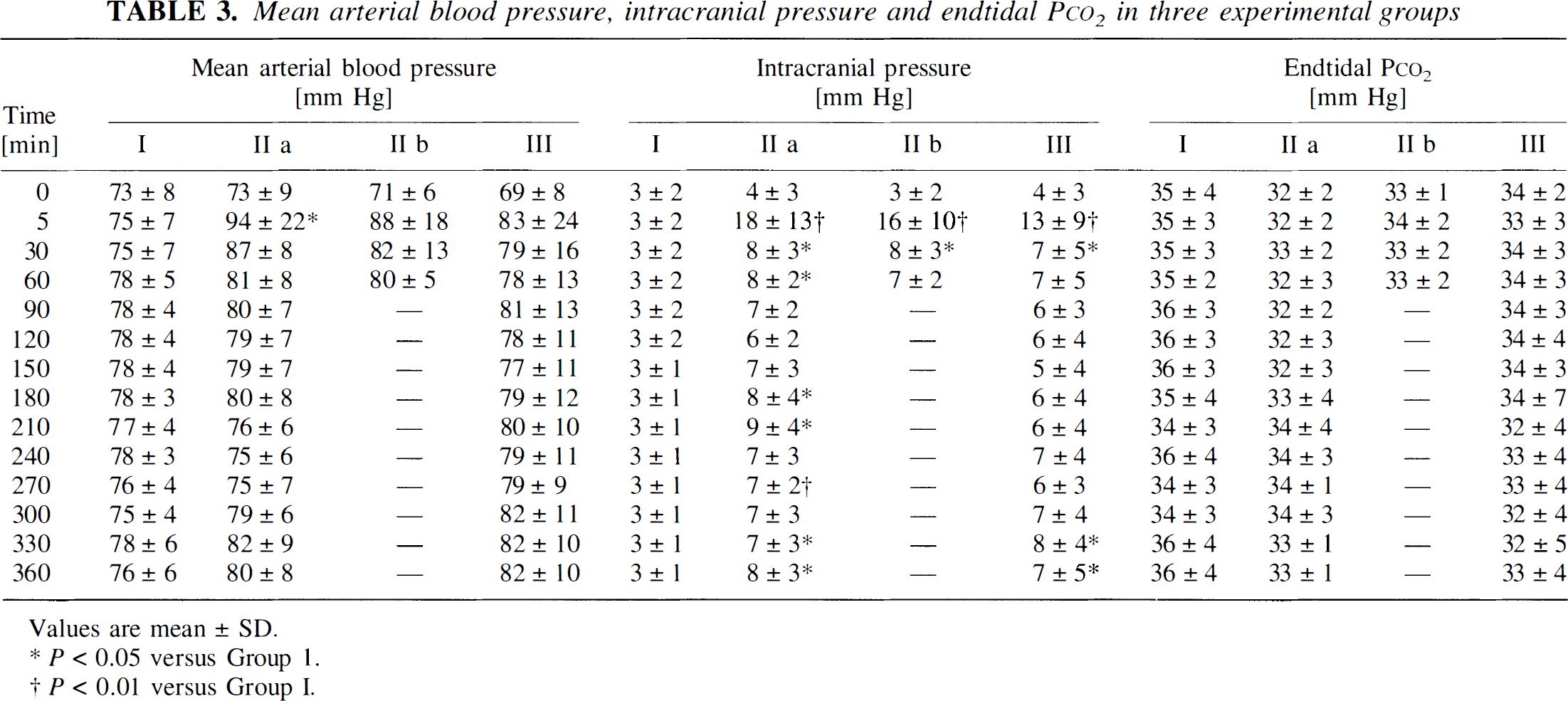
Values are mean ± SD.
P < 0.05 versus Group 1.
P < 0.01 versus Group I.
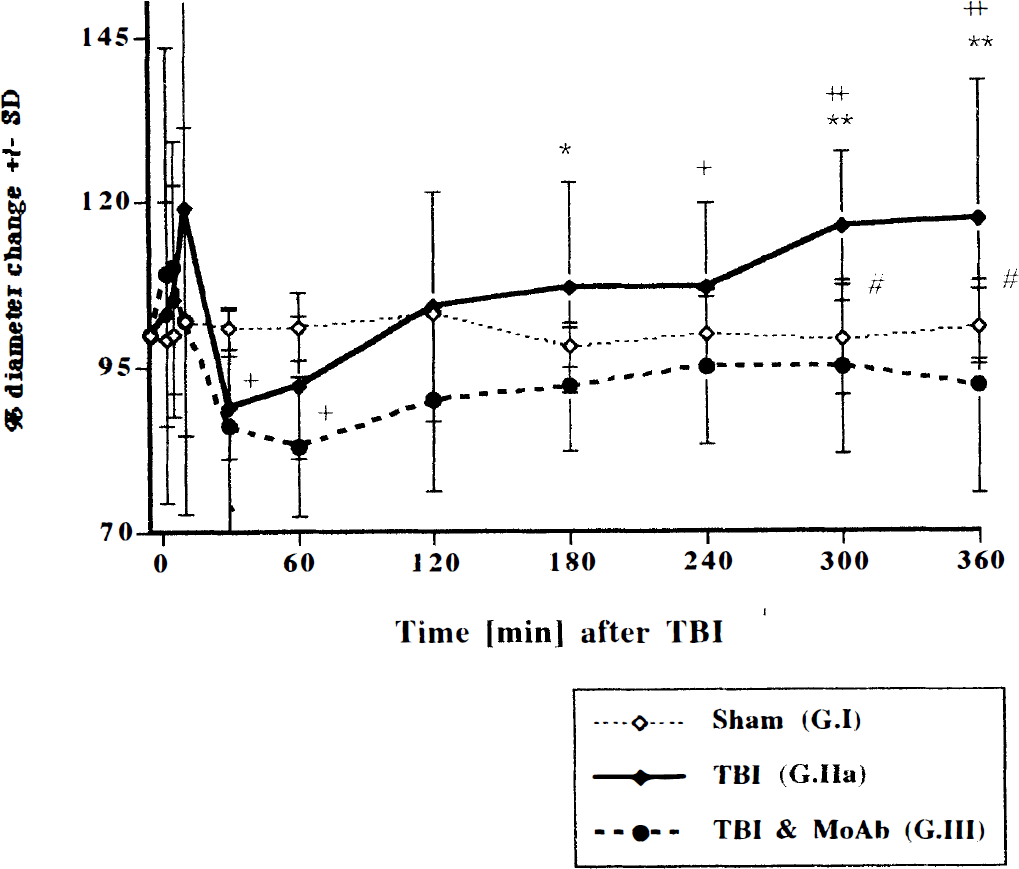
Arteriolar vessel diameters before and after 3.5 atm fluid-percussion traumatic brain injury (TBI) in rabbits. Between six and eight vessels per animal were studied. The monoclonal antibody IB4 completely abolished the secondary vasodilation seen in group IIa. Values are means ± SD. ** P < 0.01 versus group III; * P < 0.05 versus group III; # P < 0.05 group I versus group II; + P < 0.05 versus baseline; ++ P < 0.01 versus baseline.
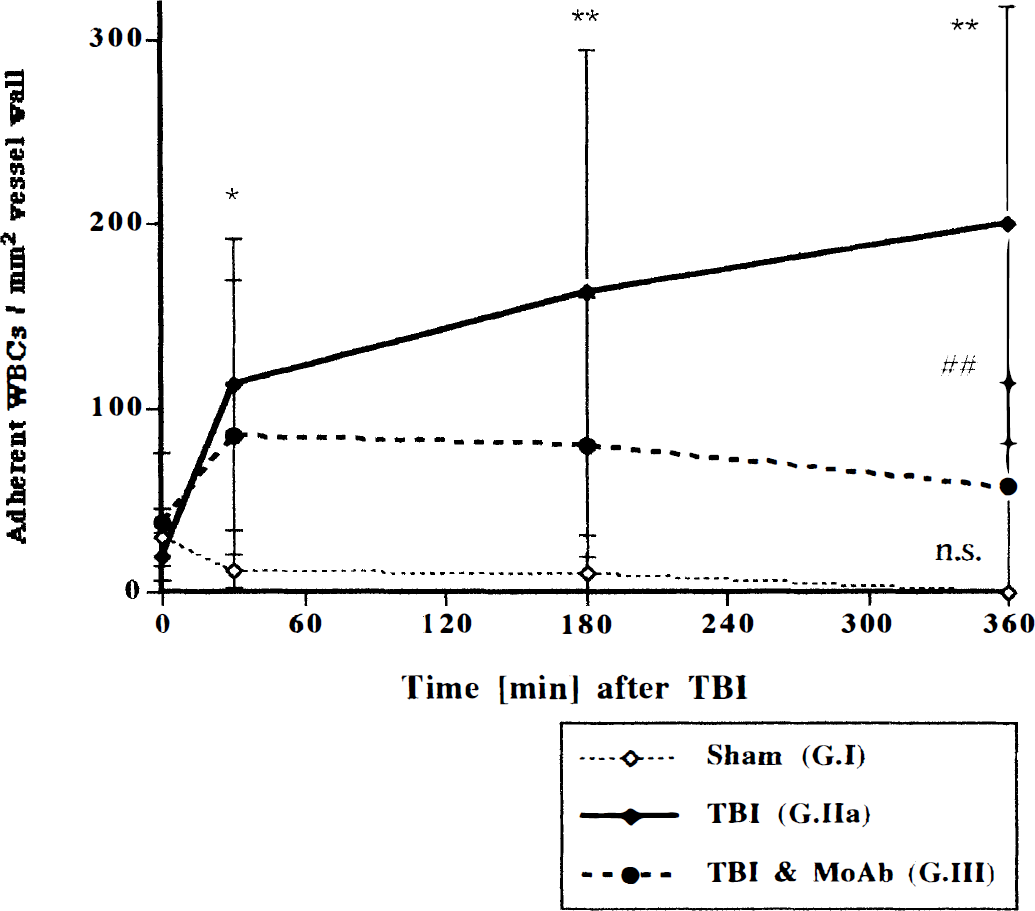
Firmly adherent white blood cells (WBC) in six to eight pial venules per animal. Adherent WBC are cells that remain stationary for more than 30 seconds. This appears to be the minimal duration of firm adhesion that is required as a prelude to emigration during an inflammatory response (Kubes and Granger, 1992). Data are given as number of WBC per mm2 of vessel wall calculated from inner vessel diameter (maximum, 60 μm) and segment length (100 to 150 μm). Values are means ± SD. n.s. not significant; ## P < 0.01 group II versus III; ** P < 0.01 versus baseline; * P < 0.05 versus baseline.
RESULTS
Implantation of chronic cranial window
Daily monitoring of the animals after implantation of the chronic windows did not reveal any signs of distress, pain, or chronic infection. One animal was excluded from the protocol because of an infection caused by an intramuscular injection. Temperatures and WBC counts were controlled at the beginning of each final experiment and were always within normal range.
Trauma severity and pathology
Trauma severity was similar in all goups (3.52 ± 0.6, 3.49 ± 0.4, and 3.50 ± 0.35 atm in groups IIa, III, and IIb, respectively). Structural abnormalities regularly seen after trauma included petechial cortical and subcortical hemorrhage with occasional eruption into the ventricles. Frank contusion with histologic signs of edema was seen directly underneath the impact site in the cerebral cortex. To a lesser degree, intraparenchymal hemorrhage was also observed contralateral to the primary injury site. Subarachnoid hemorrhage was observed ipsilateral to the injury, to a lesser degree also on the contralateral side.
MAP, ICP, and ETco2
Mean arterial blood pressure, ICP, and ET
After trauma MAP rose from 73 ± 9 to 94 ± 22 and from 69 ± 8 to 83 ± 24 mm Hg at 5 minutes after injury in groups IIa and III, respectively, and dropped to near baseline values thereafter.
Intracranial pressure in sham-operated animals remained between 2 and 3 mm Hg throughout the experiments. Traumatic brain injury was followed by increases in ICP from 4 ± 3 mm Hg to 18 ± 13 and 13 ± 9 mm Hg at 5 minutes in groups II and III, respectively. Intracranial brain pressure in groups IIa and III was significantly elevated as compared with group I at 330 and 360 minutes. No significant differences were observed between groups IIa and III.
In group IIb, MAP after trauma rose from 71 ± 6 to 88 ± 18 mm Hg at 5 minutes and dropped to 80 ± 5 mm Hg at 1 hour after injury. Mean arterial blood pressure in group IIa was not significantly different from group IIb. Intracranial pressure increased from 3 ± 2 to 16 ± 10 mm Hg immediately after trauma and was 7 ± 2 mm Hg 1 hour later. Mean ET
Pial arteriolar diameters
Percent changes in arteriolar diameters (AD) over the experimental course are given in Fig. 2. Within 10 minutes after injury, arteriolar vessel diameters transiently dilated to 119% ± 35% and 110% ± 19% of baseline in groups IIa and III, respectively. The second phase was characterized by a vasoconstriction in both groups to 89% ± 8% and 86% ± 18% at 30 minutes after TBI, respectively. Although diameters in group III remained less than 95% of baseline for the rest of the observation period, a secondary vasodilation to 117% ± 21% at 6 hours was seen in group II.
Firmly adherent WBC
The number of WBC firmly adhering to the endothelium of postcapillary pial venules increased significantly from a baseline of 19 ± 13 to 200 ± 119 per mm2 endothelium at 6 hours after injury (Figs. 3–5). In sham-operated controls it decreased from 30 ± 16 to 0 and in group III only a minor, not significant, increase was observed from 38 ± 40 to 57 ± 71 cells per mm2 at 6 hours after TBI. At 6 hours there was a highly significant difference between groups II and III.
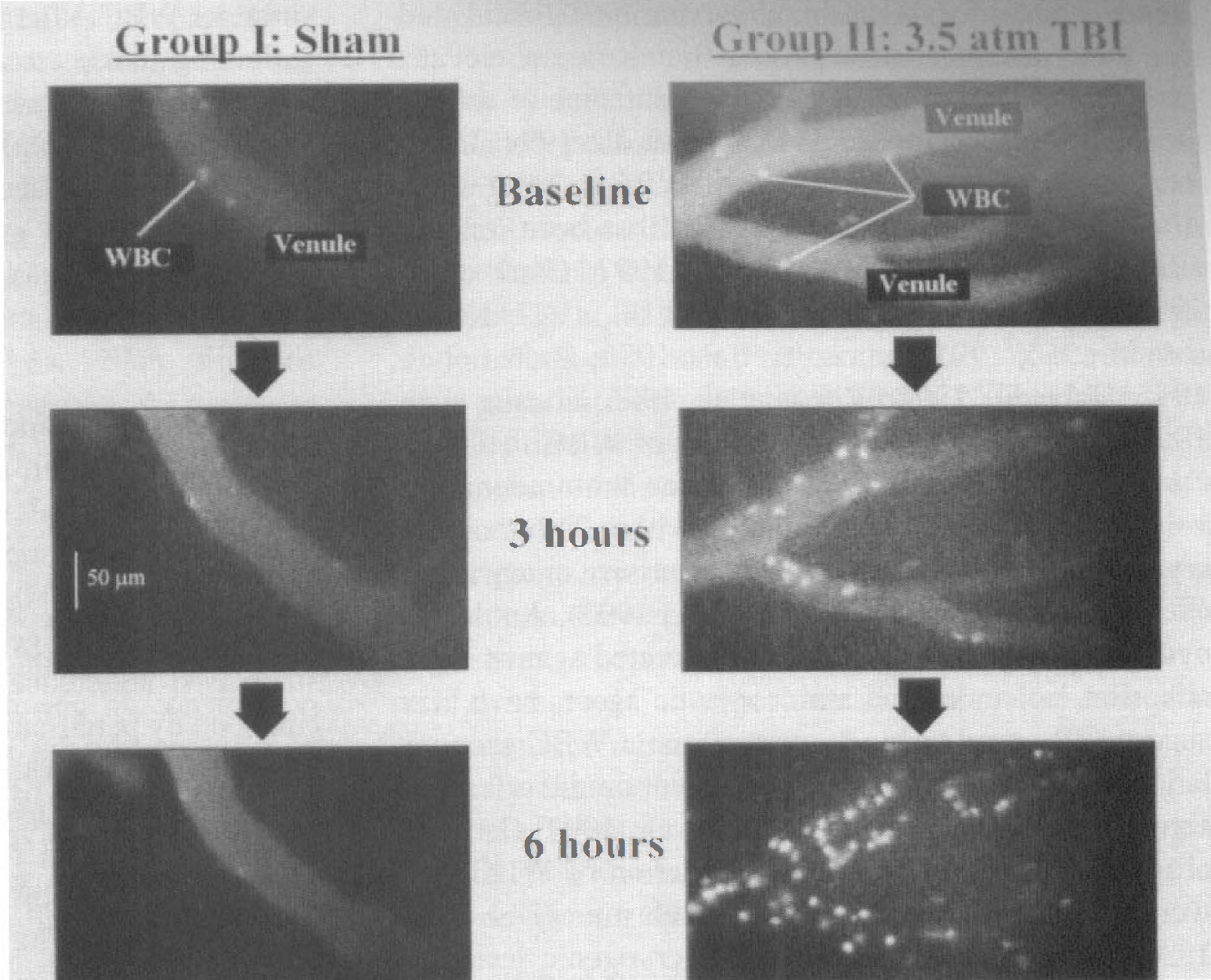
White blood cell adherence to cerebral pial venules in sham-operated animals and after TBI (groups I and IIa, respectively) at baseline and 3 and 6 hours after TBI. White blood cells were labeled with intravenous infusion of rhodamine 6G.
WBC counts
White blood cell counts were checked before fluid-percussion injury and at 6 hours after and were 7.2 ± 3.3 and 8.1 ± 2.9 (×1,000/mm3), respectively, in sham-operated animals, 7.1 ± 4.1 and 8.9 ± 3.9, respectively, after trauma, and 6.5 ± 2.3 and 8.2 ± 3.8, respectively, after trauma and MoAb.
WBC histology
Histologic analysis of brain tissue slices revealed WBC accumulation at 6 hours after TBI not only underneath the window and in close proximity to the lesion, but also in the contralateral hemisphere. Mean numbers of WBC per high-power field detected inside the vessel and in brain parenchyma are given in Table 1. Occasionally, WBC were found in vessels and tissue of sham animals. Traumatic brain injury in group IIa (for histology only, n = 4) led to a steep increase in the number of cells detected in ipsilateral brain tissue (14 ± 6 WBC per high-power field in the injured hemisphere) and inside of vessels (11 ± 4 WBC). In the contralateral hemispheres of group IIa animals the number of WBC in brain tissue was 8 ± 2 (0 WBC in vessels). In the IB4-treated group (group III, n = 5), WBC margination and extravasation were significantly reduced as compared with group IIa, with 1 ± 1 cells in the tissue and 3 ± 1 cells in vessels of the injured hemisphere and 4 ± 2 and 1 ± 1 cells in tissue and vessels of the contralateral hemisphere, respectively.
Blood–brain barrier
Blood–brain barrier as assessed qualitatively by infusion of Na+-fluorescein was found to be disrupted at 1 hour after TBI in four of five animals of group IIb (Table 2). Blood–brain barrier was preserved at 6 hours in all other animals (groups I, IIa, and III) with or without trauma and regardless of the degree of WBC margination.
DISCUSSION
TBI and the acute inflammatory response
The contribution of WBC to the development of infarction in experimental cerebral ischemia, particularly focal cerebral ischemia and reperfusion, is well established (Härtl et al., 1996; Kochanek and Hallenbeck, 1992). There is accumulating evidence that TBI also induces an acute inflammatory response that may promote and aggravate the development of secondary brain damage. Clinical and experimental studies during the past years have found elevated levels of platelet-activating factor, free radicals, and components of the arachidonic acid cascade in serum, parenchyma, and CSF early after head injury (McIntosh, 1994). Interleukin-6, tumor necrosis factor, and other cytokines have been described in the CSF of severely head-injured patients, peaking within the first days after isolated head trauma (Goodman et al., 1990; Kossmann et al., 1996; McClain et al., 1991; Ott et al., 1994). Our group found an association between elevated CSF interleukin-6 levels at 48 hours after isolated clinical severe head trauma and adverse outcome (Medary et al., 1997). Others reported that cytokines are found and produced in the brain in response to experimental trauma (DeKosky et al., 1996; Fan et al., 1995; Shohami et al., 1996; Taupin et al., 1993; Woodroofe et al., 1991; Yan et al., 1992). Inhibition of tumor necrosis factor was shown to decrease edema development at 24 hours and improve functional outcome at 4 days after insults in rats undergoing closed head injury (Shohami et al., 1996).
Hallmarks of secondary brain damage after TBI include typical features of acute inflammation, such as increased microvascular permeability, vasodilation, and blood flow changes. White blood cells could contribute to many of these observations. White blood cells are strategically located within the inflammatory cascade not only as producers of many biochemical mediators, but also as active responders to inflammatory stimuli (Weiss, 1989; Welbourn et al., 1991). White blood cells are potent cytotoxic cells in their own right and might serve as an ideal target for therapeutic intervention. Clinical studies have documented an association between elevated peripheral WBC counts and adverse outcome or secondary ICP increases after TBI (Keskil et al., 1994; Unterberg et al., 1993). Accumulation of WBC in brain within 4 to 48 hours after experimental TBI has been reported (Biagas et al., 1992; Carlos et al., 1997; Clark et al., 1996b; Clark et al., 1994; Horner et al., 1992; Kaczorowski et al., 1995; Schoettle et al., 1990; Soares et al., 1995; Uhl et al., 1994; Wilson et al., 1995; Zhuang et al., 1993). Little is known about the role of WBC in TBI. A correlation has been reported between brain edema development and WBC accumulation within 24 hours after cryogenic lesioning in swine and percussive injury in rats (Schoettle et al., 1990; Zhuang et al., 1993). Antileukocyte interventions such as MoAbs directed against WBC adhesion molecules and antineoplastic agents have been successfully used to block posttraumatic WBC accumulation, but the authors did not comment on the effects on cerebrovascular variables (Biagas et al., 1992; Carlos et al., 1997; Clark et al., 1996a). Depletion of WBC after weight drop injury in rats using vinblastine (Uhl et al., 1994) and blocking of WBC after cryogenic lesioning using antileukocyte serum (Schürer et al., 1990) did not attenuate the formation of edema or necrosis within 24 hours after insult. Taken together, this suggests that WBC migrate into brain tissue early after TBI, but does not sufficiently address the question of whether WBC actually contribute to acute posttraumatic cerebrovascular dysfunction. Against this background the present study was designed to test the hypotheses that (1) WBC accumulate in the cerebral microcirculation within the first hours after fluid-percussion injury, and (2) activated WBC affect the cerebrovascular response to TBI.
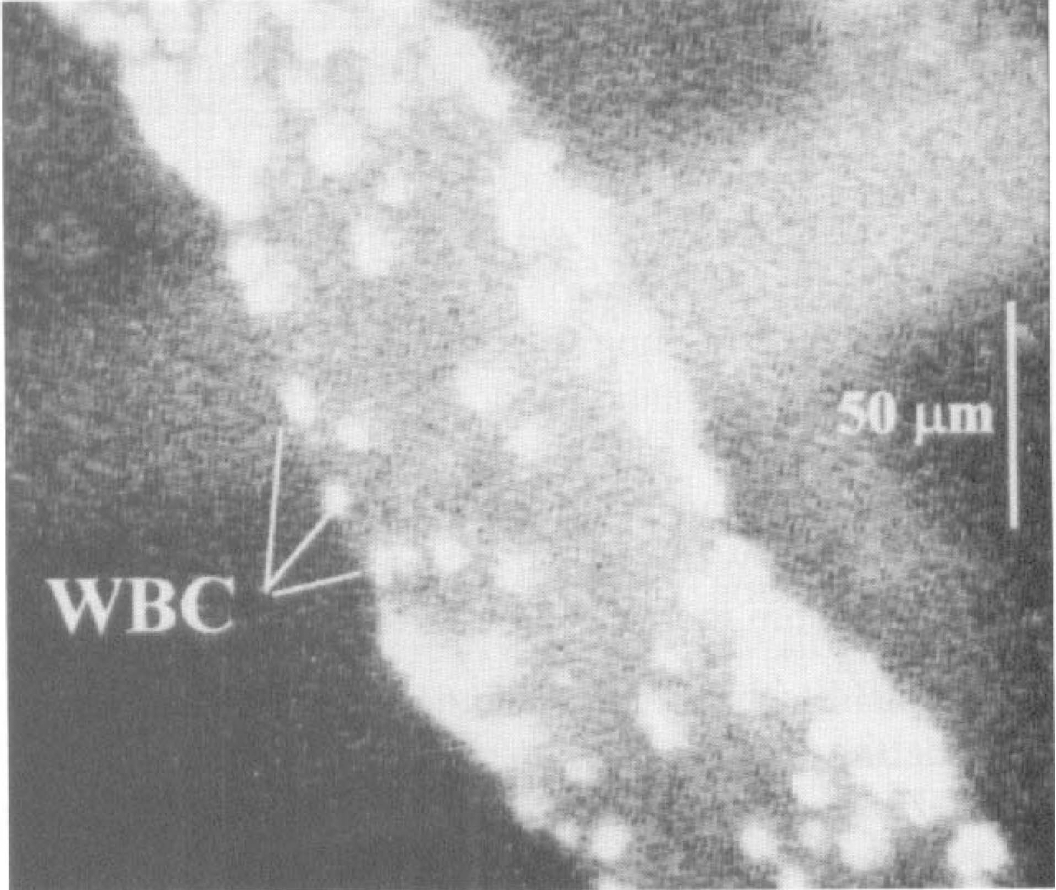
Magnified view of cerebral pial venule at 6 hours (group IIa) showing WBC sticking.
Intravital videomicroscopy
Intravital fluorescence videomicroscopy provides a tool to observe in real time microvascular responses to brain injury in vivo and to record the dynamic character of the posttraumatic response. Although WBC trafficking in experimental cerebral ischemia (Gidday et al., 1995; Villringer et al., 1994) has been studied, no intravital data are available on the early inflammatory response to TBI. In contrast to previous studies, we employed a chronic cranial window technique because pilot experiments indicated that the implantation of glass windows generated an acute local inflammatory response with increased WBC–endothelial cell interaction, which subsided within the first week after implantation.
MoAb IB4 to block WBC–endothelial cell interaction
CD18 is a subunit of the β2 integrin adhesion receptor that can be found on most WBC and that mediates firm adhesion of these cells to the vascular endothelium (Springer, 1994). The anti-CD18 MoAb IB4 effectively inhibits WBC adhesion to venular endothelium without affecting leukocyte rolling or producing cerebral vasoconstriction (Bednar et al., 1996; Carden et al., 1990; Rubin et al., 1992). We did not observe any effect of the MoAb on hemodynamic variables. Lundberg and Wright (1990) reported a greater than 95% reduction of polymorphonuclear leukocyte accumulation in the skin of rabbits after local injection of a chemoattractant and the half-life of IB4 was found to be 11.5 hours.
WBC accumulation and pathology
White blood cell activation has been studied in brain and in peripheral tissues in response to ischemia–reperfusion and superfusion with various chemotactic agents (Corvin et al., 1990; Kurose et al., 1994; Lindauer et al., 1996; Menger et al., 1992). To our knowledge, increased adherence of WBC to vascular endothelium induced by percussion–deformation trauma alone has not been reported previously. The main finding of the current paper is that TBI leads to early and significant accumulation of WBC in the cerebral microcirculation, along with arterial vasodilation and without adverse effect on BBB function. The acute inflammatory response observed in our experiments is not confined to the surface vessels directly underneath the cranial window. Histologic examination of the brains confirmed WBC accumulation in parenchymal vessels in deeper cortical layers and significant migration of WBC into brain parenchyma of both hemispheres (Table 1). Accumulation of WBC during the first 24 hours after TBI injury in rats has also been described by other groups, but was usually confined to the impacted hemisphere (Clark et al., 1994; Schoettle et al., 1990; Soares et al., 1995; Zhuang et al., 1993). The occurrence of WBC in the hemisphere opposite to the lesion may be related to the presence of pathologic alterations that occurred not only in the directly traumatized hemisphere, but to a lesser degree also contralateral. An explanation for the contralateral injury may be the modification of the trauma model: conventional fluid-percussion models produce brain injury by rapidly injecting saline into the closed cranium. This leads to diffuse mechanical loading of the brain surface and only little movement of the brain (Dixon et al., 1988; Thibault et al., 1992). We used a sterile piece of rubber fixed over the end of the injury screw to prevent the loss of potentially unsterile saline into the cranial cavity. This resulted in local contusion underneath the impact site with movement of the brain and contralateral injury.
WBC activation and cerebrovascular response to fluid-percussion injury
Little is known about the time course of WBC activation after TBI and its relationship to other variables of the cerebral microcirculation. Our data demonstrate that WBC adhesiveness increases immediately after TBI. The degree of WBC sticking in our experiments (200 WBC/mm2 endothelium at 6 hours after TBI) is comparable to data by Gidday et al. (1995) at 2 hours after 90 minutes of cerebral ischemia–reperfusion in piglets (approximately 60 WBC/mm2). Ischemia–reperfusion in striated muscle appears to trigger an earlier and more pronounced WBC response with 400 to 1500 firmly adherent WBC/mm2 endothelium after only 2 hours (Menger et al., 1992; Nolte et al., 1994). The question arises as to whether WBC activation after TBI is of any relevance for the development of posttraumatic cerebrovascular dysfunction, such as BBB disruption and intracranial hypertension.
Arteriolar diameters
The posttraumatic course in our experiments was characterized by triphasic changes in pial AD with initial vasodilation (0 to 10 minutes), followed by constriction (30 to 90 minutes) and secondary dilation to 117% ± 21% of baseline at 6 hours after TBI (Fig. 2). The late-phase vasodilation was prevented by blocking WBC using the anti-CD 18 MoAb. Increases in arteriolar vessel diameters have also been reported by others in experimental animals within the first hours after fluid-percussion injury (De Witt et al., 1986; Wei et al., 1980; Wei et al., 1981). Increases in AD immediately after fluid-percussion injury probably reflect a passive dilation caused by the posttraumatic blood pressure surge typically associated with this type of injury, overriding cerebral autoregulation. In fact, some animals displayed blood pressure peaks within the first minute after TBI as high as 200 mm Hg. The vasoconstriction observed between 30 and 90 minutes occurs as the autoregulatory function of cerebral arterioles is restored.
We cannot fully explain the delayed vasodilation after trauma, but our results indicate that WBC activation was involved in this response. Corvin et al. (1990) observed in rats equipped with cranial windows that superfusion of the brain surface with the leukotaxin n-formylmethionyl-leucyl-phenylalanine did induce WBC adherence along with arteriolar dilation (Corvin et al., 1990). Together with our study this is the only published report on a temporal and spatial association between WBC sticking and vessel behavior in the intact brain microcirculation. White blood cell sludging in the downstream microcirculation, similar to the one observed by us, can remarkably increase organ resistance in skeletal muscle, mesentery, and parenchymal organs (Helmke et al., 1996; House and Lipowsky, 1987). Compensatory arteriolar dilation in response to this elevated cerebrovascular resistance arises as a potential explanation for our findings (Kontos and Wei, 1985; Pfenninger et al., 1989). Arteriolar vasodilation after fluid-percussion injury in experimental animals has also been attributed to the production of free radicals (Kontos and Wei, 1986; Wei et al., 1981). White blood cells are a major source of oxygen free radicals, and a recent study by Matsuo et al. (1995) demonstrated that depletion of neutrophils in a model of focal cerebral ischemia in rats significantly attenuated postischemic free radical production. However, WBC accumulation occurs exclusively in venules and it is unclear how free radical production in venules affects AD. In isolated middle cerebral arterial segments Akopov et al. (1994) found acute disturbances of the endothelium-dependent relaxation after WBC stimulation with intraarterial 4b-phorbol-12b-myristate-13a-acetate, indicating that WBC cause vasoconstriction rather than dilation. These experimental results cannot be applied to the in vivo situation as it presents in the current experiments: in our experiments we did not observe an interaction between WBC and arteriolar endothelium. Moreover, Akopov et al. (1994) used large, isolated cerebral vessels in an experimental setup that did not take into account the influence of vascular resistance and pressure changes along the intact vascular tree.
These hypotheses notwithstanding, whether arterial vasodilation as observed in our experiments is a sign of cerebrovascular dysfunction remains unclear.
Intracranial pressure
A study of thromboembolic stroke combined with hemorrhagic hypotension in New Zealand rabbits treated with 1 mg/kg IB4 noted significantly reduced ICP as compared with vehicle-treated controls within 4 hours after the insult (Bednar et al., 1996). Our findings do not support a similar effect of WBC inhibition on ICP early after TBI. Traumatic brain injury in groups IIa, IIb, and III led to a significant rise in ICP. White blood cell blockade and reduction of vessel diameters in group III did not reduce ICP when compared with group IIa.
Blood–brain barrier
Tanno et al. (1992a, 1882b) reported that in rats undergoing fluid-percussion injury BBB damage was most pronounced within the first hour after TBI and the BBB was reestablished by 6 hours after injury. Others also reported that the BBB sealed within a few hours after fluid-percussion injury (Enters et al., 1992; van den Brink et al., 1994). Barzo and coworkers (1996) recently presented data from rats undergoing impact-acceleration injury demonstrating that the BBB opens rapidly and transiently after trauma for approximately 30 minutes. This correlates with our observation that the disruption of the BBB was more pronounced between 30 minutes and 1 hour as compared with 5.5 to 6 hours after trauma. The current results clearly show that WBC sticking in pial venules after fluid-percussion injury was not related to increased vessel permeability (Table 2). Data on the relationship between WBC sticking and vascular permeability in the brain are conflicting. Although cerebral ischemia studies reported a positive correlation between brain edema and WBC accumulation within 24 hours after middle cerebral artery occlusion and reperfusion in rats (Matsuo et al., 1994; Shiga et al., 1991), TBI studies failed to support this view. Schürer and coworkers (1990) demonstrated that depletion of WBC after cryogenic lesion was actually associated with more pronounced edema formation. In a weight drop model in rats, Uhl et al. (1994) failed to demonstrate an effect of pharmacologically induced neutropenia on the development of brain edema 24 hours after injury. In a preliminary study Whalen et al. (1997) found a pattern of transient BBB opening that was independent of inflammation: the BBB was leaky within the first 30 minutes to 4 hours after controlled cortical impact in rats whereas WBC accumulation peaked at 24 hours. From these and our studies, it seems that even though WBC accumulation occurs after both insults, TBI and ischemia, the effects on edema formation and BBB damage may be different. In a study similar to ours, Soares et al. (1995) found that lateral fluid percussion in rats elicited an inflammatory leukocytic recruitment only in regions experiencing concomitant BBB damage. However, the authors did not demonstrate a causal relationship between WBC extravasation and barrier breakdown and it is possible that WBC migration was a secondary event. The potential difference between cerebral ischemia and TBI deserves more attention in the future.
Mechanical trauma leads to WBC activation in peripheral tissues: increased neutrophil infiltration has been described within 30 to 180 minutes after traumatic shock in the intestines of rats (Scalia et al., 1996), increased venular WBC sticking was seen within hours after hind limb amputation–reimplantation in the muscle of rats (Siemionow et al., 1995), and WBC margination is commonly seen early after surgical trauma in rat cremaster muscle (Kunkel et al., 1996). In peripheral organs WBC sticking is associated with increased vascular permeability (Kubes et al., 1990). The degree of WBC sticking observed in the present experiments at 6 hours after TBI was less than has been reported after ischemia–reperfusion in striated muscle (400 to 1,500 sticking WBC/mm2 endothelium after only 2 hours; Menger et al., 1992; Nolte et al., 1994). It is possible that the degree of WBC activation in the present experiments was not sufficient to induce increased cerebrovascular permeability and that BBB disruption may occur more than 6 hours after TBI when more WBC are activated.
Taken together, the literature and our findings do not provide convincing evidence for a causal relationship between WBC adhesion and BBB leakage early after TBI. Structural differences between the BBB and peripheral endothelium, such as the existence of tight junctions in the former, may confer some degree of protection that renders the vascular lining in the brain less vulnerable to WBC-induced permeability changes. However, our results do not exclude the possibility that WBC may cause delayed BBB breakdown more than 6 hours after TBI.
CONCLUSION
The present experiments provide the first description of WBC dynamics in the cerebral microcirculation early after mechanical brain trauma and reveal a complex response of the cerebral microcirculation to TBI as follows: (1) Increased adherence of WBC to the vascular endothelium of pial venules in rabbits begins within 30 minutes after trauma and is associated with inflammatory cell extravasation into brain ipsilateral and contralateral to the lesion within 6 hours after injury. (2) White blood cell sticking induces dilation of cerebral arterioles that can be abolished by pharmacologic blockade of the WBC–endothelial interaction. (3) Blood–brain barrier disruption occurs within the first 60 minutes after TBI and is not related to WBC activation. White blood cell activation does not contribute to elevated ICP within 6 hours after TBI.
However, WBC are activated very early after TBI. Their possible role as mediators of secondary brain damage at a later point (more than 6 hours after TBI) and the effect of secondary insults such as hypoxia and arterial hypotension on WBC behavior should be investigated further.