
Abstract
The hypothesis was tested that hyperbaric oxygen therapy (HBO) reduced brain infarction by preventing apoptotic death in ischemic cortex in a rat model of focal cerebral ischemia. Male Sprague-Dawley rats were subjected to middle cerebral artery occlusion/reperfusion (MCAO/R) and subsequently were exposed to HBO (2.5 atmospheres absolute) for 2 h, at 6 h after reperfusion. Rats were killed and brain samples were collected at 24, 48, 72 h, and 7 days after reperfusion. Neurologic deficits, infarction area, and apoptotic changes were evaluated by clinical scores, 2,3,7-triphenyltetrazolium chloride staining, caspase-3 expression, DNA fragmentation assay, and terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL)-hematoxylin and eosin (H&E) costaining. In MCAO/R without HBO treatment animals, DNA fragmentation was observed in injured cortex at 24, 48, and 72 h but not in samples at 7 days after reperfusion. Double labeling of brain slides with NeuN and caspase-3 demonstrated neurons in the injured cortex labeled with caspase-3. TUNEL+H&E costaining revealed morphologic apoptotic changes at 24, 48, and 72 h after reperfusion. Hyperbaric oxygen therapy abolished DNA fragmentation and reduced the number of TUNEL-positive cells. Hyperbaric oxygen therapy reduced infarct area and improved neurologic scores at 7 days after reperfusion. One of the molecular mechanisms of HBO-induced brain protection is to prevent apoptosis, and this effect of HBO might preserve more brain tissues and promote neurologic functional recovery.
Cell death, either necrosis or apoptosis, occurred in the brain tissues during the first few days after cerebral ischemia (Johnson et al., 1995; Rink et al., 1995). Necrotic cell death is characterized by cellular swelling, nuclear pyknosis with karyorrhexis, and cytoplasmic eosinophilia (Searle et al., 1982; Dure et al., 1995). Apoptosis is characterized by morphologic and biochemical features, including cell shrinkage, formation of apoptotic bodies, and extensive internucleosomal fragmentation (Johnson Jr et al., 1995, 1996). Cerebral hypoxia/ischemia produces a cascade of interconnected pathologic processes, including changes in intracellular Ca2+, excitatory amino acid, oxidative stress, and inflammatory response, and leads to apoptosis in the ischemic penumbra (Okamoto et al., 1993; Johnson Jr et al., 1995; Rink et al., 1995; Leker et al., 1999). Prevention of apoptosis becomes a therapeutic strategy to preserve brain tissues and promote functional recovery (Graham and Chen, 2001).
Hyperbaric oxygen therapy (HBO) is a potent mean to increase the amount of oxygen dissolved in blood plasma and thereby delivered to the ischemic brain. However, HBO is not currently used in acute stroke management, partially due to the insufficient information of its molecular mechanisms (Kawamura et al., 1990; Anderson et al., 1991; Mink and Dutka, 1995; Nighoghossian and Trouillas, 1997; Chuba et al., 1997; Roos et al., 1998; Veltkamp et al., 2000; Badr et al., 2001a). We have found previously that HBO regulates brain metabolism (Badr et al., 2001b) and reduces the expression of COX-2 (Yin et al., 2002); it affects both neuronal injury and cell survival. In one of our recent studies, we have shown in a neonatal hypoxia rat model that apoptotic bodies appeared in the injured cortex and that HBO reduced the number of apoptotic cells (Calvert et al., 2002). However, because we used electron microscopy in that study, the data were not quantified. We postulated that HBO increases the oxygen level in ischemic brain regions (Sunami et al., 2000), decreases hypoxia, and reduces brain injury by inhibition of apoptosis. We evaluated neurologic function, infarct areas, and apoptotic changes using specific methods including caspase-3 expression, DNA fragmentation assay, and terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining. We applied HBO in the present study 6 h after ischemia and reperfusion to test HBO as a potential additional or alternative therapy for patients with acute stroke.
MATERIALS AND METHODS
These studies comply with the National Institute of Health's Guide for Care and Use of Laboratory Animals and are approved by the Institutional Animal Care and Use Committee at the University Mississippi Medical Center and at the Louisiana State University Health Science Center–Shreveport.
Preparation of middle cerebral artery occlusion/reperfusion rat model
Male Sprague-Dawley rats, weighing 300 to 350 g, were subjected to middle cerebral artery occlusion (MCAO) as described by Kittaka et al. (1997) and as described previously (Badr et al., 2001b). Briefly, the rats were anesthetized with an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg) and were allowed to breathe spontaneously. A supplemental dose of anesthetics was added if necessary. With the aid of a microscope, the left common carotid artery was exposed, and the external carotid artery was isolated and coagulated. A 3–0 nylon suture with a blunted tip was inserted into the internal carotid artery through the external carotid artery stump and was gently advanced to occlude the middle cerebral artery. After 2 h of MCA occlusion, the suture was carefully removed to restore blood flow; the skin was sutured; and the rats were allowed to wake up. To complete the surgery, the operator applied 0.25% micaine (Sigma, St. Louis, MO, U.S.A.) locally to the wound and allowed the rat to recover. Body temperature was maintained with a heating blanket at 37 ± 0.5°C during surgery and before they awoke from surgery. A successful occlusion of the left MCA is assumed when the right forelimb is paretic after filament introduction (Veltkamp et al., 2000).
Hyperbaric oxygen treatment
The rats were placed into an HBO chamber [2.5 atmospheres absolute (ATA)] for 2 h for one treatment as described previously (Badr et al., 2001a, b ; Yin et al., 2002). The HBO treatment was performed at 6 h after reperfusion. On reaching the desired pressure, the flow of oxygen (100%) was reduced to maintain constant pressure while allowing a flow out of the chamber. This constant exchange accompanied by a tray of calcium carbonate crystals was used to reduce the accumulation of CO2 in the chamber environment. After HBO, the rats were placed back into the cage until they were killed. Body temperature was not changed markedly or significantly (within 0.5°C) after HBO as shown previously (Badr et al., 2001a).
Experimental protocol
Fifty-four rats were divided randomly into the following 3 groups: control (sham surgery), middle cerebral artery occlusion/reperfusion (MCAO/R), and MCAO/R with HBO treatment. The rats in control group (n = 6) underwent left common carotid artery and external carotid exposure without MCA occlusion, and did not undergo HBO treatment. In MCAO/R group (n = 24), the MCA was occluded for 2 h and reperfused without HBO treatment. In the MCAO/R with HBO group (n = 24), HBO was applied at 6 h after reperfusion. The experimental protocol is shown in Fig. 1. The designed reperfusion duration was 24 h, 48 h, 72 h, and 7 days, respectively. The animals were not anesthetized and breathed spontaneously during HBO treatment. All animals were observed closely from 24 h to 7 days to evaluate neurologic function. On a designated time after reperfusion, the rats were killed, their brains were quickly removed, and samples were collected for infarction ratio and apoptosis.
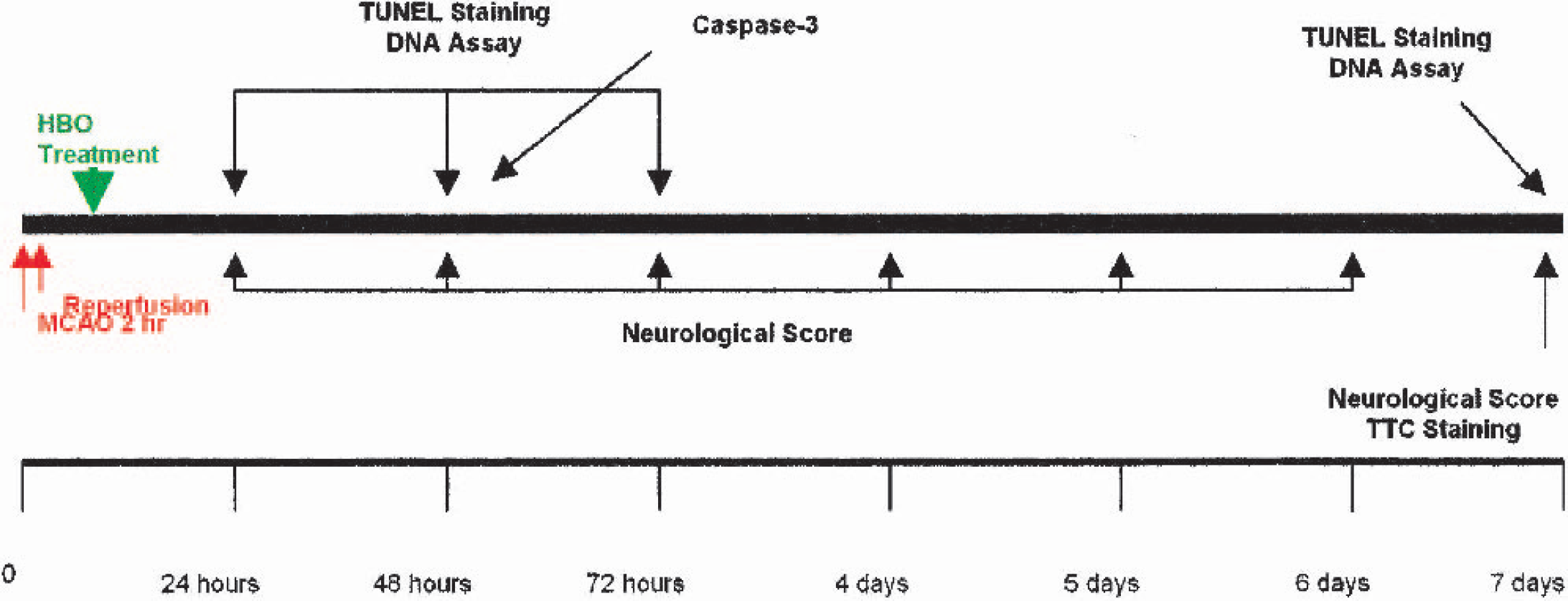
Flow chart illustrates the timing of middle cerebral artery occlusion (MCAO)/reperfusion, hyperbaric oxygen therapy (HBO) treatment, neurological score, 2,3,7-triphenyltetrazolium chloride (TTC) staining, caspase-3 expression, terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining and DNA assay.
Neurologic evaluation
At designated times, neurologic deficits of rats with and without HBO treatment were evaluated according to the method developed by Menzies et al. (1992) and as described previously (Badr et al., 2001a). A grade of 0 indicates no visible neurologic deficits. A grade of 1 is given to an animal displaying forelimb flexion. The animal is then placed on an absorbent pad and gently pulled by the tail. If the animal shows a weakness in grip in the contralateral forelimb, it receives a grade of 2. The animal is then allowed to move about freely. If it circles to the paretic side only when pulled by the tail, a grade 3 is assigned. However, if it spontaneously circles, a grade of 4 is assigned.
TTC staining
2,3,7-Triphenyltetrazolium chloride (TTC) staining was performed at 7 days after reperfusion as described previously (Badr et al., 2001b; Yin et al., 2002). Coronal sections of the brain (2 mm thick) were cut and immersed in a 2% solution of TTC for 30 minutes at 37°C. The stained slices were then fixed by immersion in 10% formaldehyde solution. The infarction area and hemisphere area of each section were traced and measured using an image analysis system [SigmaScan/Image (Image Measurement Soft), Jandel Scientific Software, San Rafael, CA, U.S.A.]. The percentage of infarction (infarct ratio) was calculated by dividing the infarct volume by the total volume of the ipsilateral hemisphere.
DNA fragmentation assay
After the brains were removed, a 4-mm-thick coronal brain slice was cut at the level of optic chiasm, and the cortex including infarction was dissected using the corpus callosum as a ventral landmark. Analysis of DNA fragmentation was performed using methods described previously (Yin et al., 1994; Meguro et al., 2001a,b). Tissues from brain cortex including infarcted area were homogenized and centrifuged briefly and washed twice with cold PBS. The pellet was lysed in 1.0 mL of a buffer consisting of 10 mmol/L Tris-HCl, 10 mmol/L ethylenediamine tetraacetic acid (EDTA) and 0.2% Triton X-100 (pH 7.5). After 10 minutes on ice, the lysate was centrifuged (13,000g) for 10 minutes at 4°C. The RNA and fragmented DNA in the supernatant were extracted, first with phenol and then with phenol-chloroform-isoamyl alcohol (25:24:1, vol:vol). The aqueous phase was added to 300 mmol/L NaCl, and the nucleic acids were precipitated with 2 volumes of ethanol. The pellet was rinsed with 70% ethanol, air dried, and then dissolved in 20 μL of 10 mmol/L Tris-HCl-1 mmol/L EDTA (pH 7.5). After digestion of the RNA with ribonuclease A (0.6 mg/mL at 37°C for 30 minutes), the samples were electrophoresed in a 2% agarose gel with Boyer's buffer (50 mmol/L Tris-HCl, 20 mmol/L sodium acetate, 2 mmol/L EDTA, and 18 mmol/L NaCl at pH 8.05). The DNA was visualized with ethidium bromide staining.
TUNEL staining
Immunohistology using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Intergen Co., Purchase, NY, U.S.A.) was employed to demonstrated apoptotic cells in ischemic brain tissue as described previously (Ogihara et al., 2000). Five-micron-thick serial coronal sections were cut from the cortex including infarction and were affixed to a slide by heating at 37°C overnight, deparaffinized, and dehydrated. After digestion with proteinase K (20 g/mL; Sigma Chemical Co., St. Louis, MO, U.S.A.) in 0.05 mol/L PBS at room temperature for 15 minutes and extensive washes in PBS, the sections were treated with 3% hydrogen peroxide in PBS to quench endogenous peroxidase activity for removing background staining. Labeling of 3′-OH terminal DNA fragments was then performed by using the above in situ apoptosis detection kit according to the manufacturer's protocol. The counterstaining was performed with 1% methyl green.
Some of the slides were costained with TUNEL and hematoxylin and eosin (H&E) for the evaluation of infarction as well as apoptosis. Quantitative (number of TUNEL-positive cells) analyses were performed at the ipsilateral cortex including both ischemic core and penumbra regions. The quantification of apoptotic cells was determined by Imaging-Pro Plus. The TUNEL-positive cells displayed a brown staining within the nucleus or cytoplasm in the apoptotic cells. Because computer automatic counting might include false-positive cells, the numbers generated only show a tendency of changes of apoptosis.
Double fluorescent immunohistochemical staining
Frozen tissue sections were cut 8 μm thick and were air dried for 30 minutes. Sections were incubated in 1% hydrogen peroxide (10 minutes), in 3% normal goat serum in PBS (30 minutes), and then incubated with mouse anti NeuN antibody (Chemicon Inc., 1:100) overnight at 4°C. Sections were later incubated with goat anti mouse-TXRED IgG 1:200 for 2 h. Between each step, sections were washed three times with PBS for 5 minutes each. Then the sections were treated with nonspecific binding with 3% bovine serum albumin in phosphate buffered saline with Tween-20 (PBST) and incubated with fluorescence-conjugated caspase-3 antibody (Cell Signaling Inc., 1:100), shielded from light with gentle shaking overnight at 4°C. The sections were washed twice for 10 minutes each time with PBS. The sections were mounted on coverslips, and the slides were visualized under the fluorescent microscope (Olympus). Color composite was done by Imaging-Pro Plus.
Data analysis
In this study, data were represented as mean ± SD. Statistical differences between the MCAO/R group (no HBO treatment) and the other groups were compared by using the one-way analysis of variance and then Scheffé F test to determine whether a significant difference was found; A P value less than 0.05 was considered statistically significant.
RESULTS
Hyperbaric oxygen therapy attenuates neurologic deficits
The results of the neurologic deficit score in each group are shown in Fig. 2. In MCAO/R group, the neurologic scores were 3.2, 2.8, 2.8, and 2.6 at 24 h, 48 h, 72 h, and 7 days after reperfusion, respectively. In MCAO/R with HBO treatment group, the neurologic scores were 2.7, 2.4, and 2.3 at 24 h, 48 h, 72 h, and 1.8 at 7 days (P < 0.05 vs. MCAO/R group) after reperfusion, respectively. Hyperbaric oxygen therapy enhanced neurologic recovery after MCAO/R at day 7 (P < 0.05). No neurologic deficit was observed in the sham-operated rats.
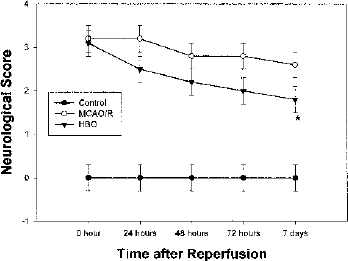
Changes of neurologic scores before and after hyperbaric oxygen therapy (HBO) treatment. Grades of 0 to 4 were used. *P < 0.05 versus no HBO treatment.
Hyperbaric oxygen therapy reduces cerebral infarction
The representative samples of TTC-stained brain sections from animals killed at 7 days after sham operation, MCAO/R, and MCAO/R with HBO treatment are shown in Fig. 3. The coronal sections were obtained by cutting the brain at a distance of 2, 4, 6, 8, and 10 mm from the rostral extremity of the frontal cortex. The white areas represent the infarction regions in these sections. Figure 4 shows the percentage of infarction in MCAO/R and MCAO/R with HBO treatment groups, respectively. In the MCAO/R group (untreated group), severe cerebral infarction was observed in all rats and the infarct ratio was 20.1% at 7 days. The infarct ratio was significantly decreased in rats treated with HBO after reperfusion at 7 days (12.6%, P < 0.05 vs. MCAO/R).
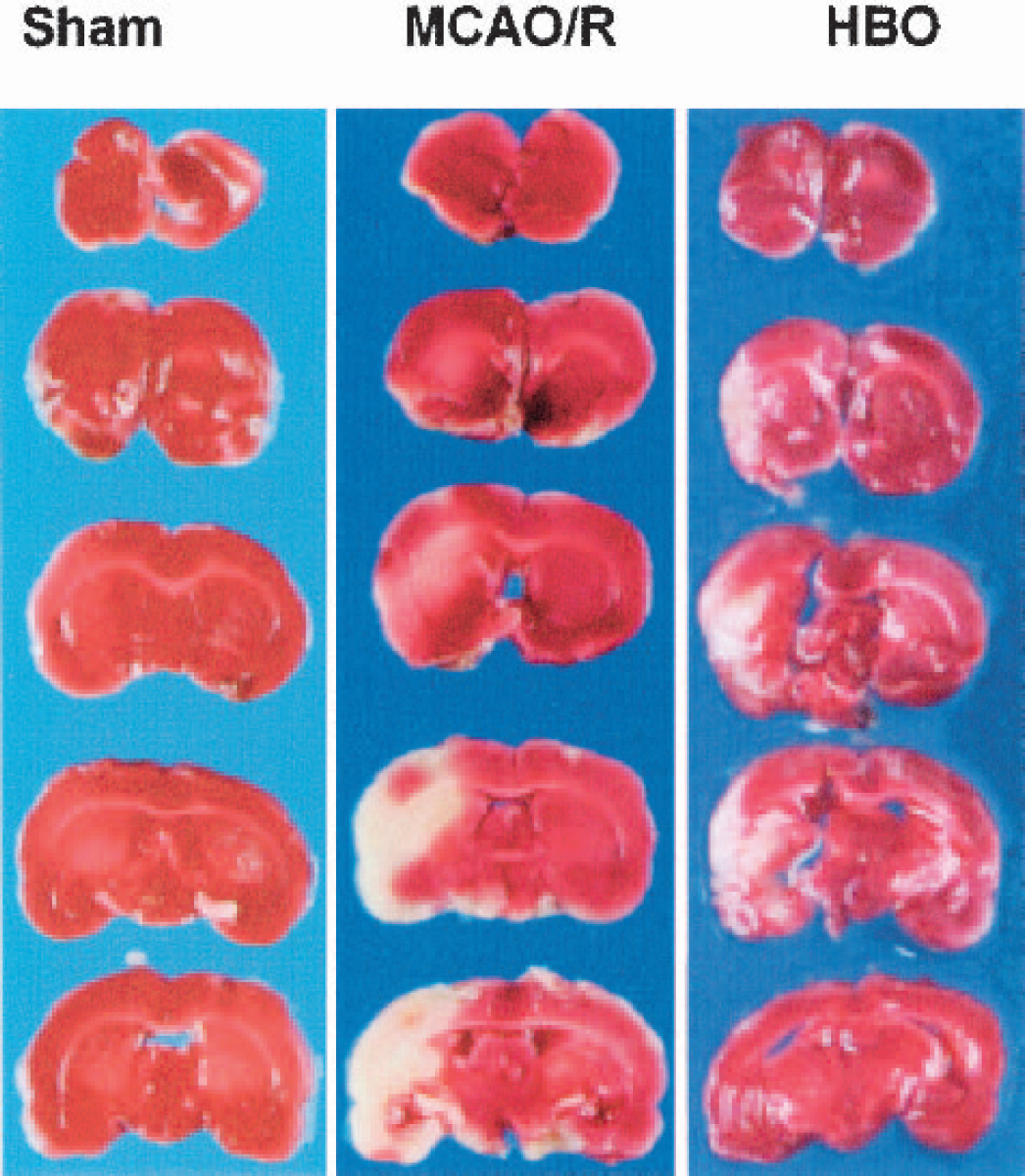
2,3,7-Triphenyltetrazolium chloride (TTC) staining. Representative sample of TTC-stained brain sections from rats killed at 7 days after ischemia. Middle cerebral artery occlusion/reperfusion (MCAO/R) animals showed severe infarctions. The white areas represent the infarct regions in these sections. In contrast, animals exposed to HBO treatment had a significant reduction in infarction.
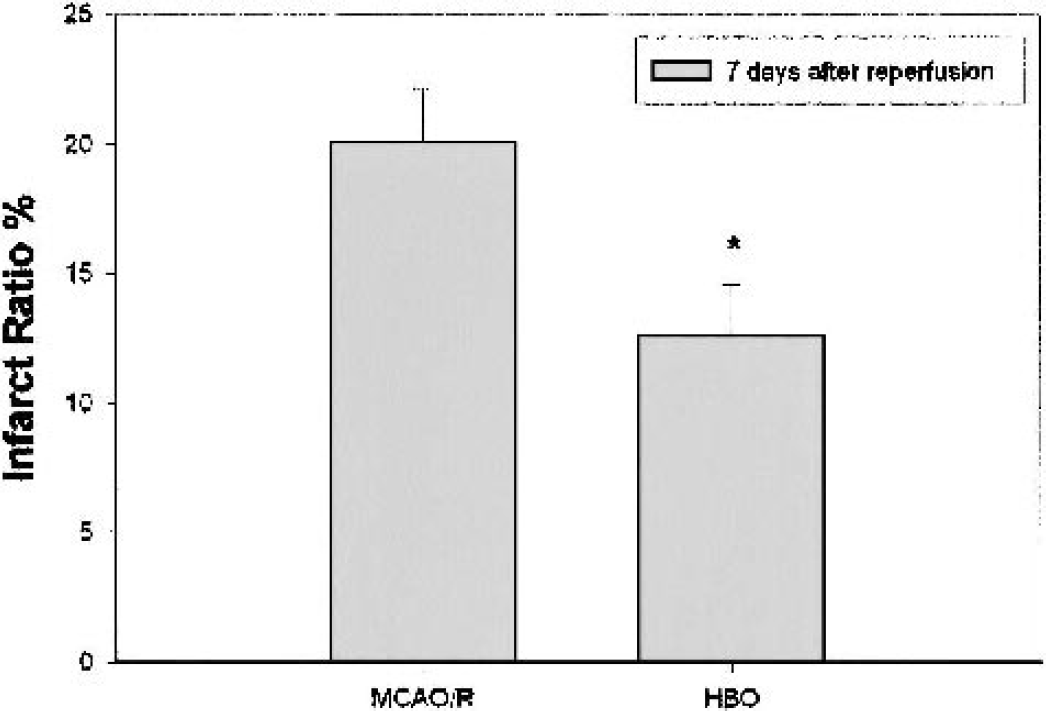
Changes of infarct volume after hyperbaric oxygen therapy (HBO) treatment. The infarct ratio was calculated as the percentage of infarcted tissue per ipsilateral hemisphere. *P <0.05 versus no HBO treatment. MCAO/R, middle cerebral artery occlusion/reperfusion.
DNA fragmentation
To evaluate the presence of apoptosis, DNA fragmentation was assessed in the injured cortex using agarose gel electrophoresis. As shown in Fig. 5, the DNA of injured cortex exhibited nucleosomal fragmentation indicative of apoptosis at 24 h, 48 h, and 72 h after MCAO/R. At 7 days after reperfusion, no DNA ladder was observed in the cortex. No clear DNA ladder was observed in the injured cortex of rats that received HBO, even though some smear patterns are shown indicating necrosis (Ogihara et al., 1999). No detectable DNA fragmentation was found in the sham-operated animals (not shown).
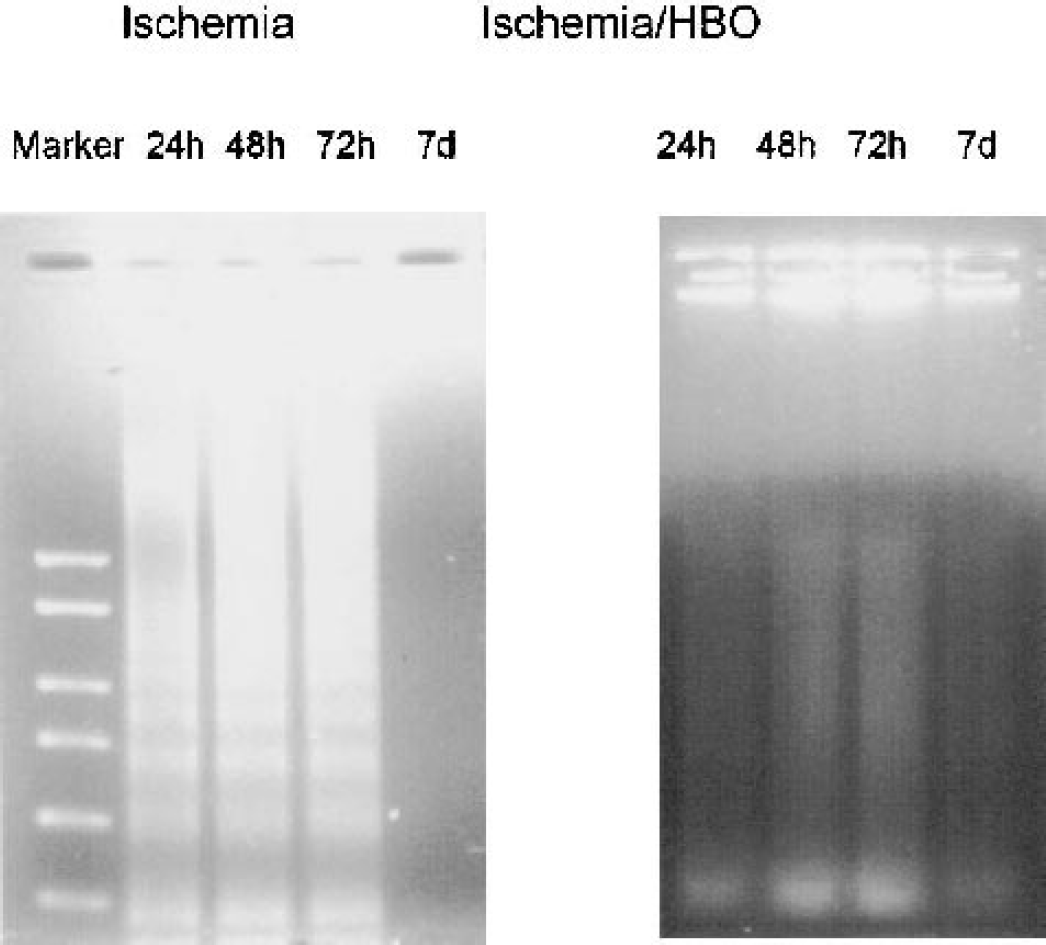
Results of agarose gel electrophoresis. DNA fragmentation was induced at 24 h, 48 h, and 72 h, but not 7 days after ischemic brain injury. No DNA fragmentation was observed after hyperbaric oxygen therapy (HBO) treatment. The left lane shows the molecular weight marker. The same experiment was performed three times with similar results.
Caspase expression
Figure 6 demonstrates that in contralateral cortex (no ischemia) of a MCAO rat reperfused for 48 h, neurons were stained to NeuN (red) but not to caspase-3 (green, insert). In the injured cortex, neurons were stained to both NeuN and caspase-3. Higher magnification shows same cells stained to both NeuN and caspase-3. Color composition indicates that NeuN was stained to the nucleus and caspase-3 around NeuN in the cytosol.
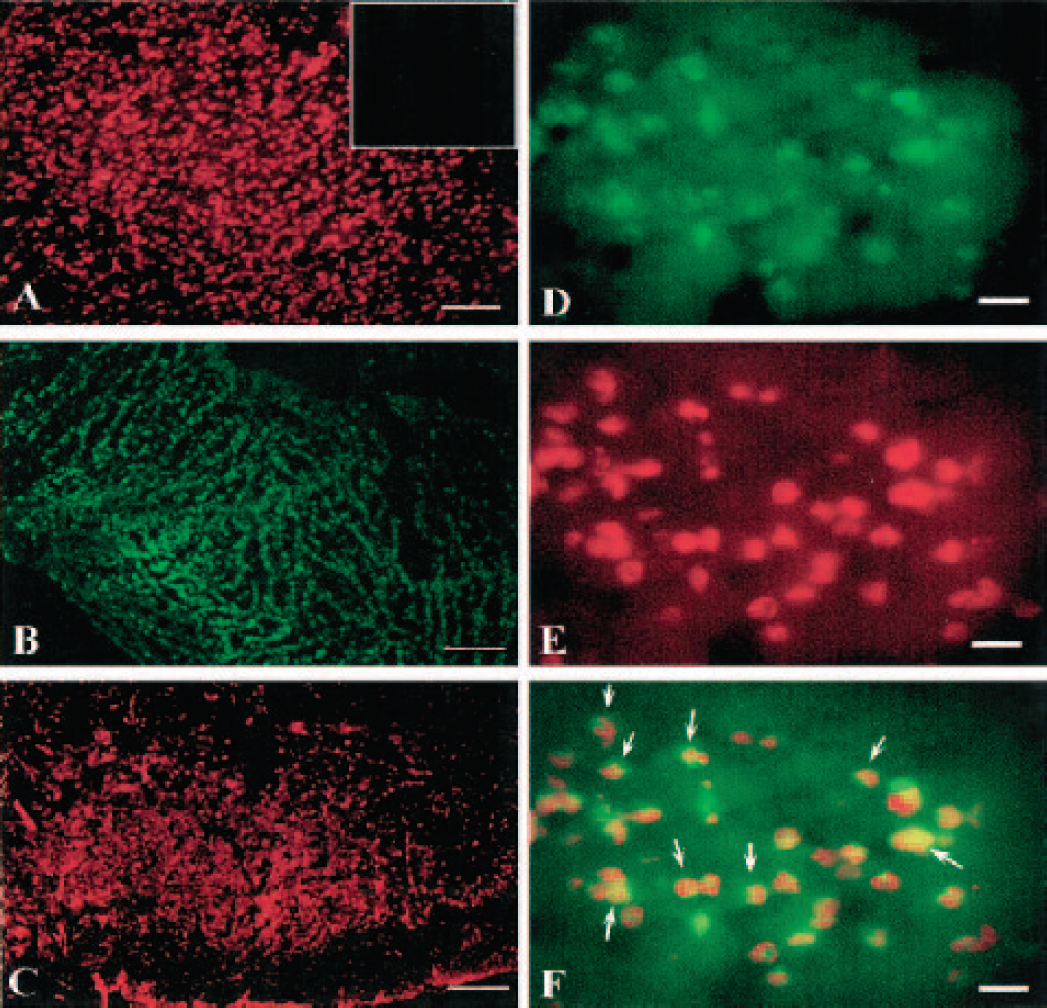
Fluorescent immunohistochemical staining of NeuN (red, Chemicon) and caspase-3 (green, Cell Signaling Tech) in samples collected from a middle cerebral artery occlusion animal killed at 48 h after reperfusion.
TUNEL staining analysis
Intense punctate or diffuse nuclear brown staining of the affected cells identified TUNEL-positive labeling. The TUNEL-positive cells were characterized by a round and shrunken morphology. The nuclei were round, condensed, and frequently fragmented into intensely TUNEL-positive nuclear fragments. The number of TUNEL-positive cells was counted in the ipsilateral cortex by Imaging-Pro Plus for the times 24 h, 48 h, 72 h, and 7 days after reperfusion, respectively. Because this counting by computer might have a degree of false positive, the numbers represent a tendency of apoptotic changes after reperfusion and after HBO treatment only. TUNEL-positive cells occurred mostly at 24 h, 48 h, and 72 h after reperfusion and disappeared at 7 days. Table 1 shows the number of TUNEL-positive cells, and HBO reduced significantly the number of positive cells. Control animals revealed no staining for TUNEL.
Percentages of TUNEL cells
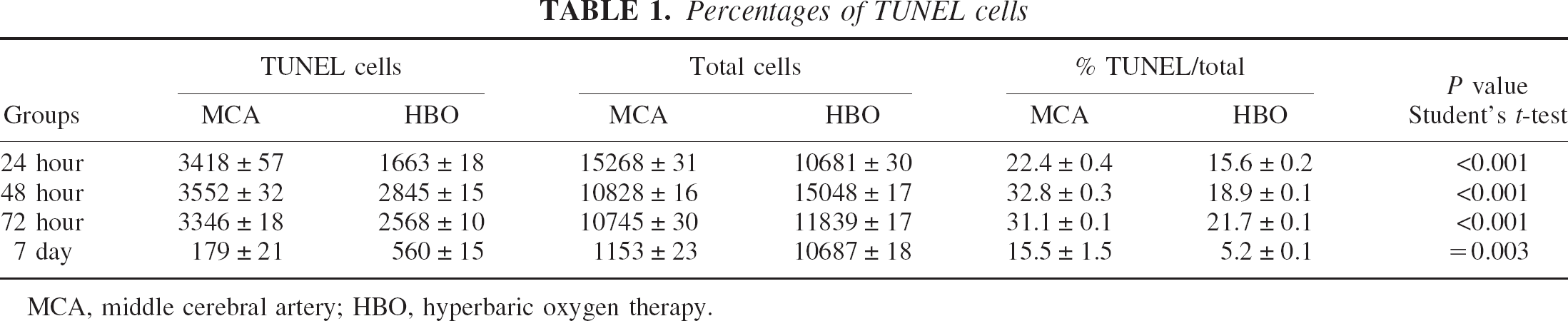
MCA, middle cerebral artery; HBO, hyperbaric oxygen therapy.
Some brain slides were costained with TUNEL and H&E to demonstrate morphologic changes as well as apoptotic changes. Figure 7 shows the infarction area and apoptotic cells in brain samples collected at 24 h after reperfusion. Even though strong staining (brown color) is shown in most cells in the ischemic cortex, the neurons and their processus kept their normal shape (Fig. 7E). At 48 h after reperfusion, the processus begins to disappear and the neuronal body becomes rounded with strong TUNEL staining in the nucleus (Fig. 8E). All cells become rounded in shape and lost their processes at 72 h after reperfusion (Fig. 9E). After 7 days, scar tissues filled ischemic regions with a few cells stained H&E without processus (Fig. 10E).
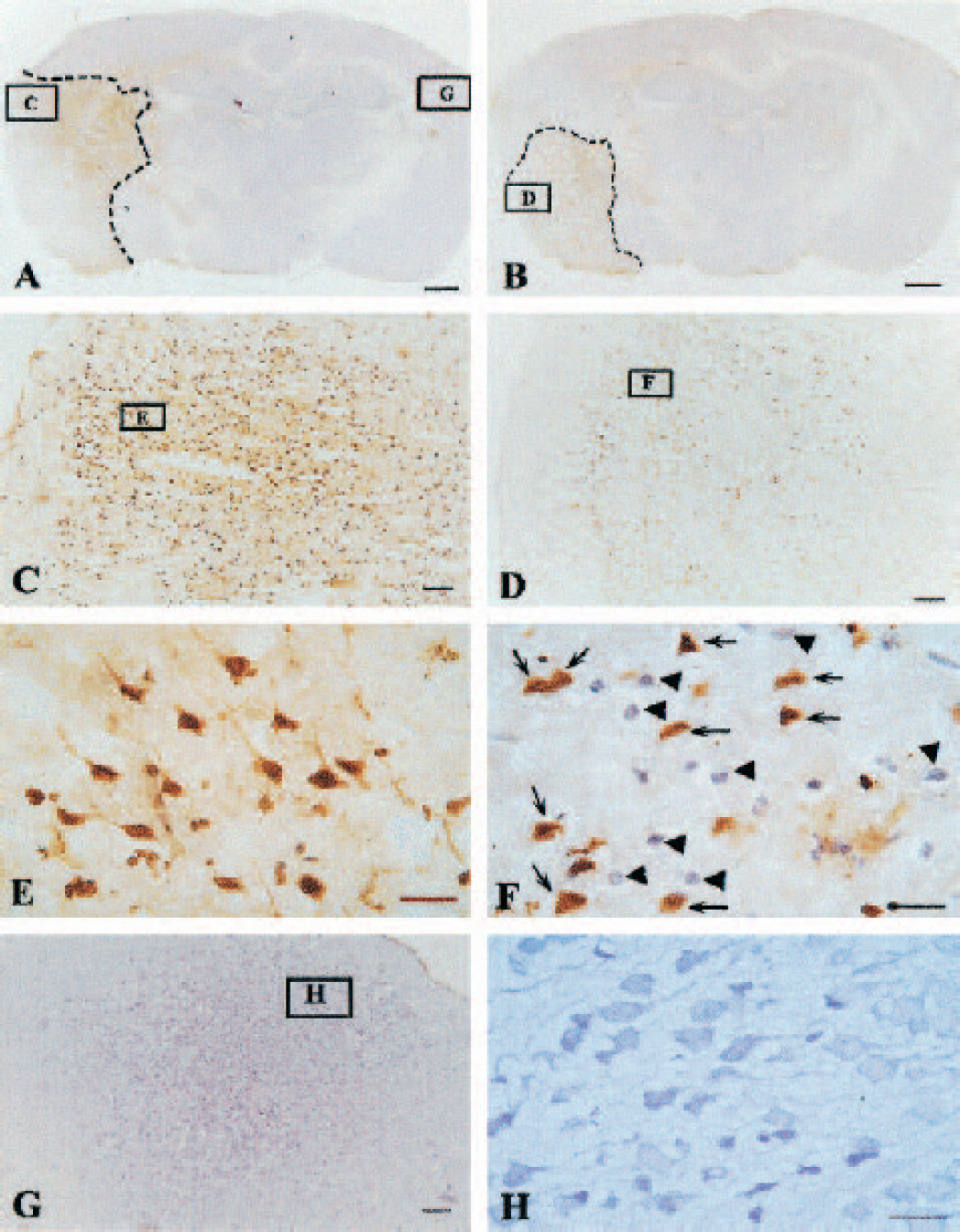
The histologic photomicrographs of brain coronal sections in animals killed at 24 h after middle cerebral artery occlusion/reperfusion (MCAO/R) with or without hyperbaric oxygen therapy (HBO). The slides were costained with terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) and hematoxylin and eosin (H&E).
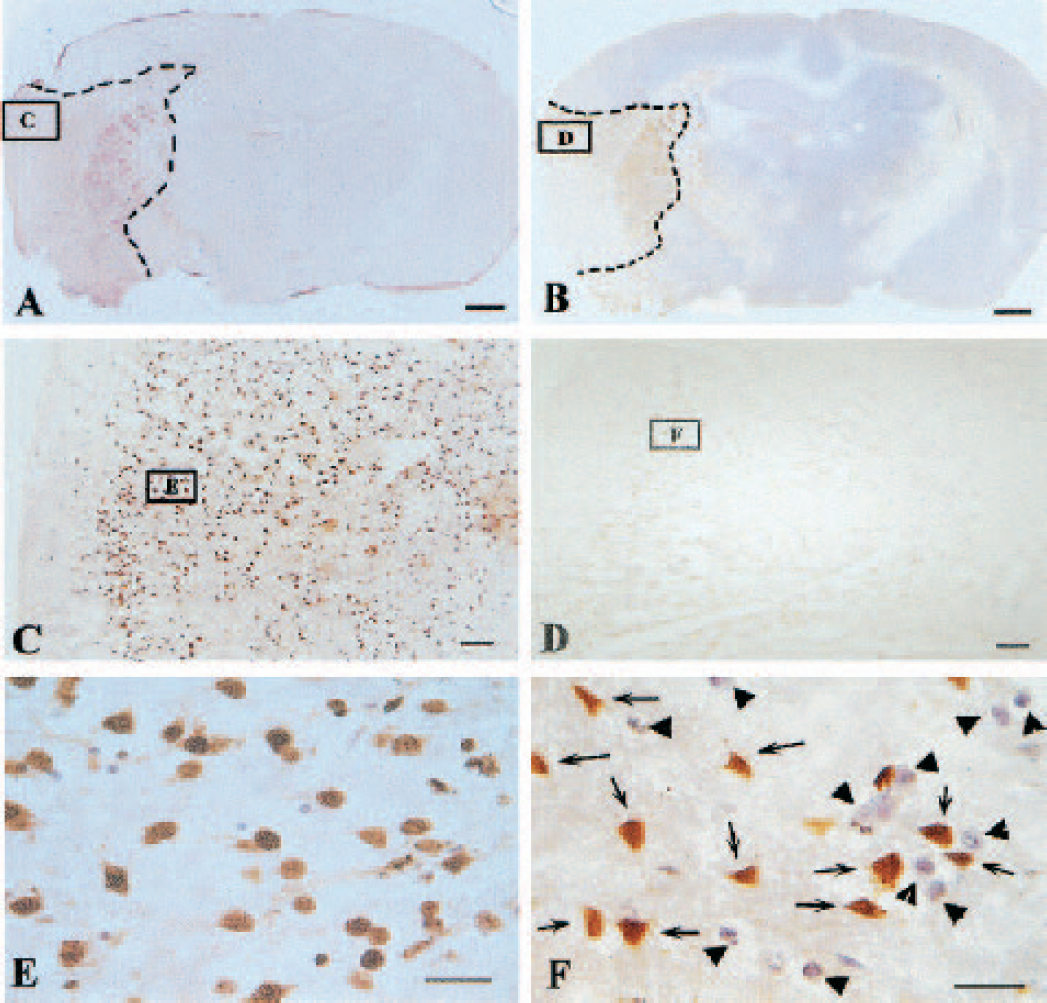
The histologic photomicrographs of brain coronal sections in animals killed at 48 h after middle cerebral artery occlusion/reperfusion (MCAO/R) with or without hyperbaric oxygen therapy (HBO). The slides were costained by terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) and hematoxylin and eosin (H&E).
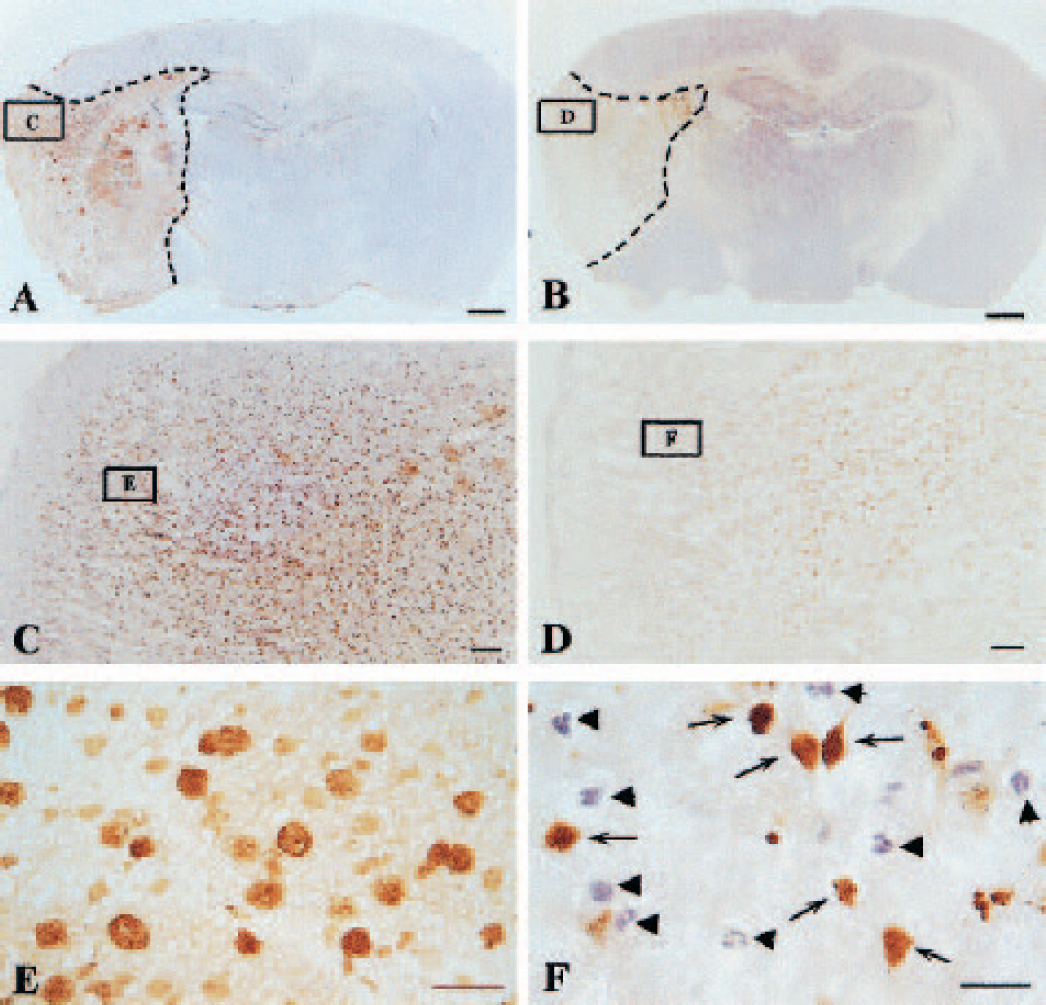
The histologic photomicrographs of brain coronal sections in animals killed at 72 h after middle cerebral artery occlusion/reperfusion (MCAO/R) with or without hyperbaric oxygen therapy (HBO). The slides were costained by terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) and hematoxylin and eosin (H&E).
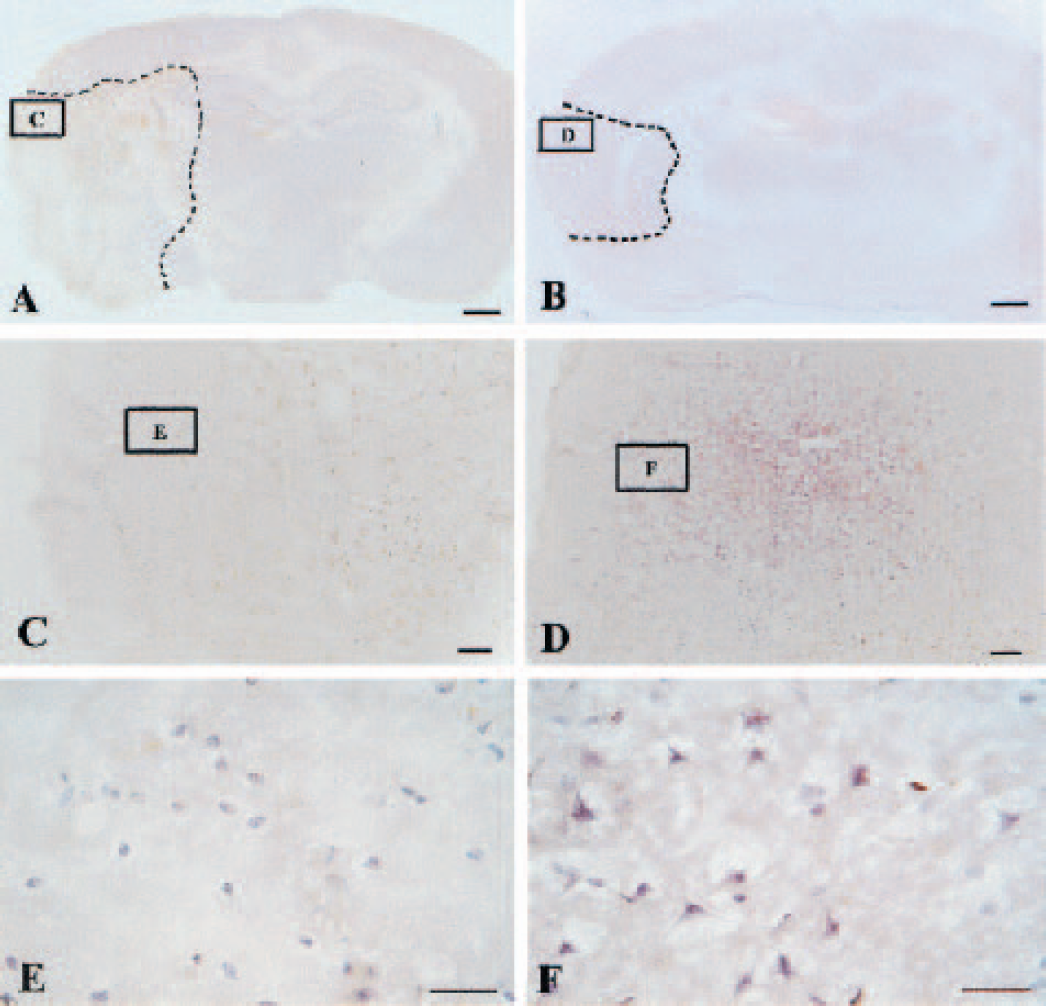
The histologic photomicrographs of brain coronal sections in animals killed at 7 days after middle cerebral artery occlusion/reperfusion (MCAO/R) with or without hyperbaric oxygen therapy (HBO). The slides were costained by terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) and hematoxylin and eosin (H&E).
Hyperbaric oxygen therapy reduced infarcted areas at 24 h (Fig. 7B), 48 h (Fig. 8B), 72 h (Fig. 9B), and 7 days (Fig. 10B); it decreased TUNEL-positive cells at 24 h (Fig. 7D), 48 h (Fig. 8D), and 72 h (Fig. 9D). In the ischemic cortex in animals treated with HBO, a significant number of TUNEL-negative cells exist and stained with H&E (Figs. 7–9F). In samples collected from HBO-treated animals at 7 days, many cells with processes exist in the ischemic region (Fig. 10E).
DISCUSSION
The major finding from our experiments is that HBO treatment reduced neuronal apoptotic death, which probably translated into protective effect against ischemic damage and decreased cerebral infarction. The effect of HBO on apoptosis was confirmed by DNA ladder and TUNEL staining in the ischemic cerebral cortex. A delayed HBO was used in the present study as an additional or alternative therapy for patients with acute stroke. The only effective treatment for acute stroke tissue plasminogen activator (tPA) can be used only in a small percentage of patients admitted into the hospital within 3 to 6 h after the onset of stroke, because the possibility of hemorrhagic transformation increases (Hamann et al., 1999; del Zoppo et al., 2000).
A rat filament occlusion and reperfusion model was used because it is a widely accepted model of reversible focal ischemia (Kittaka et al., 1997; Sharp et al., 2000). This MCAO/R rat model has been confirmed by other investigators (Singhal et al., 2002a,b) and by our previous studies (Badr et al., 2001b; Yin et al., 2002) as a reliable model for producing neurologic deficits for brain injury. The cerebral infarct and neurologic deficits after ischemia/reperfusion and the improvement after HBO are consistent with our previous publications (Badr et al., 2001a, 2001b; Yin et al., 2002).
Apoptosis in cerebral ischemia
In focal cerebral ischemia, reduction of blood flow is generally greatest in the core or center of the area supplied by the occluded vessel. Less severe but clinically important ischemia occurs in the border regions termed the penumbra (Ginsberg and Busto, 1989; Pulsinelli, 1992; Ginsberg and Pulsinelli, 1994; Pulsinelli et al., 1997). Because cerebral energy metabolism is almost entirely aerobic and the brain has only minor reserves of energy-rich compounds, bioenergetic failure occurs rapidly in the severely hypoperfused ischemic core (Katsura et al., 1994; Siesjo et al., 1995). In contrast, cell survival or death in the potentially salvageable penumbra is not only a function of residual blood flow but also the result of various cellular and molecular events trigged by ischemic stress (Hossmann, 1994). In the absence of reperfusion, the core undergoes pannecrosis, and the penumbra sustains varying degrees of injury ranging from nonselective neuronal death to pannecrosis (Neumar, 2000).
Apoptosis is an active biologic process of eliminating unwanted cells involved in the regulation of cell number under physiologic and pathologic conditions (Savitz and Rosenbaum, 1998). However, apoptosis plays an important role in the pathologic factors of delayed neuronal damage, especially in the penumbra after cerebral ischemia (Rink et al., 1995; Nicotera et al., 1997; Jiang et al., 2002). It has been shown previously that apoptosis occurs from 18 h to 3 or 4 days after ischemia (Ferrand-Drake and Wieloch, 1999; Ferrand-Drake, 2001; Culmsee et al., 2001). In the present study, using the MCAO/R rat model, we have found apoptotic changes in the injured cortex between 24 and 72 h after reperfusion. Caspase 3 expression, DNA fragmentation assay, and TUNEL staining support this observation. Apoptotic changes disappeared in samples 7 days after reperfusion in this model DNA ladder and TUNEL-positive cells were all almost eliminated. Our observation is consistent with a previous report that there was no evidence of apoptosis at 7 to 28 days after focal cerebral ischemia (Sugawara et al., 2000). For this reason, we did not conduct experiments beyond 7 days after reperfusion.
Effect of hyperbaric oxygen therapy on apoptosis
The rationale of using HBO in cerebral ischemia is to increase the oxygen delivery to the ischemic tissues (penumbra) and to prolong the functional activity of these tissues (Mink and Dutka, 1995; Nighoghossian and Trouillas, 1997; Chuba et al., 1997). In our previous studies, we have found that HBO reduces infarct volume and improves neurologic recovery in MCAO/R rats (Badr et al., 2001a). hyperbaric oxygen therapy modulates brain metabolites (Badr et al., 2001b) and reduces COX-2 expression in the penumbra regions (Yin et al., 2002). Our hypothesis generated from these previous studies is that HBO increases oxygen levels in the ischemic brain regions (Sunami et al., 2000), decreases hypoxia, and reduces cerebral infarct by reducing apoptosis in the ischemic regions. The experimental results from the present study supported this hypothesis that MCAO/R produces apoptosis and HBO reduced apoptotic changes. Abolition of DNA ladders and TUNEL-positive cells by HBO confirmed the action of HBO on apoptosis. This antiapoptosis effect may be one of the important molecular mechanisms of HBO-induced brain protection and may promote neurologic improvement.
The mechanism responsible for HBO-induced antiapoptotic effect is not clear and beyond the scope of the present study. Hyperbaric oxygen therapy increases tissue oxygen delivery, especially to areas of diminished flow, enhances neuronal viability, reduces brain edema, improves the integrity of the blood–brain barrier, and regulates postischemia metabolism (Nighoghossian and Trouillas, 1997; Veltkamp et al., 2000). Hyperbaric oxygen therapy improves brain metabolites (Badr et al., 2001b), reduces the elevated expression of COX-2 (Yin et al., 2002), inhibits the function of neutrophil β-2-integrin, a molecule involved in leukocyte adhesion and reperfusion injury, and reduces neutrophil sequestration (Atochin et al., 2000). Buras (Buras, 2000; Buras et al., 2000) suggested that HBO might affect many of the components involved in ischemic–reperfusion injury (in peripheral tissues) including polymorphonuclear leukocyte function, endothelial cellular adhesion molecules (CAM) expression, nitric oxide production, nitric oxide synthase expression, cellular energetics, lipid peroxidation, and microvascular blood flow. All of these mechanisms may contribute to the reduction of apoptosis after HBO in the ischemic penumbra. The possible apoptotic pathways such as caspases, bcl-2 family, and poly (ADP-ribose) polymerase (PARP) involved in the effect of HBO remain to be determined. We postulate that HBO interrupts apoptotic signals or biochemical cascades and reduces cerebral infarct after ischemia and reperfusion. If HBO reduces apoptosis, especially when applied at 6 h after reperfusion (8 h after initial ischemia), HBO might be used as alternative or additional therapy for patients who have had acute stroke, because no effective treatment is available and the current Food and Drug Administration–approved therapy tPA needs to be administered within 3 to 6 h after stroke.
Possible side effect of hyperbaric oxygen therapy
The side effect of pressured oxygen therapy has been noted especially for high pressure (>3 ATA) and long duration. It has been shown that HBO (4.96 ATA for 1 h) administered to rats can cause CNS toxicity (Blenkarn et al., 1969). A previous study has shown that exposing rats to 4 ATA of oxygen for 90 minutes was associated with an increased level of lipid peroxidation product, and altered enzymatic antioxidation (glutathione peroxidase) in the brain (Pablos et al., 1997). High level of HBO reduced cerebral blood flow, possibly by reducing NOS (Demchenko et al., 2000). Chavko et al. (2001) stated that 100% O2 at 5 ATA induced seizures. Long-term use of HBO results in adverse effects because of the onset of oxygen toxicity as manifested by the induction of lipid peroxidation and seizures (Nighoghossian and Trouillas, 1997).
At lower pressure, Mink and Dutka (1995) found that HBO (2.8 ATA for 75 minutes) was not associated with an increase of lipid peroxidation or with a reduction of neurophysiologic recovery, despite increased amounts of oxygen free radicals in the brain in a global cerebral ischemic model in rabbits. The authors suggested that HBO applied immediately after global cerebral ischemia does not promote early brain injury. This report is supported by other studies using similar models (Sunami et al., 2000). Thus, the Committee of the Undersea and Hyperbaric Medical Society recommends that a treatment pressure only from 2.4 to 3.0 ATA should be used at the lowest effective pressure to avoid O2 convulsions (Chavko et al., 2001). Our treatment protocol of using 2.5 ATA is similar to that of Mink and Dutka (1995), and our protocol may not enhance lipid peroxidation in the injured brain tissues.